")
Back to Journals » International Journal of Nanomedicine » Volume 13
Tumor-targeting delivery of herb-based drugs with cell-penetrating/tumor-targeting peptide-modified nanocarriers
Authors Kebebe D , Liu Y , Wu Y, Vilakhamxay M, Liu Z, Li J
Received 9 November 2017
Accepted for publication 24 January 2018
Published 9 March 2018 Volume 2018:13 Pages 1425—1442
DOI https://doi.org/10.2147/IJN.S156616
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Dereje Kebebe,1–4 Yuanyuan Liu,1–3 Yumei Wu,1–3 Maikhone Vilakhamxay,1–3 Zhidong Liu,1–3 Jiawei Li1–3
1Tianjin State Key Laboratory of Modern Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China; 2Institute of Traditional Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China; 3Engineering Research Center of Modern Chinese Medicine Discovery and Preparation Technique, Ministry of Education, Tianjin University of Traditional Chinese Medicine, Tianjin, China; 4School of Pharmacy, Institute of Health Sciences, Jimma University, Jimma, Ethiopia
Abstract: Cancer has become one of the leading causes of mortality globally. The major challenges of conventional cancer therapy are the failure of most chemotherapeutic agents to accumulate selectively in tumor cells and their severe systemic side effects. In the past three decades, a number of drug delivery approaches have been discovered to overwhelm the obstacles. Among these, nanocarriers have gained much attention for their excellent and efficient drug delivery systems to improve specific tissue/organ/cell targeting. In order to enhance targeting efficiency further and reduce limitations of nanocarriers, nanoparticle surfaces are functionalized with different ligands. Several kinds of ligand-modified nanomedicines have been reported. Cell-penetrating peptides (CPPs) are promising ligands, attracting the attention of researchers due to their efficiency to transport bioactive molecules intracellularly. However, their lack of specificity and in vivo degradation led to the development of newer types of CPP. Currently, activable CPP and tumor-targeting peptide (TTP)-modified nanocarriers have shown dramatically superior cellular specific uptake, cytotoxicity, and tumor growth inhibition. In this review, we discuss recent advances in tumor-targeting strategies using CPPs and their limitations in tumor delivery systems. Special emphasis is given to activable CPPs and TTPs. Finally, we address the application of CPPs and/or TTPs in the delivery of plant-derived chemotherapeutic agents.
Keywords: cancer, nanocarriers, cell-penetrating peptide, targeting drug delivery, herb-based drug, tumor targeting
Introduction
Despite great advancements in novel drug delivery systems, cancer nevertheless remains one of the main causes of morbidity and mortality worldwide. In 2015, cancer was accountable for 8.8 million deaths. Globally, for every six deaths, one is due to cancer. Within the coming two decades, this is estimated to increase by about 70%. In addition, the economic burden due to cancer has been significantly increasing. In 2010, the overall annual economic cost of cancer was estimated at approximately US$1.16 trillion.1
Chemotherapy, radiation, and surgery with chemotherapy are the major treatment protocols for cancer. Utilization of chemotherapy has been demonstrated to enhance the survival rate of patients with malignant cancer slightly. Surgery and radiotherapy are the best treatments for local tumors and nonmetastatic cancers, and they are also helpful in cancer that has not been disseminated throughout the body. Therefore, chemotherapy is the treatment of choice for metastatic malignancies, since they are well distributed to every organ in the body.2 However, most conventional chemotherapeutic agents currently in clinical use are limited, due to several undesirable properties, including poor solubility and bioavailability, rapid elimination from systemic circulation, narrow therapeutic index, and unselective site of action after intravenous/oral administration and cytotoxicity to normal tissues, which can be the main obstacles to success of cancer therapy.3
Furthermore, chemotherapeutic agents are often unable to penetrate and reach the internal parts of solid tumors, leading to inefficient cytotoxicity to cancerous cells.4 The other limitation of traditional chemotherapy is associated with Pgp, a multidrug-resistant (MDR) protein that is overexpressed in cancer cells, acts as an efflux pump and decreases accumulation of drug inside the tumor, and plays a key role in the development of resistance to anticancer drugs. Therefore, the drugs administered remain unsuccessful in exerting the desired effect.3,5 Though different drug delivery approaches consisting of antibodies, hormones, and growth factors have been designed and administered, significant internalization the cancerous cells cannot be achieved, due to the reticuloendothelial system and intracellular enzymes.6 Consequently, in recent decades, much work has been devoted to overcoming the major drawbacks of conventional cancer chemotherapy.
Nanocarrier-based drug delivery systems have been shown to have great potential in reducing the aforementioned problems of cancer therapy.7–13 Moreover, decorating nanocarrier surfaces with different ligands targeting specific receptors that are overexpressed on tumor cells has been exploited for active targeting of chemotherapeutic agents to cancer cells.14
Most importantly, nanocarriers functionalized with cell-penetrating peptides (CPPs) and/or tumor-targeting peptides (TTPs) have been a highly promising strategy and attracted the attention of researchers. These peptides are very advantageous, as they efficiently deliver a broad variety of cargo intracellularly and extracellularly.15–17 CPP-mediated drug delivery is achieved either by the formation of stable, noncovalent complexes or by a covalent bond with the cargo.16 However, most of the first-generation CPPs suffered from a lack of cell-type specificity and in vivo stability, which led to the emergence of activable CPPs (ACPPs) and TTPs. In this review, we focus on recent advances in CPPs and TTPs, and the tumor-targeting approach of herb-based anticancer bioactive substances using CPP- and/or TTP-modified nanocarriers.
Overview of nanocarrier-based tumor-targeting drug delivery approaches
Targeted cancer therapy is viewed as an irreplaceable component of current anticancer drug development.18 The best strategy to improve the efficacy and reduce the toxic effects of an anticancer drug is delivering it to a specific target and keeping sufficient concentrations for enough time, which allows the drug to produce the desired therapeutic effect. Cancer cells can be targeted by two approaches: passive targeting and active targeting.
Passive targeting involves the extravagation of drug formulations through leaky vasculature/capillaries of tumors that result from abnormal angiogenesis at sites of tumors, resulting in accumulation and retention. This phenomenon is known as the enhanced permeability and retention effect3,19 (Figure 1). In addition, the release of drug from pH-sensitive formulations in the acidic microenvironment inside the cancer cell,6,20 phagocytosis of the particulate carrier by mononuclear phagocytosis systems, and privileged localization in the organs of the reticuloendothelial system are also considered passive targeting.21
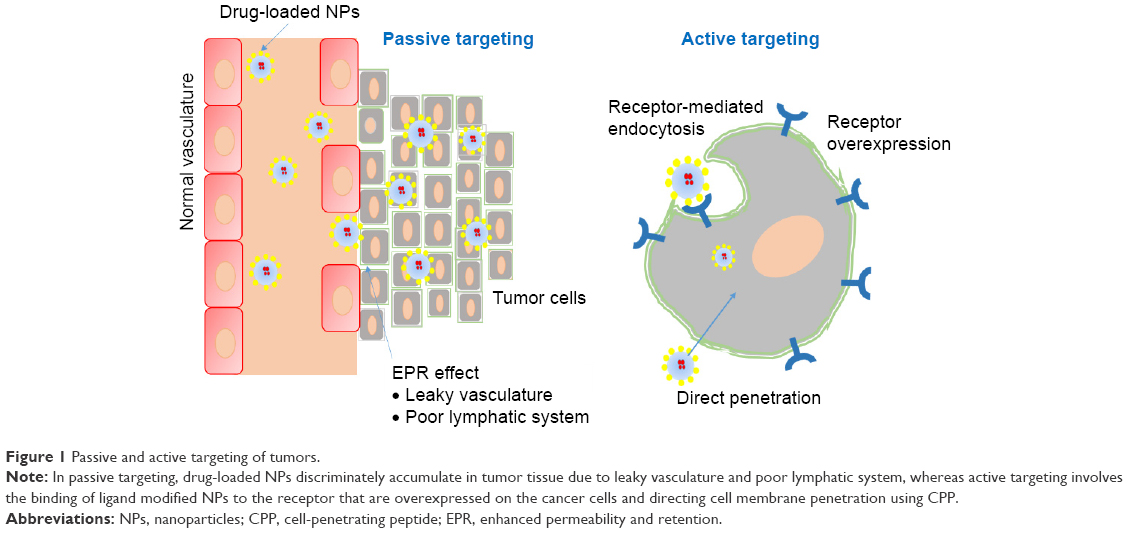 | Figure 1 Passive and active targeting of tumors. |
Nanoparticle (NP)-based drug delivery plays a crucial role in the passive targeting of tumors.22 NPs possess unique properties, such as large surface area, small size, high efficiency in encapsulating a wide variety of drugs, and modifiable surface chemistry. These benefits make NPs a potentially superior treatment approach to traditional cancer therapies. The size of drug carriers and the abnormal and permeable vasculature of tumors are the base for passive targeting.23 Most tumors manifest an abnormally dense and leaky vasculature formed via stimulation by VEGF. In normal vasculature, particles >2 nm are prevented from crossing through endothelial cells due to tight junctions.24 However, in tumor vasculature, tight junctions and the basement membrane are disordered, allowing passage of particles of 10–200 nm through the leaky neovasculature of the tumor and then retention in the tumor. Furthermore, the poor venous and lymphatic system in the tumor creates an opportunity for the NPs to accumulate therein at high concentrations for a long time.3 The size and surface charge of NPs are known to affect the half-life and biodistribution of NPs significantly. Larger NPs (>100 nm) are usually cleared from the circulation by phagocytosis. Similarly, very small NPs (<10 nm) have a high rate of clearance. The positive charge on the surface of particles is known to assist internalization into cancer cells. Moreover, surface modification of NPs with such polymers as polyethylene glycol (PEG) can increase the circulation time of particles via inhibition of clearance by the reticuloendothelial system and increase the accumulation of NPs at tumor sites but can affect cellular uptake by cancer cells.25
In addition to targeting potential, nanocarriers offer protection of the drug from degradation, decrease renal elimination, increase its half-life in the circulation, allow controlled release kinetics, and improve solubility. Furthermore, NPs have known to have the potential to bypass MDR mechanisms by various approaches.2,10,26
Several types of nanocarrier have emerged for the drug delivery of chemotherapeutic agents, including magnetic and metallic NPs, such as iron oxide or gold NPs, silver NPs, nanodiamonds, carbon-based structures (graphene sheets and carbon nanotubes), polymeric NPs, dendrimers, quantum dots, hydrogel-based delivery systems, silica-based NPs, lipid-based NPs (liposomes), solid-lipid NPs, nanostructured lipid carriers (NLCs), viral NPs, and hybrid NPs.27–31
Passive targeting of almost all nanocarriers occurs with poor selectivity and insufficient tumor cell uptake. A more advanced approach of targeting for oncology applications is the modification of the surface of NPs with specific tumor-homing ligands.32 The ligands are known to bind to receptors that can be overexpressed on the surface of a cancer cell. Ligands with selective affinity for a specific receptor or molecule differentially expressed at the target site are presented on the surface of nanocarriers, resulting in selective accumulation and cellular uptake at the site of action.33 This strategy significantly increases accumulation and retention of NPs in the tumor vasculature and specific and successful internalization by target tumor cells, which is known as active tumor targeting.34 Ligands that have been used to modify NPs include monoclonal antibodies, folic acid, hyaluronic acid, albumin, vitamins (folate, vitamin B12, thiamine, and biotin), transferrin, lectins, aptamers, and peptides.6,21,23,35
Most recently, peptide-based targeting ligands have been attracting the attention of researchers and are the main concern of this review. CPPs provide several advantages over other ligands.36 Peptide ligands are highly selective for target tissue/cells, and multiple ligands can be conjugated to a single drug carrier to offer multivalent conjugation, thus increasing binding affinity to the target.37 CPP details are presented in the following section.
