")
Back to Journals » International Journal of General Medicine » Volume 15
Tumor DNA Methylation Profiles Enable Diagnosis, Prognosis Prediction, and Screening for Cervical Cancer
Authors Tu J, Chen S, Wu S, Wu T, Fan R, Kuang Z
Received 25 December 2021
Accepted for publication 16 June 2022
Published 27 June 2022 Volume 2022:15 Pages 5809—5821
DOI https://doi.org/10.2147/IJGM.S352373
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Jiannan Tu,1,* Shengchi Chen,1,* Shizhen Wu,1,* Ting Wu,1,* Renliang Fan,1 Zhixing Kuang2
1Department of Oncology, Nanping First Hospital Affiliated to Fujian Medical University, Nanping, 353000, People’s Republic of China; 2Department of Radiation Oncology, Nanping First Hospital Affiliated to Fujian Medical University, Nanping, 353000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhixing Kuang, Department of Radiation Oncology, Nanping First Hospital Affiliated to Fujian Medical University, Nanping, 353000, People’s Republic of China, Email [email protected]
Background: DNA-methylation-based machine learning algorithms have demonstrated powerful diagnostic capabilities, and these tools are currently emerging in many fields of tumor diagnosis and patient prognosis prediction. This work aimed to identify novel DNA methylation diagnostic biomarkers for differentiating cervical cancer (CC) from normal tissues, as well as a prognostic prediction model to predict survival of CC patients.
Methods: The methylation profiles with the available clinical characteristics were downloaded from the Gene Expression Omnibus (GEO) database and The Cancer Genome Atlas (TCGA) program. We first screened out the differential methylation sites in CC and normal tissues and performed multiple statistical analyses to discover DNA methylation diagnostic markers that are used to distinguish CC and normal control. Then, we developed a methylation-based survival model to improve risk stratification.
Results: A diagnostic prediction panel consists of five CpG markers that could predict cervical cancer versus normal tissue with highly correct rate of 100%, and cg16428251, cg22341310, and cg23316360 which in diagnostic prediction panel all could yield high sensitivity and specificity for detection of CC and normal in six cohorts (area under curve [AUC] > 0.8), in addition to excellent performance in discriminating between CC and normal sample. The diagnostic marker panel also effectively predicted the CIN3 versus normal tissue with high accuracy in two datasets (AUC = 0.80, 0.789, respectively). Furthermore, a prognostic prediction model aggregated two CpG markers that effectively stratified the prognosis of high-risk and low-risk groups (training cohort: hazard ratio [HR] 4, 95% CI: 1.7– 9.6, P = 0.0021; testing cohort: hazard ratio [HR] 1.9, 95% CI: 1.2– 3.1, P = 0.0072).
Conclusion: The findings of our study showed that DNA methylation markers are of great value in the diagnosis and prognosis of CC.
Keywords: cervical cancer, DNA methylation, markers, diagnosis, prognosis
Introduction
Cervical cancer (CC) is a kind of gynecological malignant disease that accounts for 570,000 new cases and 311,000 deaths worldwide each year and continues to be listed among the top gynecological cancers worldwide, only behind breast cancer worldwide.1,2 Despite the efforts in vaccination that have been made to reduce the incidence of CC, it is estimated that approximately 13,800 new cases of CC will be diagnosed among the most developed countries like the United States in 2020, and nearly 4290 women will die of this disease.3 The situation in low-income areas still needs improvement, and CC is far from being eradicated. The overall 5-year survival rate of all CC patients is 68% and in patients with recurrent or metastatic CC survival remains poor, with a 5-year survival rate of 17%.4,5 CC can be easily treated if detected at early stages,6 thus there is a need for reliable approaches that will allow early diagnosis for this type of cancer to improve prognosis. Testing for human papillomavirus (HPV) DNA is most widely utilized for detection and surveillance of CC due to persistent infections of high-risk human papillomavirus (hrHPV). It is a causative agent that leads to high-grade precancerous cervical lesions and can progress to CC,7 but not all patients infected with HPV will progress to invasive CC. Thus, there is an urgent need to look for novel biomarkers beyond HPV detection which relate to the stepwise progression from cervical intraepithelial neoplasia (CIN2/CIN3) to invasive CC. The influence of epigenetic changes which encompass DNA methylation, histone modifications, and non-coding RNAs on tumor progression and clinical outcomes has been widely described for different cancers.8 Epigenetic alterations occur in the early or precancerous process of carcinogenesis and are considered as the precursor of malignant CC.9,10 Therefore, the discovery of new epigenetic biomarkers can be effective in blocking the progress of tumor development and reduces mortality and morbidity.