CPP/TTP-based drug delivery
CPPs and classification
A recent advancement in the field of molecular biology that appears to have had an enormous impact on cancer therapy is the innovation of the tissue- or cell-penetration system.15,38 CPPs are short peptides that are able to cross through tissue and cell membranes in an energy-dependent or -independent manner and are used to transport a wide variety of bioactive conjugates (cargoes), including proteins, peptides, DNA, siRNA, small drugs, fluorescent compounds, NPs, and other substances, into cells.39 CPPs are extremely beneficial, because they are biocompatible and the peptide sequence can be altered to fine-tune hydrophobicity, affinity, charge, solubility, and stability. They can also be readily synthesized in sufficient quantity.17
Besides the use of CPPs as inert vectors for the transport of cargo molecules, dual-acting CPPs that have properties of both cell permeation and bioactivity have been emerging. Selected CPPs are known to produce bioactivity, including neuroprotecting effects,40 induce apoptosis in cancer cells,41,42 and suppress breast tumorigenesis.43 More recently, a CPP obtained from azurin (p28) was found to prevent phosphorylation of VEGFR2, FAK, and Akt, leading to inhibition of tumor growth and angiogenesis.44
CPPs can be conjugated to cargoes by either noncovalent complex formation or covalent bonds. Covalent conjugation of a CPP may be obtained chemically through disulfide bonds, amine bonds, or specific linkers that enable the release of the cargo when internalized into the cell.42 However, the possibility of changing the bioactivities of the conjugates is the main risk of the covalent conjugation of CPPs.45 Furthermore, covalent methods are associated with such problems as lack of suitable reactive groups on the polymer, unstable intermediates, and inefficient coupling and purification. In these cases, the noncovalent approach seems more appropriate.46 The noncovalent complex is formed by electrostatic and/or hydrophobic interactions between positively charged CPPs and negatively charged drug carriers.47
CPPs can be classified by their origin, function, sequence, or mechanism of uptake. According to their physicochemical properties, CPPs can be cationic, hydrophobic, or amphiphilic. Cationic CPPs have highly positive net charges at physiological pH. They originate from the basic short strands of arginines and lysines, eg, TAT48–60 (GRKKRRQRRRPPQ), penetratin (RQIKIWFQNRRMKWKK), and DPV1047 (VKRGLKLRHVRPRVTRMDV). Hydrophobic CPPs primarily contain apolar residues, which have amino-acid groups that are vital for cellular uptake, along with a low net charge, eg, Pep7 (SDLWEMMMVSLACQY) and C105Y (CSIPPEVKFNKPFVYLI) are some of the hydrophobic peptides. The third class of CPPs is the amphiphilic CPPs, which contain both hydrophilic and apolar hydrophobic regions of amino acid sequences. Pep1 (KETWWETWWTEWSQPKKKRKV) and pVEC (LLIILRRRIRKQAHAHSK) are typical examples. Based on their origin, peptides can be classified as derived CPPs, chimeric CPPs, and synthetic CPPs.48,49 Detailed reviews on CPP classification can be found elsewhere.39,49,50
Limitations of CPPs
Despite the great potential of CPPs as carriers for drug delivery to cancer, most conventional CPPs suffer from a number of limitations, including lack of cell specificity and in vivo instability, which are the main obstacles to their further clinical development.51
Lack of cell specificity
In vivo applications of CPP are limited because of their aspecific tissue/cell penetration. To date, several approaches have been suggested to target cancer cells selectively using CPPs. Among these, ACPPs and the combined use of CPPs with TTPs are discussed in the following sections.
In vivo degradation
In vivo stability should be taken into account for CPPs to be beneficial as vectors for the delivery of any therapeutic agent. Due to their peptidic nature, CPPs are degraded both extracellularly and intracellularly by proteases.52,53 In vivo CPP stability depends on factors that influence proteolytic cleavage, including amino acid sequence, conformation, chemistry (unnatural amino acids, D-isomers, chemical bridges, amino acid modifications), routes of administration, type of conjugate cargo, and method of conjugation of CPPs with this cargo.53
Several strategies have been employed to improve the in vivo stability of CPPs. One approach is the synthesis of retro inverso (RI) CPPs, introduced by Chorev and Goodman.54 RI CPPs are peptides consisting in D-amino acids in the reverse sequence of the naturally occurring L isoforms. Subsequently, RI transformation has extensively been used as an approach of producing a proteolytic resistant peptide analogues.55 However, RI peptides are reported to cause cytotoxicity, decline in metabolic activity, morphological changes, and induction of apoptosis.56 In addition, end-to-end cyclization of CPPs have been proposed to attain improved metabolic stability. End-to-end cyclization is known to offer greater proteolytic resistance and higher internalization efficiency as a result of improved structural or conformational rigidity/stability. Cyclization through an intramolecular disulfide bond has also been reported to improve in vivo stability.57,58
Moreover, the half-life of CPPs can be prolonged via backbone stabilization by the inclusion of β- and γ-peptide residue in the CPP sequence. In addition to increasing chemical stability, backbone stabilization may also enhance physical stability.42,59,60 Furthermore, there have been a number of strategies suggested to improve the stability of CPPs, and the reader is referred to a review done by Reissmann.60
Toxicity and immunogenicity
Although CPPs are generally considered safe in terms of toxicity and immunogenicity at effective concentrations,61,62 though toxic and immunoinducing effect of some CPPs have been reported. Verdurmen and Brock reviewed CPPs and drug carriers and found a variety of side effects that might have been related or unrelated to their mechanisms of internalization.63 The toxicity of CPPs depends mainly on peptide concentration, type of cargo molecule, and conjugation strategy.64 Moreover, amphiphilic CPPs are known to be more toxic than cationic ones.65
Despite some in vitro reports on cytotoxic effects of CPPs, it is difficult to compare results from different studies. However, in one comparative study by El-Andaloussi et al on delivery efficiency and cytotoxicity of three well-known CPPs – Tat, TP10, and penetratin – were characterized using different cargoes. The result of this study demonstrated that the cytotoxicity of these peptides was cargo dependent. In addition, the extent of cytotoxicity and internalization of TP10 varied significantly depending on the position of cargo coupling within the peptide. Among the three peptides, TP10 exhibited long-term toxic effects in HeLa and CHO cells at 20 μM. Penetratin and TAT had no effect on membrane integrity, whereas TP10 induced LDH leakage (~20%) at 10 μM in HeLa cells.64 On the other hand, Suhorutsenko et al evaluated cytotoxic and immunogenic responses of TP10 and its chemically modified analogues – PepFect peptides (PF3, PF4, and PF6) – in monocytic leukemia and peripheral blood mononuclear cell lines. All peptides were found to be nontoxic and nonimmunogenic in-vitro at concentration of 10 μM and 5 μM, and in-vivo at a dose of 5 mg/kg.66
Kilk et al conducted a metabolomic approach to a CPP toxicity study on five representatives of the most common CPPs: transportan (TP), penetratin, HIV TAT-derived peptide, nona-arginine (R9), and model amphiphilic peptide. The study showed that the intracellular metabolome was mostly affected by TP, followed by HIV TAT-derived peptide and model amphiphilic peptide. Only minor changes were observed with penetratin or R9 treatment. The cells recovered from 5 μM TP treatment, but no recovery was observed at higher concentrations. This metabolomic study demonstrated that TP affected cellular redox potential and depleted energy and pools of purines and pyrimidines.67
In general, few in vivo studies are available on toxic and immunological responses of CPPs. One in vivo toxicity study of CPPs and ACPPs showed that systemically administered CPPs (R9) resulted in acute toxicity in mice at a dose fourfold lower than MMP-cleavable ACPPs. The study also reported that CPPs bound to the vasculature at the site of injection, then redistributed, mostly to the liver. However, ACPPs were distributed among tissue more broadly, showed prolonged circulation, were eliminated by both renal and hepatobiliary routes, were less toxic, and enabled targeting of tumor-expressing enzymes that cleaved the linker.68
An in vivo study of immunological response to delivery of a p38 siRNA using the penetratin and HIV-TAT to mouse lungs was conducted by Moschos et al.69 The study showed that penetratin coupled with siRNA induced an innate immunoresponse, but siRNA coupled with HIV-TAT and penetratin (not complexed with siRNA) did not. Similarly, an in vitro study reported more recently by Carter et al showed that HIV-TAT, Antennapedia and TP failed to induce a significant increase in the release of the inflammatory cytokines IL8 and IL6 in epithelial cells.70
The presence and origin of cargo molecules in CPP–cargo conjugates also influences toxicity and may exert a different effect from that produced by the peptide alone. Research has indicated that toxic effects may be derived from conjugation of different cargoes to CPPs.65 For example, penetratin alone and TAT coupled with siRNA do not cause an immunoreaction, but when penetratin is conjugated to siRNA, the complex does provoke an innate immunoresponse.69 The conjugation of carboxyfluorescein to CPPs is known to increase CPP cytotoxicity.64 Cardozo et al showed that the cytotoxicity of TAT48–57 and penetratin was higher for CPP conjugates with larger cargo peptides than for those unlabeled or with smaller cargo.71 In addition to CPP–cargo conjugates, nanomaterials used as carriers elicit an immunoresponse and cytotoxicity.72
Activable CPPs
The lack of cell specificity of CPPs is mainly due to their electrostatic interaction. Generally, cationic CPPs bind to anionic components of the plasma membrane, and this is principally accountable for their membrane-transporting properties.73 To overwhelm the problem of aspecificity, researchers introduced ACPPs. ACPPs are novel targeting agents comprising of a polycationic CPP attached to a neutralizing polyanion unit via a cleavable linker (Figure 2). Adsorption and uptake of CPPs into cells are prevented until the linker is cleaved.74 In ACPPs, stimulus-responsive materials are exploited to stimulate the selective display of CPPs within the pathological environment of a tumor, such as lower pH caused by buildup of lactic acid or overexpression of extracellular matrix development-remodeling proteases, or may be the external application of heat or light to a disease site.75
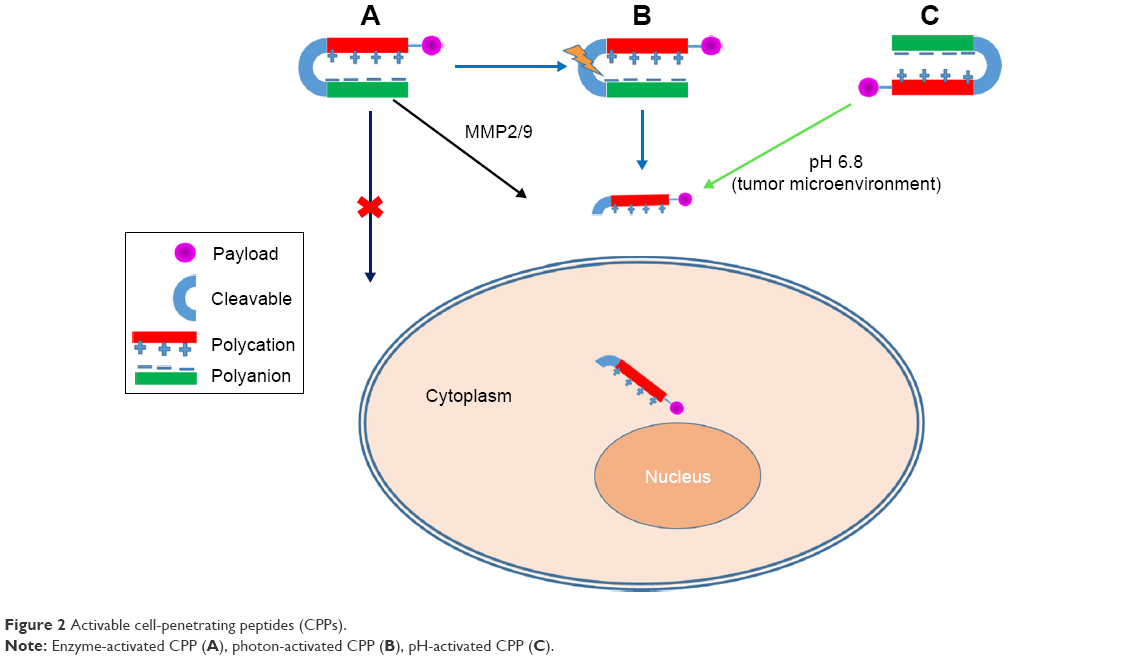 | Figure 2 Activable cell-penetrating peptides (CPPs). |
Acid-activated CPPs
The lower pH of the tumor microenvironment compared with normal tissue can be used as a targeting strategy.76 This approach has been known to improve the intracellular delivery of cargo molecules functionalized with CPPs.50 A number of studies involving different approaches toward acid-activated CPPs for tumor-targeted delivery have been reported. For instance, Jin et al investigated acid-activated peptide by amidizing TAT lysine residue amines to succinyl amides (aTAT), completely inhibiting TAT-aspecific interactions in the blood compartment. The succinyl amides in the aTAT were rapidly hydrolyzed, fully reestablishing TAT functions in acidic tumor tissue or inside cell lysosomes/endosomes. As such, aTAT-functionalized PEG-block-poly(ε-caprolactone) micelles result in long circulation in the blood compartment and efficiently accumulate and transport doxorubicin (Dox) to tumor tissue, offering high antitumor activity and low toxicity to the cardiovascular system.77 Cheng et al also used ACPPs with 2,3-dimethylmaleic anhydride (DMA) as a shielding group functionalized with the anticancer drug Dox to produce a novel prodrug (Dox-ACPP-DMA) for tumor-targeting drug delivery. DMA (shielding group) conjugated to ACPPs through an amide bond between DMA and the primary amines of K6, which was used to inhibit the cell-penetrating activity of the polycationic CPPs (R8) by intramolecular electrostatic attraction at physiological pH 7.4. However, at tumor extracellular pH 6.8, the removal of the shielding group by hydrolysis resulted in charge reversal, activating and restoring the original function of CPPs for better cellular uptake by tumor cells. After cell internalization, drug release in cells was further triggered by overexpressed intracellular proteases.78 In another example, Regberg et al demonstrated that peptides (PF3) modified with a leucine–histidine sequence was found to be a pH responsive CPP. The modified PF3 analogue showed significantly improved cellular bioactivity over the unmodified PepFect.79 Similarly, a tumor-specific pH-responsive peptide H7K(R2)2 (RRK[HHHHHHH]RR) has been reported to have the ability to respond to acidic pH in the tumor microenvironment.80
TH(AGYLLGHINLHHLAHL[Aib]HHIL-NH2) is another promising pH-responsive CPP derived by complete replacement of all lysines in the TK(AGYLLGKINLKKLAKL[Aib]LLIL-NH2) sequence of by histidines. TH does not show cell-penetrating efficacy in normal tissue or during blood circulation because of the neutral pH under those environments. However, at lower tumor environment pH, the cell-penetrating capacity of TH is activated as a result of histidine protonation in TH peptides and conversion of the surface charge of TH from negative to positive. Paclitaxel (Ptx)-loaded TH-modified liposomes show significantly greater inhibition against tumor cell growth than that of PEG liposomes and free Ptx both in vivo and in vitro at pH of 6.3 as compared with at pH 7.4.81
Similarly, Yao et al developed a modified form of TH by the introduction of electron-donating group, such as ethyl, isopropyl, and butyl to the C2 position of histidine TH to form corresponding TH analogues (ethyl-TH, isopropyl-TH, and butyl-TH). The TH analogues formed were conjugated to a camptothecin (Cpt)- and butyl-TH-modified conjugate and displayed a pronounced cytotoxicity effect on cancer cells more strongly in a pH-dependent manner compared to TH and other conjugates.82
Very recently, new pH-controllable CPPs (PCCPPs) were reported on by Lee et al.83 According to this study, synthesized poly-L-lysine-based PCCPPs were capable of undergoing pH-dependent conformational transition, thereby displaying specific cell-penetrating properties at the target site. At a physiological pH, the PCCPPs contained a low helical tendency due to electrostatic attractions between carboxylate and protonated amine groups in each side chain. Therefore, unselective cellular internalization was hindered. In contrast with an acidic environment (pH 5–6), the pH-inactivated motif quickly changed to an intact helical structure, whereby electrostatic interactions between repulsions and attractions were well balanced throughout the side chains. Accordingly, the conformation of PCCP was able change to a helix that possessed greater cell-penetrating properties specifically at cancer sites.
Enzyme-activated CPPs
Specific enzymes, such as proteases, glycosidases, or esterases, are overexpressed in tumor cells, and this fact can be used as a tool for developing tumor-targeting drug delivery.84 Quenching of the cell-penetrating activity of polycationic peptides by electrostatic interactions with the polyanionic domain can block cellular uptake. However, in tumor tissues, MMP2/9 are overexpressed, cleave the substrate, and release polycationic peptides from the polyanionic domain, thereby stimulating cellular adhesion and subsequent uptake of peptides (Figure 2).76,85 Shi et al developed a conjugate of ACPPs with Dox, which is sensitive to MMP2/9, to form ACPP–Dox conjugates consisting of the polycationic domain (CPPs), cleavable MMP2/9-sensitive substrate, the polyanionic domain, and Dox. Activation of ACPP–Dox was found to occur via MMP2/9 in an enzyme-concentration-dependent manner. Flow cytometry and laser confocal microscopy demonstrated that higher cellular uptake of ACPP–Dox occurred with HT1080 cells (overexpressed MMPs) than MCF7 cells (underexpressed MMPs) after enzyme-triggered activation.86
Similarly, Gao et al developed NPs functionalized with ACPPs: EEEEEEEE(E8)-6-aminohexanoyl-PLGLAG-RRRRRRRR(R8) to deliver cargoes effectively to glioma cells and further improve glioma treatment. In the blood, cationic R8 is shielded by E8 via electrostatic forces, and the cell-penetrating capability of R8 is blocked. In the glioma site, PLGLAG is cleaved by MMP2. Therefore, E8 was able to be detached from R8 at the glioma site, leading to a regaining of penetration ability of R8.87
Xia et al also constructed MMP-based activable low-molecular weight protamine (ALMWP; CVSRRRRRRGGRRRR)-functionalized NPs. Ptx-loaded ALMWP NPs demonstrated superior antitumor efficacy over unmodified NPs and LMWP-functionalized NPs.88 Other research has also found that MMP2-based ACPP nanocarriers show a greater cellular uptake, cytotoxicity, tumor targeting, and antitumor efficacy in vitro and in vivo than non-MMP 2-based and free drugs.89
Cancer cells produce high levels of intracellular reactive oxygen species (ROS), such as O2−, H2O2, and HO−, compared to the normal ROS environment. Using this principle very recently, Yoo et al developed novel protease ACPPs containing an ROS-responsive methionine, a cell-permeable lysine chain (poly[L-methionine-block-L-lysine]) (ML), and a MMP-cleavable linker (PLGLAG). The ML–PLGLAG conjugate (MLMP) and Dox were incorporated into the micelle core. MLMP demonstrated MMP-sensitive cleavage and ROS-induced Dox release, while MLMP (Dox) exhibited pronounced tumor inhibition efficiency with no toxicity compared to free Dox. The findings of this study suggesting that dual-stimuli-based delivery systems have great potential in cancer therapy.90
Although MMP-based ACPPs are known to target tumors effectively in vivo, ACPP substrates are not perfectly selective for MMPs, and MMP is not exclusively expressed in tumors. Therefore, in order to improve the targeting potential of ACPPs, Whitney et al explored other classes of proteases and substrates that might offer better specificity and flexibility than MMP-based approaches by using phage display. Accordingly, a unique ACPP (RLQLKL) was identified. This ACPP was not cleaved by MMP or various coagulation factors but was efficiently cleaved by elastases and plasmin, both of which have been shown to be overexpressed in tumor cells.91 Similarly, cathepsin B enzyme-responsive92 and urokinase-type plasminogen activator-based93 ACPPs have been reported for their potential in tumor-specific drug delivery systems.
Photon-sensitive CPPs
The two aforementioned ACPP types involve the activation of CPPs in vivo in tumor tissue. However, another approach is the external illumination of tumors by near-infrared or ultraviolet (UV) light to stimulate dissociation of photosensitive groups from CPP NPs, thus controlling the release of drugs at the tumor site.94 Shamay et al developed photon-sensitive CPPs using polymers bearing light-activated caged CPPs for selective cellular uptake upon UV-light illumination (365 nm), which may offer a promising approach to the delivery of payloads to target cells.95 Lin et al also employed this strategy to take advantage of both photosensitive peptides (PSPs) and asparagine–glycine–arginine (NGR) to develop an siRNA delivery system for the treatment of CD13-overexpressing exterior tumors with the aid of UV illumination. The PSPs consisted of two units: CPPs (CKRRMKWKK) and a photolabile-protective group (Nvoc). Nvoc masked the positive charges of lysine residues on CPPs temporarily. In circulation, the penetration capability of the CPPs was shielded, but upon reaching the tumor environment, the uncaging of the PSPs was stimulated by the external application of UV light, resulting in activated CPPs.96
However, the use of UV light has been limited, due to its harmful effect on tissue and low penetrability.97 Near-IR light is able to penetrate tissues deeply and is less harmful to cells in contrast to UV light. With this in mind, Yang et al designed NLCs conjugated with photon-sensitive CPPs (psCPPs; CGRRMKWKK) and NGR in an attempt to improve targeted delivery of Ptx to tumor cells. The psCPP unit facilitated specific cellular uptake after cleavage of the photon-sensitive protective group, whereas NGR moiety discriminately bound to CD13-positive tumors. The results of the study demonstrated that the tumor-growth-inhibition rate and cellular uptake of the psCPP/NGR-NLC group were significantly higher than the rest of the Ptx groups.97 Similarly, Yang et al developed near-IR light-activated psCPP/NGR liposomes that provide a reasonable approach to selective targeted delivery of siRNA.98
Thermally activated CPPs
In this approach, a biopolymer elastin-like polypeptide, a heat-sensitive carrier able to undergo phase transition upon reaching an externally heated tumor environment is used. This carrier is tumor specific, because it aggregates only at heated tumor sites at 39°C–42°C.50,99
Tumor-targeting peptides
TTPs are also known as tumor homing peptides and are small peptides shorter than CPPs by three to ten residues that have strong affinity and specificity for tissue targets or tumor cells.51 The overexpression of specific receptors in most tumors and their vasculature is utilized by TTPs, for which they show high binding ability.17 Among a number of different receptors that are known to be overexpressed in tumor cells, integrins are the most attractive target for drug delivery, because they have crucial roles in the process of tumor cell proliferation, migration, invasion, and survival.100
Peptides-targeting tumor vasculature
As mentioned earlier, tumor vasculature varies from normal blood vessels both structurally and morphologically. For example, the tumor blood vessels are leaky and porous, unlike normal vasculature.101,102 In addition to the altered morphology, tumor vasculature differs from that of normal by its molecular composition.37
Arginine–glycine–aspartic acid (RGD) and NGR peptides are the most extensively studied peptides for targeting tumor vasculature.101 RGD peptides can selectively target tumor vasculature expressing αγβ3 and αγβ5 integrins, and an NGR peptide (CNGRC) binds to CD13 (aminopeptidase N), which is expressed in the tumor vasculature specifically.103 RGR is another peptide that was selected from phage display in pancreatic tumors that have exhibited superior affinity to angiogenic vessels in insulinomas and recognize various αβ integrins. RGR has been exploited as a carrier for delivery of therapeutic proteins (TNFα and IFNγ) to targeted sites in cancer therapy.104
Another peptide that is targeting to angiogenic vasculature is F3. F3 (KDEPQRRSARLSAKPAPPKPEPKPKKAPAKK) is a 31-amino acid peptide that is able to target blood vessels and tumor tissue. F3 is known to target nucleolin, which is selectively expressed on the surface of endothelial cells and tumor cells.37 After binding, F3 is internalized through receptor-mediated endocytosis, and then translocated from the cytoplasm to the nucleus, where it distributes itself throughout organelles.105
Such peptides as CREKA, CLT1 (CGLIIQKNEC), and CLT2 (CNAGESSKNC) are also known to target tumor blood vessels and represent as a novel type of homing peptide.101 They have an affinity to bind with clotted plasma proteins that exist on the walls of tumor vasculature as well as in tumor stroma.106 Li et al identified TCP1 (CTPSPFSHC), a vasculature-homing peptide, by in vivo phage library screening in an orthotopic colorectal cancer model. The TCP1 peptide was found to recognize selectively the vasculature of orthotopic colorectal cancer in normal BALB/c mice induced by syngeneic colon cancer cells (colon 26).107 Recently, a number of more peptides homing to tumor vasculature have been identified.