DNA methylation refers to the biochemical process that methylates the 5-carbon of cytosine under the action of DNA methyltransferase,11 and accumulating evidence has shown that aberrant methylation is a central epigenetic modification involved in sophisticated pathogenesis of numerous tumorigenesis and other diseases.12 Aberrant methylation generally causes transcriptional repression through the prevention of the binding of transcription factor or local conformational changes of chromatin,13 but a DNA-methylation-based biomarker can play a role in preventing gene activation rather than changing the level of gene expression.14 Traditionally, the research on DNA-methylation-based biomarkers has mainly concentrated on the effects of promoter CpG with hypermethylation in tumor suppressor genes. However, not all CpG islands in a single promoter region can contribute to the same function,14 and that is why detecting methylation sites can sometimes be more valuable and effective than detecting expression levels. On the other hand, lots of tumor suppressor genes are silenced by hypermethylation of DNA, indicating high methylation is an early event in carcinogenesis of many kinds of tumor and suggesting that the level of some specific DNA methylation site could be the earliest detectable tumor changes related to tumorigenesis.15,16 Therefore, detecting DNA methylation of a specific genomic location may be a promising tool for early diagnosis, prognosis prediction, and therapeutic application. Previous research on diagnosis and prediction of liver cancer reported by Xu et al found that the sensitivity of 10 CpG marker diagnostic prediction panels for hepatocellular carcinoma ranged from 83.3% to 85.7%, with specificity ranging from 95.8% to 97.5% and AUC up to 0.969.17 Apart from this, Huiyan Luo et al also reported 9 methylation markers to predict the diagnosis of colorectal cancer; the sensitivity of 9 methylation marker diagnostic prediction panels is 87.5%, with 89.9% specificity and 0.96 AUC,15 demonstrating the excellent performance in predictive value of methylation CpG location. However, the potential value of DNA methylation biomarkers for screening and early detection of CC is far from complete and requires further investigation.
The development and utilization of a CpG microarray constituted a landmark in the field of cancer research, and has been one of the main driving forces behind the development of diagnosis and prognosis stratification. Publicly available databases which store large amounts of DNA methylation profiling data, such as The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) have already demonstrated powerful value for epigenetic research. In this study, we performed a comprehensive study of CC DNA methylation microarray, with a focus on identifying novel diagnostic and prognostic methylation markers (Figure S1). A panel of five CpG methylation markers (cg10635061, cg16428251, cg22341310, cg23316360, cg24019564) shows excellent capability in diagnosis of CC. Finally, we propose a 2 CpG methylation prognosis predictive model which could be used to predict the survival in CC.
Methods
Data Sources and Data Preprocessing
Level 3 DNA methylation data of CC samples which are based on the Illumina Infinium Human Methylation 450 platform (450K array) were downloaded from the TCGA data portal (https://portal.gdc.cancer.gov/). Furthermore, seven DNA methylation matrix profiles of GSE99511 (28 normal samples, 36 CIN3, 4 tumor samples), GSE30759 (15 normal samples, 48 tumor samples), GSE30760 (167 normal samples, 48 tumor samples), GSE41384 (10 normal samples, 9 tumor samples), GSE46306 (20 normal samples, 17 CIN3, 6 tumor samples), and GSE38266 (21 HPV+ tumor samples, 21 HPV- tumor samples) were collected from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/). GSE30759, GSE30760, and GSE41384 are based on the GPL8490 Platform (Illumina Human Methylation 27 Beadchip), and GSE99511, GSE46306, and GSE38266 are based on the Platform of GPL13534 (Illumina Human Methylation 450 Beadchip).
Screening of Methylation Biomarkers Using LASSO and Random Forest Analysis
A valuable tool called GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/) was developed for GEO users to analyze microarray data. In our study, GEO2R was used to perform difference analysis to find differentially methylated genes (DMGs) in GSE30759, only if the differentially methylated site meets the criteria of |t|≥2.5 and adj P<0.01 were screened out. Then we ranked the list by adjusted P-value and selected the top 1000 candidate markers for the analysis of LASSO and random forest. The LASSO algorithm is a regression analysis that is used to achieve regularization and feature selection, and it constructs a penalty function to get a more refined model with the aim of enhancing the prediction accuracy and interpretability of the statistical model. The optimal values of the penalty parameter λ were determined by the results of 10 times cross-validations. Random forest are collections of decision trees, which are generated by recursive binary division of different random sub-samples of training data. In analysis of random forest, we first chose the best number of methylation sites based on cross-validation results, which can be visualized by a plot of the error rate curve, then, according to the mean decrease in accuracy, we selected the optimal methylated site. Mean decrease in accuracy can be regarded as the degree to which the classification accuracy decreases without this feature, which is equivalent to our commonly used concept of classification contribution. Lastly, five overlapping markers between LASSO and random forest analysis were screened out. LASSO analysis was performed with the package “glmnet”, and random forest analysis was performed with the package “randomForest”.