Peptides-targeting tumor lymphatics
LyP1 (CGNKRTRGC) and LyP2 (CNRRTKAGC) are peptides that home to tumor lymphatics.37 Laakkonen et al identified LyP1 (CGNKRTRGC) by screening breast carcinoma xenografts (MDA-MB435).108 LyP1 binds selectively to p32, which is a mitochondrial protein that shows unusual expression on surface tumor lymphatics, tumor cells, and a subset of myeloid cells.109 LyP1 has been found to distinguish lymphatics and tumor cells in MDA-MD435 and MMTV-PyMT breast carcinoma and KRIB osteosarcoma xenografts and their metastatic lesions; however, it is unable to recognize C8161 melanomas.109 Simultaneously, the LyP2 peptide (CNRRTKAGC) homes to lymphatics of C8161 melanomas and cervical carcinomas and K14-HPV16 skin, but not to MDA-MB435 tumors, showing heterogeneity in the molecular markers of tumor cells and lymphatics. Another peptide, RMS-II (CMGNKRSAKRPC), which has some sequence similarity with LyP1, was also identified in an in vitro screen for peptides binding to RMS cell lines. Better targeting ability to RMS xenografts was observed by RMS-II than LyP1 in vivo. Furthermore, RMS-II recognized tumor blood vessels, but not tumor lymphatic vessels, which showed the different specificities of these two peptides.101
Peptides homing to tumor cells
Some homing peptides are known to possess cell-penetrating properties. For example, F3 and LyP1 peptides are cell-type-specific CPPs. These peptides have the ability to enter tumor cells and blood (F3) or lymphatic endothelial cells (LyP1) in tumors to which they home.101 CGNKRTR (tLyP1) is also a homing and tumor-penetrating peptide via NRP1-mediated endocytosis via the C-end rule internalization pathway.110
CRGRRST (RGR) and CGKRK are other homing peptides that are conveyed to a nucleus of the targeted cell after cellular uptake. These peptides consist of a number of basic amino acids, which are considered to be accountable for intracellular and intranuclear transportation. He et al synthesized a novel tumor-homing peptide, CSNIDARAC (4R22), which bound strongly to H460 cells and efficiently internalized into the cells. It also selectively bound to other lung tumor cell lines compared to cells of different cancer types.111
A number of endothelial cell-targeting peptides have been found to act as tumor-penetrating peptides to enable the internalization of a conjugated drug to the cancer cell. These peptides share a specific C-terminal C-end rule sequence, (R/K)XX(R/K), which is responsible for tissue penetration and cell internalization.112 For example, internalizing RGD (iRGD; CRGDKGPDC), one of the most innovative TTPs is a 9-amino acid cyclic peptide, where the lysine residue can also be an arginine and the aspartic acid a glutamic acid. In addition to targeting the αvβ3 integrin receptor, it is able to penetrate tumors. Compared to other RGD peptides, iRGD can distribute widely in extravascular tumor tissue.113
Synergistic use of CPPs with TTPs in tumor-targeting delivery
TTPs bind with receptors that are upregulated on tumor cells selectively but may not be capable of reaching the target by themselves. On the other hand, CPPs penetrate plasma membrane effectively but lack target specificity. Target-specific CPPs able both to penetrate the plasma membrane and selectively transport the drug to the desired target of action is considered an ideal type of CPP.17
Combining a TTP with a suitable CPP is known to facilitate the translocation of the conjugate moieties to target tumor sites with better selectivity and specificity.51 Studies have shown that TTPs conjugated with CPPs have dramatically increased efficiency in translocating drug molecules specifically to cancer cells compared to TTPs alone.114 Myrberg et al used the cyclic peptide cCPGPEGAGC (PEGA), which is a homing peptide that has been known to target breast tumor tissue in mice; however, the PEGA peptide was not able to cross the plasma membrane. The combined use of PEGA with pVEC by conjugation displayed specific targeting to breast cancer cells in vitro and in vivo. In addition, the conjugated PEGA–pVEC chimeric peptide was found to increase the efficacy of chlorambucil more than fourfold.114 A further study by the same group used the combination of CREKA with pVEC as a chimeric peptide for delivery of chlorambucil intracellularly. The results revealed that the chlorambucil–CREKA–pVEC conjugate produced significantly enhanced anticancer activity in vitro over the anticancer drug alone. The study also showed that CREKA–pVEC was better in translocating cargo molecules inside cancer cells compared to the PEGA–pVEC peptid.115,116
More recently, Fan et al evaluated the combined effect of SP90 (SMDPFLFQLLQL) with C peptide (GPGLWERQAREHSERKKRRRESECKAA) in breast cancer homing ability. SP90 is able to bind selectively to breast cancer cells (MDA-MB231), but C peptide has no cell specificity. However, the combined use of SP90 with C-peptide (SP90–C) showed a 10- and 12-fold increase in efficiency of intracellular delivery compared to C peptide and SP90 alone, respectively. Furthermore, SP90 and SP90–C conjugate were utilized for the delivery of HIV1 VPR (potential novel anticancer protein drug) into breast cancer cells. SP90–VPR–C has demonstrated improved apoptosis-inducing and antiproliferative activity of HIV1 VPR, without affecting normal breast cells.117
Nowadays, sequences of peptide exhibiting both cell-penetrating and specific targeting properties are designed using mRNA-display technology. For instance, RLW (RLWMRWYSPRTRAYG) is known to exhibit both properties and can target A549 cells specifically. Gao et al conjugated RLW with NPs to form RNPs for targeted delivery into lung cancer cells (A549), and a conventional CPP (R8 [RRRRRRRR]) was used as a control. In vitro cell uptake study shown that RNPs specifically improved uptake by A549 cells compared to human umbilical vein endothelial cells. However, conjugation with R8 increased uptake by both cell types, indicating better targeting specificity for RNPs. RNPs loaded with docetaxel (Dtx) demonstrated RLW selectively targeted A549 cells and enhanced the cytotoxicity of the drug in vitro.118 In addition, Lim et al developed a novel nontoxic and cancer-specific CPP, BR2 (BR2RAGLQFPVGRLLRRLLR [17 amino acids]), based on the cell-penetrating motif of buforin IIB. BR2 was fused to a single-chain variable fragment (scFv) antibody against mutated Kras. BR2 exhibited cancer-specific activity without toxicity to normal cells. Moreover, BR2 displayed greater membrane translocation efficiency than TAT49–57. Similarly, BR2 fussed with scFv was found to induce a higher degree of apoptosis than TAT-fused scFv in Kras-mutated HCT116 cells.119
Peptides for targeting MDR cancer
Most recently, anticancer drug–CPP/TTP conjugates have been one of the promising strategies to overcome tumor MDR. Sheng et al designed a dual-targeting hybrid peptide – HAIYPRHGGCGMPKKKPTPIQLNP (T10-ERK) – composed of an ERK-peptide inhibitor (MPKKKPTPIQLNP), a thiol linker (GGCG), and transferrin receptor-binding peptide (HAIYPRH) to overwhelm the problem of drug resistance. T10-ERK was conjugated to DoxO-EMCH (a prodrug of Dox), resulting T10-ERK–Dox. The efficacy of T10-ERK–Dox in reversing drug resistance compared with free Dox, and T10–Dox was determined using MCF7/ADR cells and nude mice-bearing MCF7/ADR xenografts. The results of the study indicated that T10-ERK–Dox efficiently inhibited drug resistance and improve the cytotoxicity of Dox by blocking Pgp-mediated drug efflux and inducing apoptosis.120
Furthermore, Lelle et al formulated a conjugate consisting of octaarginine CPP (Ac-CRRRRRRRR-NH2) and Dox dimer with high DNA affinity. The cytotoxicity of the peptide-drug conjugate was evaluated against drug-sensitive and Dox-resistant cancer cells, and showed that the conjugate overcame drug resistance in neuroblastoma cells efficiently.121 Feng et al also studied a Dox–SAPSP (slightly acidic pH-sensitive peptide) conjugate in order to interfere with drug resistance in cancer therapy. In this study, Dox was attached to SAPSP (CHGAHEHAGHEHAAGEHHAHE-NH2) to achieve a Dox–SAPSP prodrug. Cell uptake studies showed that Dox–SAPSP selectively accumulated in both Dox-sensitive and Dox-resistant cancer cells and was 26-fold less toxic toward noncancerous MCF10A cells than free Dox.122
Application of CPP/TTP nanocarriers for herb-based anticancer drugs
Herb-based anticancer drugs
The use of plants or phytoconstituents has a long history in cancer therapy and has played a significant role in the discovery of effective anticancer agents. Approximately 60% of presently used anticancer agents are derived from natural products in one way or another.123 Recently, more than 3,000 plants globally were reported to have anticancer properties.124 Some examples of important anticancer drugs that are derived from plant sources including Ptx (Taxol), vinblastine, capsaicin, vincristine, the Cpt derivatives, topotecan, irinotecan, and etoposide (Table 1 and Figure 3).125
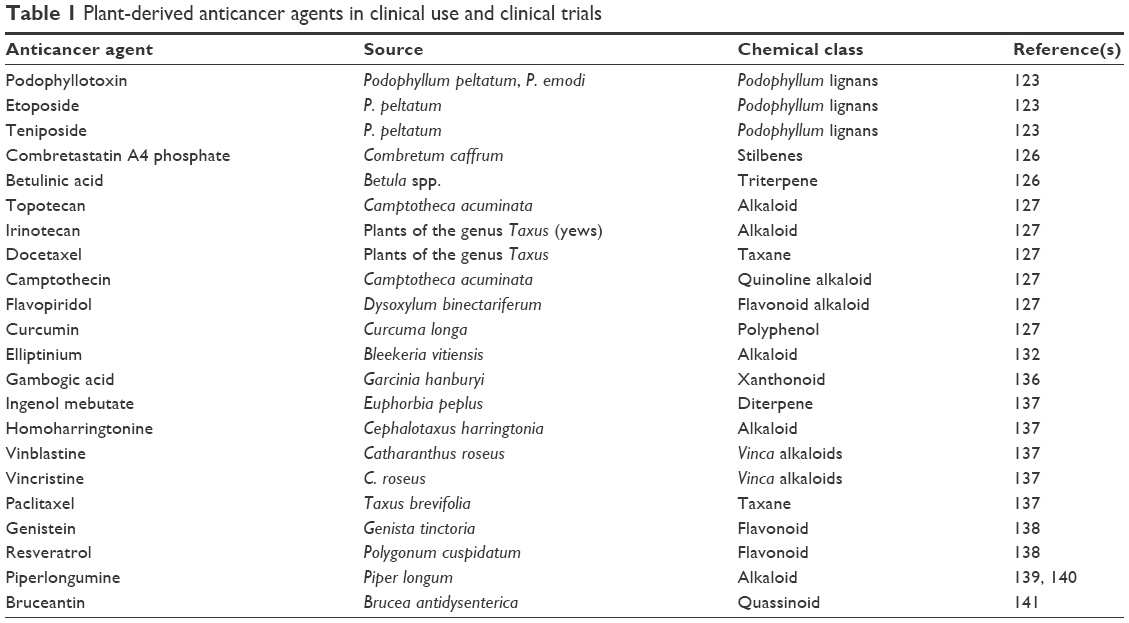 | Table 1 Plant-derived anticancer agents in clinical use and clinical trials |
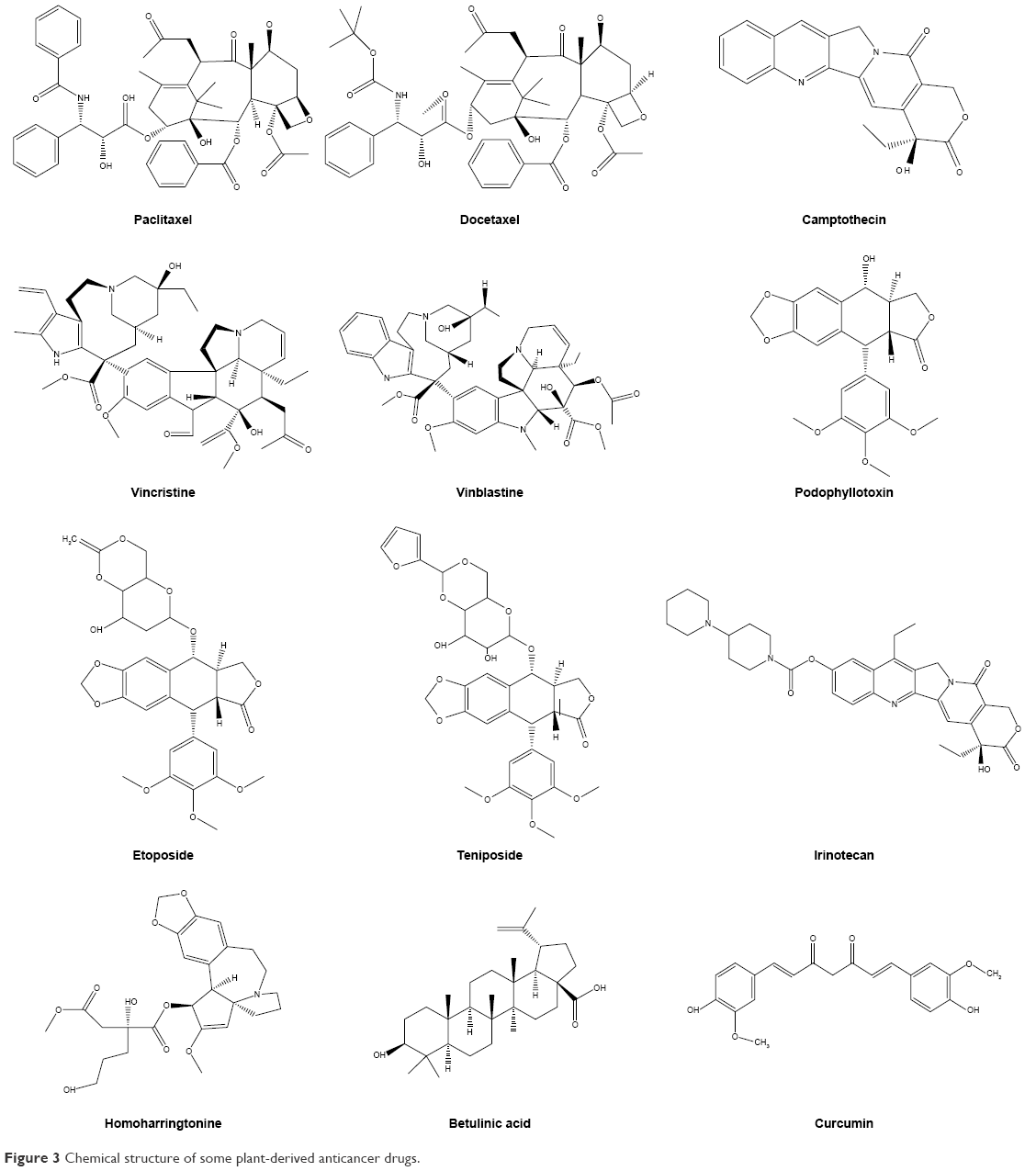 | Figure 3 Chemical structure of some plant-derived anticancer drugs. |
Vinca alkaloids (vinblastine and vincristine) are obtained from the Madagascar periwinkle, Catharanthus roseus (Apocynaceae), and were the first plant source used clinically as anticancer agents for leukemias, lymphomas, breast, testicular, and lung cancers, and Kaposi’s sarcoma.126 In recent years, semisynthetic derivatives of vinca alkaloids, such as vindesine and vinorelbine, have received approval from the US Food and Drug Administration, and vinflunine has been approved by the European Medicines Agency as a second-line chemotherapeutic agent in the treatment of metastatic urothelial cancer.127 Moreover, vinflunine and vinorelbine have shown minimized toxicity in comparative animal models.128,129
Taxanes are a class of herbal drugs that are commonly utilized in the treatment of breast cancer and initially isolated from the plant (Taxus brevifolia). Ptx (Taxol) and docetaxel (Taxotere) are the derivatives of taxanes that are clinically very essential in combination with synthetic chemotherapeutic agents such as Dox, capecitabine, and gemcitabine in order to achieve better clinical effects.130 Cpt is a quinoline alkaloid with topoisomerase I-inhibitor activity obtained from the bark and stem of Camptotheca acuminate.131 However, it was withdrawn from clinical trials due to its poor water solubility and severe toxic effects. In order to solve these limitations of Cpt, a number of Cpt analogues, such as topotecan, irinotecan, and belotecan, have been derived and approved for clinical use.127
Podophyllotoxin is another essential active antitumor agent extracted from the roots and rhizomes of Podophyllum spp. (Berberidaceae). The two semisynthetic derivatives of podophyllotoxin that are used in the treatment of lymphomas and bronchial and testicular cancers are etoposide and teniposide.126 Homoharringtonine and elliptinium are the other herb-derived agents in clinical use. Homoharringtonine was originally derived from the Chinese plant Cephalotaxus harringtonia var. drupacea (Cephalotaxaceae) and exhibits excellent anticancer activity against different types of leukemias, including some resistant to standard treatment. A racemic mixture of harringtonine and homoharringtonine is being utilized effectively in China to treat both acute and chronic myelogenous leukemia. Elliptinium was isolated from species of many genera of the family Apocynaceae, including Bleekeria vitiensis, a Fijian medicinal plant with anticancer properties.132
There are a number of promising anticancer drugs currently in clinical trials. For instance, Piperlongumine is a natural product extracted from Piper longum (Piperaceae) known to have potent anticancer activity.133 Curcumin is a polyphenol extracted from Curcuma longa.127 Despite curcumin’s poor absorption, a Phase II trial has shown anticancer activity in some patients with pancreatic cancer, and it is known to be well tolerated.134 Among bioactive compounds from medieval European plants, resveratrol, garlic compounds, and hypericin are under clinical evaluation.135 Besides these, a large number of other bioactive compounds isolated from herbs are under clinical investigation. In addition, several bioactive agents isolated from Chinese herbs have recently been found to exhibit strong anticancer activities, and are undergoing preclinical and clinical studies. Among these natural products with anticancer potential are flavonoids (gambogic acid, curcumin, silibinin, and wogonin), alkaloids (berberine), terpenes (artemisinin, β-elemene, ursolic acid, triptolide, and oridonin), quinones (emodin and shikonin), and saponins (ginsenoside Rg3).136
Recent examples of herb-based anticancer-bioactive CPP/TTP nanocarriers
Though a large number of potent plant-derived anticancer drugs have been investigated, their clinical use is limited, due to the fact that most herbal bioactive extracts are hydrophobic in nature, resulting in poor bioavailability and therapeutic failure. To circumvent this challenges, different nanocarriers, such as NPs, nanocapsules, liposomes, quantum dots, phytosomes, dendrimers, and nanoemulsions, have been exploited to deliver herbal anticancer bioactive agents. As mentioned in the previous sections, nanochemotherapeutic agents exhibit improved bioavailability, enhanced bioactivity, and stability with minimal systemic toxic effects.142
Moreover, surface modification of these nanocarriers with various ligands has been found to improve the targeting of bioactive agents to a cancer cell and then increase therapeutic efficacy and reduce toxic effects. Among these, CPPs/TTPs provide a highly promising approach for the intracellular delivery of anticancer drugs. In this section, we discuss the delivery of herb-based anticancer drugs using CPP/TTP-modified nanocarriers. Several studies have reported that CPP/TTP–herb-based anticancer conjugates demonstrate promising antitumor activity (Table 2).
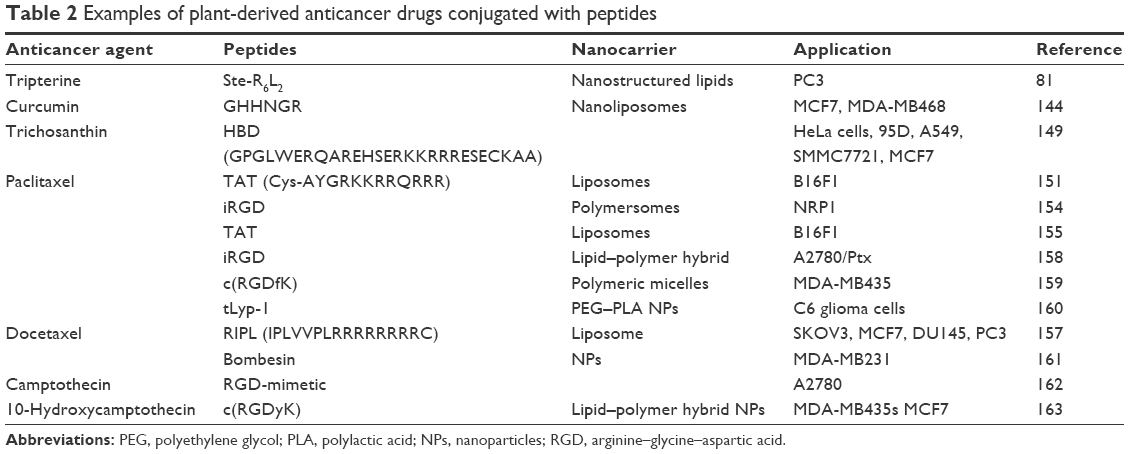 | Table 2 Examples of plant-derived anticancer drugs conjugated with peptides |
Curcumin is an extensively investigated active flavonoid isolated from the rhizome of C. longa with broad-spectrum anticancer properties.136 However, its low water solubility and poor bioavailability have limited its clinical use.143 In the last few decades, it has been studied in various delivery systems to circumvent these limitations. Kangarlou et al synthesized linear tumor-homing peptides (GHHNGR) and conjugated them with curcumin-loaded nanoliposomes. The conjugated curcumin-loaded liposomes showed significant cytotoxicity around sevenfold that of an aqueous curcumin suspension in MCF7 (IC50 3.8 μM) and MDA-MB468 (IC50 5.4 μM). Furthermore, the entrapped curcumin exhibited a prolonged half-life and reduced degradation, in contrast to the free drug, in aqueous suspensions.144 Simion et al also demonstrated that lipid nanoemulsion-loaded curcumin (CmLN) conjugated with a nona-arginine peptide (R9–CmLN) exhibited significantly higher uptake and internalization of R9–CmLN compared to nonfunctionalized CmLN in human endothelial cells.145 Furthermore, Das et al synthesized RGDK–lipopeptides and functionalized these noncovalently with single-walled carbon nanotubes (SWNTs) to form RGDK–SWNT. RGDK–SWNT was capable of delivering the anticancer drug curcumin to B16F10 melanoma cells more efficiently than NIH3T3 cells (noncancerous), leading to the selective killing of B16F10 cells.146
Tripterine, also known as celastrol, is a Chinese herbal medicine extracted from the thunder-god vine (Tripterygium wilfordii) and demonstrates promising bioactivity against a number of tumor cell lines.147 Yuan et al studied the antitumor activity of tripterine using CPP (Ste-R6L2)-modified NLCs in a human prostate cancer cell line (PC3) and a prostate tumor-bearing mouse model. CPP–tripterine (CT) NLCs showed dramatically enhanced antitumor activity in vivo and in vitro in comparison with free tripterine. In vivo tumor inhibition rates of CT-NLCs (54.5%) were higher than cyclophosphamide (37.07%) at low doses (2 mg/kg) in the PC3 cell line. Moreover, significantly higher amounts of TNFα and IL6 cytokine content were observed following the administration of CT-NLCs and T-NLCs than high-dose tripterine.81
Trichosanthin (Tcs) is a protein extracted from the root tubers of Trichosanthes kirilowii and has been used as an abortifacient for centuries in China. It is a type I RIP and well-known traditional Chinese medicine for various types of tumor cells.148 Lu et al used a human-derived CPP (HBD) (GPGLWERQAREHSERKKRRRESECKAA) to improve the delivery of Tcs. In this study, HBD was fused with the C terminus of recombinant Tcs (rTcs) to improve the translocation efficiency of Tcs. The IC50 of rTcs-HBD in the tested tumor cells was much lower than rTcs, showing that HBD delivered the rTcs into tumor cells efficiently.149
Dual-modified nanocarriers with more than one ligand are gaining much attention in anticancer drug research. Chen et al developed bifunctional NPs (BF-NPs) based on PLGA–PEG and modified them with CPP (R7) and folic acid simultaneously. The vincristine sulfate-loaded BF-NPs were prepared by an emulsion solvent evaporation method. Higher cellular uptake was found for BF-NPs than NPs modified by folic acid or R7 alone. In vitro cytotoxicity, cell apoptosis, and cell cycle also exhibited better potency of BF-NPs compared to those NPs merely modified by folic acid or R7.150
Ptx is a bioactive agent that has attracted much attention over the last three decades.151 It is a member of the taxane family, and one of the most important and effective antineoplastic agents for the treatment of many forms of advanced and refractory cancers. Ptx is a unique chemotherapeutic agent that is available on the market in three nanoplatforms – liposomes (Lipusu), polymeric NPs (Abraxane), and polymeric micelles (Genexol, Nanoxel, and Paclical) – for parenteral administration.152
Several reports have demonstrated that Ptx conjugates with different types of CCP/TTP show better anticancer activity. Fu et al used the well-known CPP TAT (Cys–AYGRKKRRQRRR) and functionalized it with Ptx-loaded liposomes containing PEG for prolonged circulation. PEG was detached at the tumor site in the presence of an exogenous reducing agent (glutathione), and this allowed cell internalization of TAT. Ptx–TAT liposomes with glutathione demonstrated higher cellular uptake and better penetration to three-dimensional tumor spheroids in vitro compared with analogous stable, shielded liposomes. Moreover, improved tumor distribution and superior delivery efficiency were exhibited by Ptx–TAT liposomes in vivo.153 Simón-Gracia et al developed Ptx-loaded iRGD polymersomes that showed enhanced peritoneal tumor growth inhibition and suppression of local dissemination effects, in contrast to Ptx polymersomes or Abraxane.154 Duan et al also formulated Ptx-CPPs using TAT and the LMWP VSRRRRRRGGRRRR. Ptx-CPPs showed considerably enhanced cellular uptake, high cytotoxicity, elevated cell apoptosis, and minimized mitochondrial membrane potential against both A549 and A549T cells compared with free Ptx. Ptx–LMWP displayed stronger tumor growth inhibition than Ptx–TAT in A549T cells.155 There have been a number of more recent reports on the role of CPPs in the delivery of Ptx, presented in Table 2.
Docetaxel (Dtx) is a semisynthetic derivative of Ptx that is considered one of the most promising antitumor drugs and is effective against a wide range of cancer, such as prostate, breast, pancreatic, lung, gastric, and hepatic carcinomas.156 Recently, Yoon et al designed an RIPL peptide (IPLVVPLRRRRRRRRC) and conjugated it with liposomes loaded with Dtx (Dtx–RIPL) in order to deliver the drug into hepsin-expressing cancer cells selectively. Dtx–RIPL liposomes were able to suppress tumor growth and elongate survival time significantly in BALB/c nude mice with ovarian tumors (SKOV3 cells).157
Conclusion and future perspectives
In the treatment of cancer, drug targeting is indispensable in the selective and quantitative accumulation of a drug in the target organ or tissue. NP-based targeting is of core importance in both passive- and active-targeting systems. NPs have been found to have enormous advantages in terms of reducing aspecific cellular uptake and side effects, elongating circulation, and offering controlled release and encapsulation of multiple drugs for combination therapy.
NPs modified with CPPs/TTPs could have potentially increased cellular uptake and cytotoxicity to cancer cells. A number of studies have demonstrated that CPP-NPs are able to enhance extracellular and intracellular internalization of various small drug molecules and biomolecules. However, lack of cell specificity and in vivo degradation were found to be the main limitations of further clinical development of CPPs. The use of ACPPs and/or the use of CPPs with TTPs in combination have been known to reduce problems associated with aspecificity of CPP. More recently, researchers have developed a CPP that exhibits both cell-penetrating properties and tumor cell-targeting efficiency using mRNA-display technology. Moreover, dual-targeting hybrid–peptide conjugates and multifunctionalized NPs have been among promising strategies to overcome tumor MDR.
Despite the promising potential of CPP/TTP-modified nanocarriers for cancer therapy, there have been no adequate reports on their in vivo toxicity or immunogenicity. In addition, the issue of detailed mechanisms of penetration and pharmacokinetics and development of a sensitive method of detection of CPPs/TTPs should be thoroughly addressed for further preclinical and clinical study. Finally, we hope that more advanced and optimized CPPs will be discovered and play a significant role in shifting the paradigm of cancer therapy.
Acknowledgment
This study was financially supported by the Tianjin Natural Science Foundation (16JCYBJC28200).
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. Cancer. 2018. Available from: http://www.who.int/mediacentre/factsheets/fs297/en. Accessed February 13, 2018. | ||
Pérez-Herrero E, Fernández-Medarde A. Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015;93:52–79. | ||
Ghaz-Jahanian MA, Abbaspour-Aghdam F, Anarjan N, Berenjian A, Jafarizadeh-Malmiri H. Application of chitosan-based nanocarriers in tumor-targeted drug delivery. Mol Biotechnol. 2015;57:201–218. | ||
Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ. Limited penetration of anticancer drugs through tumor tissue: a potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res. 2002;8:878–884. | ||
Sutradhar KB, Amin ML. Nanotechnology in cancer drug delivery and selective targeting. ISRN Nanotechnology. 2014;2014:939378. | ||
Gupta A, Kaur CD, Saraf S, Saraf S. Targeting of herbal bioactives through folate receptors: a novel concept to enhance intracellular drug delivery in cancer therapy. J Recept Signal Transduct Res. 2017;37:314–323. | ||
Lundstrom K. Cancer therapy applying viral nanoparticles. In: Khudyakov Y, Pumpens P, editors. Viral Nanotechnology. Boca Raton: Taylor & Francis; 2015:455–466. | ||
Baker JR. Dendrimer-based nanoparticles for cancer therapy. Hematology Am Soc Hematol Educ Program. 2009:708–719. | ||
Jain S, Hirst DG, O’Sullivan JM. Gold nanoparticles as novel agents for cancer therapy. Br J Radiol. 2012;85:101–113. | ||
Pandey H, Rani R, Agarwa V. Liposome and their [sic] applications in cancer therapy. Braz Arch Biol Technol. 2016;59:e16150477. | ||
Bondì ML, Craparo EF, Giammona G, et al. Nanostructured lipid carriers-containing anticancer compounds: preparation, characterization, and cytotoxicity studies. Drug Deliv. 2007;14:61–67. | ||
Oerlemans C, Bult W, Bos M, Storm G, Nijsen JF, Hennink WE. Polymeric micelles in anticancer therapy: targeting, imaging and triggered release. Pharm Res. 2010;27:2569–2589. | ||
Parveen S, Sahoo SK. Polymeric nanoparticles for cancer therapy. J Drug Target. 2008;16:108–123. | ||
Gao H, Yang Z, Zhang S, et al. Ligand modified nanoparticles increases [sic] cell uptake, alters endocytosis and elevates glioma distribution and internalization. Sci Rep. 2013;3:2534. | ||
Torchilin VP, Benson H, Toth I. Cell penetrating peptide-modified pharmaceutical nanocarriers for intracellular drug and gene delivery. Biopolymers. 2008;90:604–610. | ||
Zhang XX, Eden HS, Chen X. Peptides in cancer nanomedicine: drug carriers, targeting ligands and protease substrates. J Control Release. 2012;159:2–13. | ||
Dissanayake S, Denny WA, Gamage S, Sarojini V. Recent developments in anticancer drug delivery using cell penetrating and tumor targeting peptides. J Control Release. 2017;250:62–76. | ||
Liu Z, Delavan B, Roberts R, Tong W. Lessons learned from two decades of anticancer drugs. Trends Pharmacol Sci. 2017;38:852–872. | ||
Prabhakar U, Maeda H, Jain RK, et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73:2412–2417. | ||
Ulbrich K, Holá K, Subr V, Bakandritsos A, Tuček J, Zbořil R. Targeted drug delivery with polymers and magnetic nanoparticles. Chem Rev. 2016;116:5338-5431. | ||
Thanki K, Kushwah V, Jain S. Recent advances in tumor targeting approaches. In: Devarajan PV, Jain S, editors. Targeted Drug Delivery: Concepts and Design. Heidelberg: Springer; 2015:41–112. | ||
Yu X, Trase I, Ren M, Duval K, Guo X, Chen Z. Design of nanoparticle-based carriers for targeted drug delivery. J Nanomater. 2016;2016:1087250. | ||
Xu X, Ho W, Zhang X, Bertrand N, Farokhzad O. Cancer nanomedicine: from targeted delivery to combination therapy. Trends Mol Med. 2015;21:223–232. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–151. | ||
Bahrami B, Hojjat-Farsangi M, Mohammadi H, et al. Nanoparticles and targeted drug delivery in cancer therapy. Immunol Lett. 2017;190:64–83. | ||
Kunjachan S, Rychlik B, Storm G, Kiessling F, Lammers T. Multidrug resistance: physiological principles and nanomedical solutions. Adv Drug Deliv Rev. 2013;65:1852–1865. | ||
Yildiz I, Shukla S, Steinmetz NF. Applications of viral nanoparticles in medicine. Curr Opin Biotechnol. 2011;22:901–908. | ||
Gupta C, Prakash D, Gupta S. Cancer treatment with nano-diamonds. Front Biosci (Schol Ed). 2017;9:62–70. | ||
Yao VJ, D’Angelo S, Butler KS, et al. Ligand-targeted theranostic nanomedicines against cancer. J Control Release. 2016;240:267–286. | ||
de Oliveira RO, de Santa Maria LC, Barratt G. Nanomedicine and its applications to the treatment of prostate cancer. Ann Pharm Fr. 2014;72:303–316. | ||
Babaei M, Eshghi H, Abnous K, Rahimizadeh M, Ramezani M. Promising gene delivery system based on polyethylenimine-modified silica nanoparticles. Cancer Gene Ther. 2017;24:156–164. | ||
Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–627. | ||
Yameen B, Choi W, Vilos C, Swami A, Shi J, Farokhzad OC. Insight into nanoparticle cellular uptake and intracellular targeting. J Control Release. 2014;190:485–499. | ||
Zhong Y, Meng F, Deng C, Zhong Z. Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules. 2014;15:1955–1969. | ||
Jaracz S, Chen J, Kuznetsova LV, Ojima I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg Med Chem. 2005;13:5043–5054. | ||
Thayer AM. Improving peptides. Chem Eng News. 2011;89:13–20. | ||
David A. Peptide ligand-modified nanomedicines for targeting cells at the tumor microenvironment. Adv Drug Deliv Rev. 2017;119:120–142. | ||
Karra N, Benita S. The ligand nanoparticle conjugation approach for targeted cancer therapy. Curr Drug Metab. 2012;13:22–41. | ||
Guidotti G, Brambilla L, Rossi D. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol Sci. 2017;38:406–424. | ||
Meloni BP, Craig AJ, Milech N, Hopkins RM, Watt PM, Knuckey NW. The neuroprotective efficacy of cell-penetrating peptides TAT, penetratin, Arg-9, and Pep-1 in glutamic acid, kainic acid, and in vitro ischemia injury models using primary cortical neuronal cultures. Cell Mol Neurobiol. 2014;34:173–181. | ||
Ma C, Yin G, You F, et al. A specific cell-penetrating peptide induces apoptosis in SKOV3 cells by down-regulation of Bcl-2. Biotechnol Lett. 2013;35:1791–1797. | ||
Kristensen M, Birch D, Nielsen HM. Applications and challenges for use of cell-penetrating peptides as delivery vectors for peptide and protein cargos. Int J Mol Sci. 2016;17:E185. | ||
Hsieh TH. A novel cell-penetrating peptide suppresses breast tumorigenesis by inhibiting β-catenin/LEF-1 signaling. Sci Rep. 2016;6:19156. | ||
Mehta R, Yamada T, Taylor BN, et al. A cell penetrating peptide derived from azurin inhibits angiogenesis and tumor growth by inhibiting phosphorylation of VEGFR-2, FAK and Akt. Angiogenesis. 2011;14:355–369. | ||
Kristensen M, de Groot AM, Berthelsen J, Franzyk H, Sijts A, Nielsen HM. Conjugation of cell-penetrating peptides to parathyroid hormone affects its structure, potency, and transepithelial permeation. Bioconjug Chem. 2015;26:477–488. | ||
Sun W, Fletcher D, van Heeckeren CR, Davis PB. Non-covalent ligand conjugation to biotinylated DNA nanoparticles using TAT peptide genetically fused to monovalent streptavidin. J Drug Target. 2012;20:678–690. | ||
Copolovici DM, Langel K, Eriste E, Langel U. Cell-penetrating peptides: design, synthesis, and applications. ASC Nano. 2014;8:1972–1994. | ||
Jafari S, Dizaj SM, Adibkia K. Cell-penetrating peptides and their analogues as novel nanocarriers for drug delivery. Bioimpacts. 2015;5:103–111. | ||
Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today. 2012;17:850–860. | ||
Wang F, Wang Y, Zhang X, Zhang W, Guo S, Jin F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J Control Release. 2014;174:126–136. | ||
Regberg J, Srimanee A, Langel U. Applications of cell-penetrating peptides for tumor targeting. Pharmaceuticals (Basel). 2012;5:991–1007. | ||
Palm C, Jayamanne M, Kjellander M, Hällbrink M. Peptide degradation is a critical determinant for cell-penetrating peptide uptake. Biochim Biophys Acta. 2007;1768:1769–1776. | ||
Fominaya J, Bravo J, Rebollo A. Strategies to stabilize cell penetrating. Ther Deliv. 2015;6:1171–1194. | ||
Chorev M, Goodman M. Recent developments in retro peptides and proteins: an ongoing topochemical exploration. Trends Biotechnol. 1995;13:438–445. | ||
Vaissière A, Aldrian G, Konate K, et al. A retro-inverso cell-penetrating peptide for siRNA delivery. J Nanobiotechnology. 2017;15:34. | ||
Holm T, Räägel H, Andaloussi SE, et al. Retro-inversion of certain cell-penetrating peptides causes severe cellular toxicity. Biochim Biophys Acta. 2011;1808:1544–1551. | ||
Horn M, Reichart F, Natividad-Tietz S, Diaz D, Neundorf I. Tuning the properties of a novel short cell-penetrating peptide by intramolecular cyclization with a triazole bridge. Chem Commun (Cab). 2016;52:2261–2264. | ||
Lättig-Tünnemann G, Prinz M, Hoffmann D, et al. Backbone rigidity and static presentation of guanidinium groups increases cellular uptake of arginine-rich cell-penetrating peptides. Nat Commun. 2011;2:453. | ||
Friedler A, Friedler D, Luedtke NW, Tor Y, Loyter A, Gilon C. Development of a functional backbone cyclic mimetic of the HIV-1 Tat arginine-rich motif. J Biol Chem. 2000;275:23783–23789. | ||
Reissmann S. Cell penetration: scope and limitations by the application of cell-penetrating peptides. J Pept Sci. 2014;20:760–784. | ||
Dietz GP, Bahr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci. 2004;27:85–131. | ||
Durzyńska J, Przysiecka L, Nawrot R, et al. Viral and other cell-penetrating peptides as vectors of therapeutic agents in medicine. J Pharmacol Exp Ther. 2015;354:32–42. | ||
Verdurmen WP, Brock R. Biological responses towards cationic peptides and drug carriers. Trends Pharmacol Sci. 2011;32:116–124. | ||
El-Andaloussi S, Järver P, Johansson HJ, et al. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: a comparative study. Biochem J. 2007;407:285–292. | ||
Uusna J, Langel K, Langel U. Toxicity, Immunogenicity, uptake, and kinetics. In: Langel U, editor. Cell-Penetrating Peptides: Methods and Protocols. 2nd ed. Heidelberg: Springer; 2015:133–148. | ||
Suhorutsenko J, Oskolkov N, Arukuusk P, et al. Cell-penetrating peptides, PepFects, show no evidence of toxicity and immunogenicity in vitro and in vivo. Bioconjug Chem. 2011;22:2255–2262. | ||
Kilk K, Mahlapuu R, Soomets U, Langel U. Analysis of in vitro toxicity of five cell-penetrating peptides by metabolic profiling. Toxicology. 2009;265:87–95. | ||
Aguilera TA, Timmers MM, Olson ES, Jiang T, Tsien RY. Systemic in vivo distribution of activatable cell penetrating peptides is superior to that of cell penetrating peptides. Integr Biol (Camb). 2009;1:371–381. | ||
Moschos SA, Jones SW, Perry MM, et al. Lung delivery studies using siRNA conjugated to TAT48–60 and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjug Chem. 2007;18:1450–1459. | ||
Carter E, Lau CY, Tosh D, Ward SG, Mrsny RJ. Cell penetrating peptides fail to induce an innate immune response in epithelial cells in vitro: implications for continued therapeutic use. Eur J Pharm Biopharm. 2013;85:12–19. | ||
Cardozo AK, Buchillier V, Mathieu M, et al. Cell-permeable peptides induce dose- and length-dependent cytotoxic effects. Biochim Biophys Acta. 2007;1768:2222–2234. | ||
Yen HJ, Hsu SH, Tsai CL. Cytotoxicity and immunological response of gold and silver nanoparticles of different sizes. Small. 2009;5:1553–1561. | ||
He X, Li J, An S, Jiang C. pH-sensitive drug-delivery systems for tumor targeting. Ther Deliv. 2013;4:1499–1510. | ||
Olson E. In vivo characterization of activatable cell penetrating peptides for targeting protease activity in cancer. Integr Biol (Camb). 2009;1:382–393. | ||
MacEwan SR, Chilkoti A. Harnessing the power of cell-penetrating peptides: activatable carriers for targeting systemic delivery of cancer therapeutics and imaging agents. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2013;5:31–48. | ||
Kurrikof K, Gestin M, Langel U. Recent in vivo advances in cell-penetrating peptide-assisted drug delivery. Expert Opin Drug Deliv. 2015;13:373–387. | ||
Jin E, Zhang B, Sun X, et al. Acid-active cell-penetrating peptides for in vivo tumor-targeted drug delivery. J Am Chem Soc. 2013;135:933–940. | ||
Cheng H, Zhu JY, Xu XD, et al. Activable cell-penetrating peptide conjugated prodrug for tumor targeted drug delivery. ACS Appl Mater Interfaces. 2015;7:16061–16069. | ||
Regberg J, Vasconcelos L, Madani F, Langel U, Hällbrink M. pH-responsive PepFect cell-penetrating peptides. Int J Pharm. 2016;501:32–38. | ||
Zhao BX, Zhao Y, Huang Y, et al. The efficiency of tumor-specific pH-responsive peptide-modified polymeric micelles containing paclitaxel. Biomaterials. 2012;33:2508–2520. | ||
Zhang Q, Tang J, Fu L, et al. A pH-responsive α-helical cell penetrating peptide-mediated liposomal delivery system. Biomaterials. 2013;34:7980–7993. | ||
Yao J, Ma Y, Zhang W, et al. Design of new acid-activated cell-penetrating peptides for tumor drug delivery. PeerJ. 2017;5:e3429. | ||
Lee D, Noh I, Yoo J, Rejinold NS, Kim YC. pH-controllable cell-penetrating polypeptide that exhibits cancer targeting. Acta Biomater. 2017;57:187–196. | ||
He H, Sun L, Ye J, et al. Enzyme-triggered, cell penetrating peptide-mediated delivery of anti-tumor agents. J Control Release. 2016;240:67–76. | ||
van Duijnhoven SM, Robillard MS, Nicolay K, Grüll H. Tumor targeting of MMP-2/9 activatable cell-penetrating imaging probes is caused by tumor-independent activation. J Nucl Med. 2011;52:279–286. | ||
Shi NQ, Gao W, Xiang B, Qi XR. Enhancing cellular uptake of activable cell-penetrating peptide-doxorubicin conjugate by enzymatic cleavage. Int J Nanomedicine. 2012;7:1613–1621. | ||
Gao H, Zhang S, Cao S, Yang Z, Pang Z, Jiang X. Angiopep-2 and activatable cell-penetrating peptide dual-functionalized nanoparticles for systemic glioma-targeting delivery. Mol Pharm. 2014;11:2755–2763. | ||
Xia H, Gu G, Hu Q, et al. Activatable cell penetrating peptide-conjugated nanoparticles with enhanced permeability for site-specific targeting delivery of anticancer drug. Bioconjug Chem. 2013;24:419–430. | ||
Zhu L, Wang T, Perche F, Taigind A, Torchilin VP. Enhanced anticancer activity of nanopreparation containing an MMP2-sensitive PEG-drug conjugate and cell-penetrating moiety. Proc Natl Acad Sci U S A. 2013;110:17047–17052. | ||
Yoo J, Rejinold NS, Lee D, Jon S, Kim YC. Protease-activatable cell-penetrating peptide possessing ROS-triggered phase transition for enhanced cancer therapy. J Control Release. 2017;264:89–101. | ||
Whitney M, Crisp JL, Olson ES, et al. Parallel in vivo and in vitro selection using phage display identifies protease-dependent tumor-targeting peptides. J Biol Chem. 2010;285:22532–22541. | ||
Li J, Liu F, Shao Q, et al. Enzyme-responsive cell-penetrating peptide conjugated mesoporous silica quantum dot nanocarriers for controlled release of nucleus-targeted drug molecules and real-time intracellular fluorescence imaging of tumor cells. Adv Healthc Mater. 2014;3:1230–1239. | ||
Braun GB, Sugahara KN, Yu OM, et al. Urokinase-controlled tumor penetrating peptide. J Control Release. 2016;232:188–195. | ||
Zhang D, Wanga J, Xu D. Cell-penetrating peptides as noninvasive transmembrane vectors for the development of novel multifunctional drug-delivery systems. J Control Release. 2016;229:130–139. | ||
Shamay Y, Adar L, Ashkenasy G, David A. Light induced drug delivery into cancer cells. Biomaterials. 2011;32:1377–1386. | ||
Lin W, Xie X, Yang Y, et al. Enhanced small interfering RNA delivery into cells by exploiting the additive effect between photo-sensitive peptides and targeting ligands. J Pharm Pharmacol. 2015;67:1215–1231. | ||
Yang Y, Xie X, Yang Y, Zhang H, Mei X. Photo-responsive and NGR-mediated multifunctional nanostructured lipid carrier for tumor-specific therapy. J Pharm Sci. 2015;104:1328–1339. | ||
Yang Y, Yang Y, Xie X, et al. Dual-modified liposomes with a two-photon-sensitive cell penetrating peptide and NGR ligand for siRNA targeting delivery. Biomaterials. 2015;48:84–96. | ||
Walker LR, Ryu JS, Perkins E, et al. Fusion of cell-penetrating peptides to thermally responsive biopolymer improves tumor accumulation of p21 peptide in a mouse model of pancreatic cancer. Drug Des Devel Ther. 2014;8:1649–1658. | ||
Mitchell K, Svenson KB, Longmate WM, et al. Suppression of integrin α3β1 in breast cancer cells reduces cyclooxygenase-2 gene expression and inhibits tumorigenesis, invasion, and cross-talk to endothelial cells. Cancer Res. 2010;70:6359–6367. | ||
Laakkonen P, Vuorinen K. Homing peptides as targeted delivery vehicles. Integr Biol (Camb). 2010;2:326–337. | ||
Li ZJ, Cho C. Peptides as targeting probes against tumor vasculature for diagnosis and drug delivery. J Transl Med. 2012;10:S1. | ||
Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377–380. | ||
Lu L, Qi H, Zhu J, et al. Vascular-homing peptides for cancer therapy. Biomed Pharmacother. 2017;92:187–195. | ||
Christian S, Pilch J, Akerman ME, Porkka K, Laakkonen P, Ruoslahti E. Nucleolin expressed at the cell surface is a marker of endothelial cells in angiogenic blood vessel. J Cell Biol. 2003;163:871–878. | ||
Wang C, Wang X, Zhong X, et al. The antitumor activity of tumor-homing peptide-modified thermosensitive liposomes containing doxorubicin on MCF-7/ADR: in vitro and in vivo. Int J Nanomedicine. 2015;10:2229–2248. | ||
Li ZJ, Wu WK, Ng SS, et al. A novel peptide specifically targeting the vasculature of orthotopic colorectal cancer for imaging detection and drug delivery. J Control Release. 2010;148:292–302. | ||
Laakkonen P, Akerman ME, Biliran H, et al. Antitumor activity of a homing peptide that targets. Proc Natl Acad Sci U S A. 2004;101:9381–9386. | ||
Fogal V, Zhang L, Krajewski S, Ruoslahti E. Mitochondrial/cell surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res. 2008;68:7210–7218. | ||
Seleci DA, Seleci M, Stahl F, Scheper T. Tumor homing and penetrating peptide-conjugated niosomes as multi-drug carriers for tumor-targeted drug delivery. RSC Adv. 2017;7:33378–33384. | ||
He X, Na MH, Kim JS, et al. A novel peptide probe for imaging and targeted delivery of liposomal doxorubicin to lung tumor. Mol Pharm. 2011;8:430–438. | ||
Teesalu T, Sugahara K, Kotamraju V, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci U S A. 2009;106:16157–16162. | ||
Ruoslahti E. Tumor penetrating peptides for improved drug delivery. Adv Drug Deliv Rev. 2017;110–111:3–12. | ||
Myrberg H, Zhang L, Mäe M, Langel U. Design of a tumor-homing cell-penetrating peptide. Bioconjug Chem. 2008;19:70–75. | ||
Bolhassani A. Potential efficacy of cell-penetrating peptides for nucleic acid and drug delivery in cancer. Biochim Biophys Acta. 2011;1816:232–246. | ||
Mäe M, Myrberg H, El-Andaloussi S, Langel U. Design of a tumor homing cell-penetrating peptide for drug delivery. Int J Pept Res Ther. 2009;15:11–15. | ||
Fan LQ, Du GX, Li PF, Li MW, Sun Y, Zhao LM. Improved breast cancer cell-specific intracellular drug delivery and therapeutic efficacy by coupling decoration with cell penetrating peptide and SP90 peptide. Biomed Pharmacother. 2016;84:1783–1791. | ||
Gao H, Zhanga Q, Yanga Y, Jiang X, He Q. Tumor homing cell penetrating peptide decorated nanoparticles used for enhancing tumor targeting delivery and therapy. Int J Pharm. 2015;478:240–250. | ||
Lim KJ, Sung BH, Shin JR, et al. A cancer specific cell-penetrating peptide, BR2, for the efficient delivery of an scFv into cancer cells. PLoS One. 2013;8:e66084. | ||
Sheng Y, You Y, Chen Y. Dual-targeting hybrid peptide-conjugated doxorubicin for drug resistance reversal in breast cancer. Int J Pharm. 2016;512:1–13. | ||
Lelle M, Freidel C, Kaloyanova S, et al. Overcoming drug resistance by cell-penetrating peptide-mediated delivery of a doxorubicin dimer with high DNA-binding affinity. Eur J Med Chem. 2017;130:336–345. | ||
Feng C, Rui M, Shen H, et al. Tumor-specific delivery of doxorubicin through conjugation of pH-responsive peptide for overcoming drug resistance in cancer. Int J Pharm. 2017;528:322–333. | ||
Cragg GM, Newman DJ. Plants as a source of anti-cancer agents. J Ethnopharmacol. 2005;100:72–79. | ||
Solowey E, Lichtenstein M, Sallon S, Paavilainen H, Solowey E, Lorberboum-Galski H. Evaluating medicinal plants for anticancer activity. ScientificWorldJournal. 2014;2014:721402. | ||
Ahmad R, Ahmad N, Naqvi AA, Shehzad A, Al-Ghamdi MS. Role of traditional Islamic and Arabic plants in cancer therapy. J Tradit Complement Med. 2017;7:195–204. | ||
Shah U, Shah R, Acharya S, Acharya N. Novel anticancer agents from plant sources. Chin J Nat Med. 2013;11:16–23. | ||
Khazir J, Mir BA, Pilcher L, Riley DL. Role of plants in anticancer drug discovery. Phytochem Lett. 2014;7:173–181. | ||
Okouneva T, Hill BT, Wilson L, Jordan MA. The effects of vinflunine, vinorelbine, and vinblastine on centromere dynamics. Mol Cancer Ther. 2003;2:427–436. | ||
Simoens C, Lardon F, Pauwels B, et al. Comparative study of the radiosensitising and cell cycle effects of vinflunine and vinorelbine, in-vitro. BMC Cancer. 2008;8:65. | ||
Thapa RK, Khan GM, Parajuli-Baral K, Thapa P. Herbal medicine incorporated nanoparticles: advancements in herbal treatment. Asian J Biomed Pharm Sci. 2013;3:7–14. | ||
Wall ME, Wani MC, Cook CE, et al. Plant antitumor agents – I: the isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J Am Chem Soc. 1966;88:3888–3890. | ||
Prakash O, Kumar A, Kumar P. Anticancer potential of plants and natural products. Am J Pharmacol Sci. 2013;1:104–115. | ||
Saeidnia S, Abdollahi M. Perspective study on anticancer drugs from natural origin. Int J Pharmacol. 2014;10:90–108. | ||
Dhillon N, Aggarwal BB, Newman RA, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14:4491–4499. | ||
Teiten MH, Gaascht F, Dicato M, Diederich M. Anticancer bioactivity of compounds from medicinal plants used in European medieval traditions. Biochem Pharmacol. 2013;86:1239–1247. | ||
Tan W, Lu J, Huang M, et al. Anti-cancer natural products isolated from Chinese medicinal herbs. Chin Med. 2011;6:27. | ||
Bishayee A, Sethi G. Bioactive natural products in cancer prevention and therapy: progress and promise. Semin Cancer Biol. 2016;40–41:1–3. | ||
Herst PM, Davis JE, Neeson P, Berridge MV, Ritchie DS. The anticancer drug, phenoxodiol, kills primary myeloid and lymphoid leukemic blasts and rapidly proliferating T cells. Haematologica. 2009;94:928–934. | ||
Saeidnia S, Abdollahi M. Perspective study on anticancer drugs from natural origin. Int J Pharmacol. 2014;10:90–108. | ||
Aodah A, Pavlik A, Karlage K, Myrdal PB. Preformulation studies on piperlongumine. PLoS One. 2016;11:e0151707. | ||
Saklani A, Kutty SK. Plant-derived compounds in clinical trials. Drug Discov Today. 2008;13:161–171. | ||
Wani W, Tarawadi K, Kaul-Ghanekar R. Nanocarriers for delivery of herbal based drugs in breast cancer: an overview. J Nano Res. 2015;34:29–40. | ||
Anand P, Kunnumakkara A, Newman R, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4:807–818. | ||
Kangarlou S, Ramezanpour S, Balalaie S, Mohammadi SR, Haririan I. Curcumin-loaded nanoliposomes linked to homing peptides for integrin targeting and neuropilin-1-mediated internalization. Pharm Biol. 2017;55:277–285. | ||
Simion V, Stan D, Constantinescu CA, et al. Conjugation of curcumin-loaded lipid nanoemulsions with cell-penetrating peptides increases their cellular uptake and enhances the anti-inflammatory effects in endothelial cells. J Pharm Pharmacol. 2016;68:195–207. | ||
Das K, Nimushakavi S, Chaudhuri A, Das PK. An integrin-targeting RGDK-tagged nanocarrier: anticancer efficacy of loaded curcumin. ChemMedChem. 2017;8:738–750. | ||
Li Z, Wu X, Li J, et al. Antitumor activity of celastrol nanoparticles in a xenograft retinoblastoma tumor model. Int J Nanomedicine. 2012;7:2389–2398. doi:2310.2147/IJN.S29945. | ||
Shaw PC, Lee KM, Wong KB. Recent advances in trichosanthin, a ribosome-inactivating protein with multiple pharmacological properties. Toxicon. 2005;45:683–689. | ||
Lu YZ, Li PF, Li YZ, et al. Enhanced anti-tumor activity of trichosanthin after combination with a human-derived cell-penetrating peptide, and a possible mechanism of activity. Fitoterapia. 2016;112:183–190. | ||
Chen J, Li S, Shen Q. Folic acid and cell-penetrating peptide conjugated PLGA–PEG bifunctional nanoparticles for vincristine sulfate delivery. Eur J Pharm Sci. 2012;47:430–443. | ||
Amin A, Gali-Muhtasib H, Ocker M, Schneider-Stock R. Overview of major classes of plant-derived anticancer drugs. Int J Biomed Sci. 2009;5:1–11. | ||
Bernabeu E, Cagel M, Lagomarsino E, Moretton M, Chiappetta DA. Paclitaxel: what has been done and the challenges remain ahead. Int J Pharm. 2017;526:474–495. | ||
Fu H, Shi K, Hu G, et al. Tumor-targeted paclitaxel delivery and enhanced penetration using TAT-decorated liposomes comprising redox-responsive poly(ethylene glycol). J Pharm Sci. 2015;104:1160–1173. | ||
Simón-Gracia L, Hunt H, Scodeller P, et al. iRGD peptide conjugation potentiates intraperitoneal tumor delivery of paclitaxel with polymersomes. Biomaterials. 2016;104:247–257. | ||
Duan Z, Chen C, Qin J, et al. Cell-penetrating peptide conjugates to enhance the antitumor effect of paclitaxel on drug-resistant lung cancer. Drug Deliv. 2017;24:752–764. | ||
Ganesh T. Improved biochemical strategies for targeted delivery of taxoids. Bioorg Med Chem. 2007;15:3537–3545. | ||
Yoon HY, Kwak SS, Jang MH, et al. Docetaxel-loaded RIPL peptide (IPLVVPLRRRRRRRRC)-conjugated liposomes: drug release, cytotoxicity, and antitumor efficacy. Int J Pharm. 2017;523:229–237. | ||
Zhang JM, Wang L, Chan HF, et al. Co-delivery of paclitaxel and tetrandrine via iRGD peptide conjugated lipid-polymer hybrid nanoparticles overcome multidrug resistance in cancer cells. Sci Rep. 2017;7:46057. | ||
Shahin M, Ahmed S, Kaur K, Lavasanifar A. Decoration of polymeric micelles with cancer-specific peptide ligands for active targeting of paclitaxel. Biomaterials. 2011;32:5123–5133. | ||
Hu Q, Gao X, Gu G, et al. Glioma therapy using tumor homing and penetrating peptide-functionalized PEG–PLA nanoparticles loaded with paclitaxel. Biomaterials. 2013;34:5640–5650. | ||
Kulhari H, Pooja D, Shrivastava S, Naidu VG. Peptide conjugated polymeric nanoparticles as a carrier for targeted delivery of docetaxel. Colloids Surf B Biointerfaces. 2014;117:166–173. | ||
Alloatti D, Giannini G, Vesci L, et al. Camptothecins in tumor homing via an RGD sequence mimetic. Bioorg Med Chem Lett. 2012;22:6509–6512. | ||
Yang Z, Luo X, Zhang X, Liu J, Jiang Q. Targeted delivery of 10-hydroxycamptothecin to human breast cancers by cyclic RGD-modified lipid-polymer hybrid nanoparticles. Biomed Mater. 2013;8:025012. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.