Identification of DNA Methylation Markers That Discriminate Between CC and Normal Sample
The dataset of GSE30759 was randomly divided into training cohort and validation cohort with a 6:4 ratio to evaluate the categorical accuracy of five overlapping markers obtained from the analysis results of LASSO algorithms and the random forest algorithm by building a random forest classifier. The out-of-bag (OOB) error represents an unbiased estimate of the current classification error, and we observed a perfect consistency between predicted results and true diagnosis results in both the training and validation datasets. Finally, these five DNA methylation markers are identified as hub markers for verification by evaluating area under ROC in GSE30759, GSE30760, GSE41384, GSE99511, GSE46306, and TCGA dataset. Combinatory diagnosis was performed by combined diagnosis score which was calculated by logistic regression model by using the 5 markers. The Wilcoxon test was performed to explore methylation level differences in normal, CIN3, and cancer tissue; in addition to this, we also compared the methylation levels between HPV- and HPV+ patients in GSE38266.
Building and Validation of a Predictive Model for Prognosis and Survival
Univariate Cox proportional hazards regression analysis was used to evaluate the relationship between 1000 candidate DNA methylation markers and the overall survival (OS) of CC patients within datasets of GSE30759. The methylation markers with P < 0.05 were identified and used to perform LASSO analysis. The LASSO-selected candidate methylation sites were chosen to perform multivariate Cox regression analyses and to build a methylation predictive model. The risk score for each CC patient was constructed as follows:
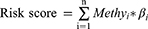
The beta value of methylation site i is represented by 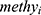 , and the coefficient of methylation marker i in the multivariate Cox regression analysis is represented by
, and the coefficient of methylation marker i in the multivariate Cox regression analysis is represented by 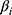 . All CC patients were split into two groups according to the median value of risk score, and survival analysis was performed by Kaplan–Meier plot. Then, the predictive model was validated in the TCGA dataset. Moreover, we also performed univariate and multivariate Cox analyses on the predictive model and clinicopathological features. In addition, clinicopathological factors and the prognostic prediction model were used to generate nomographs. Calibration curves were used to evaluate the degree of agreement between predicted outcomes and actual outcomes.
. All CC patients were split into two groups according to the median value of risk score, and survival analysis was performed by Kaplan–Meier plot. Then, the predictive model was validated in the TCGA dataset. Moreover, we also performed univariate and multivariate Cox analyses on the predictive model and clinicopathological features. In addition, clinicopathological factors and the prognostic prediction model were used to generate nomographs. Calibration curves were used to evaluate the degree of agreement between predicted outcomes and actual outcomes.
Statistical Analysis
The Wilcoxon test was applied to compare the degree of methylation in normal tissues, CIN3, and CC tissues. A logistic regression model was used to build a combinatory classifier. The ROC curve was adopted to assess the performance of the classifier. Evaluation of receiver operating characteristic (ROC) for diagnostic prediction of all datasets was calculated and visualized by MedCalc software.18 Survival curves were calculated and visualized by Kaplan–Meier, and log rank test was applied to detect survival difference between high-risk group and low-risk group. Univariate and multivariate Cox proportional hazard models were performed to estimate the hazard ratios of prognostic factors, with the P value <0.05 considered to be statistically significant. The C-index was used to assess the accuracy of the nomogram. All analysis was conducted in R software (version 3.5.1), except analysis of diagnostic sensitivity and specificity.
Results
Screening Candidate Methylated Site
There were 3887 hypermethylated genes and 3702 hypomethylated genes in GSE30759 identified by the gene methylation microarray analysis. The 1000 top markers ranked by adjusted P-value were selected the for LASSO and random forest analysis to further narrow down methylation site (Figure S2). According to the cross‐validation results in random forest analysis which were depicted by the error rate curve graph, only if the number of methylation markers is greater than 10 can the error be minimized and the disease is well classified (Figures S3 and 1A). So we chose 30 methylation sites ranked by mean decrease in accuracy for further analysis (Figure 1B); at the same time, 13 markers were obtained by using a LASSO analysis (Figure 1C and D). There were five overlapping methylation markers between the two methods of LASSO and random forest analysis.
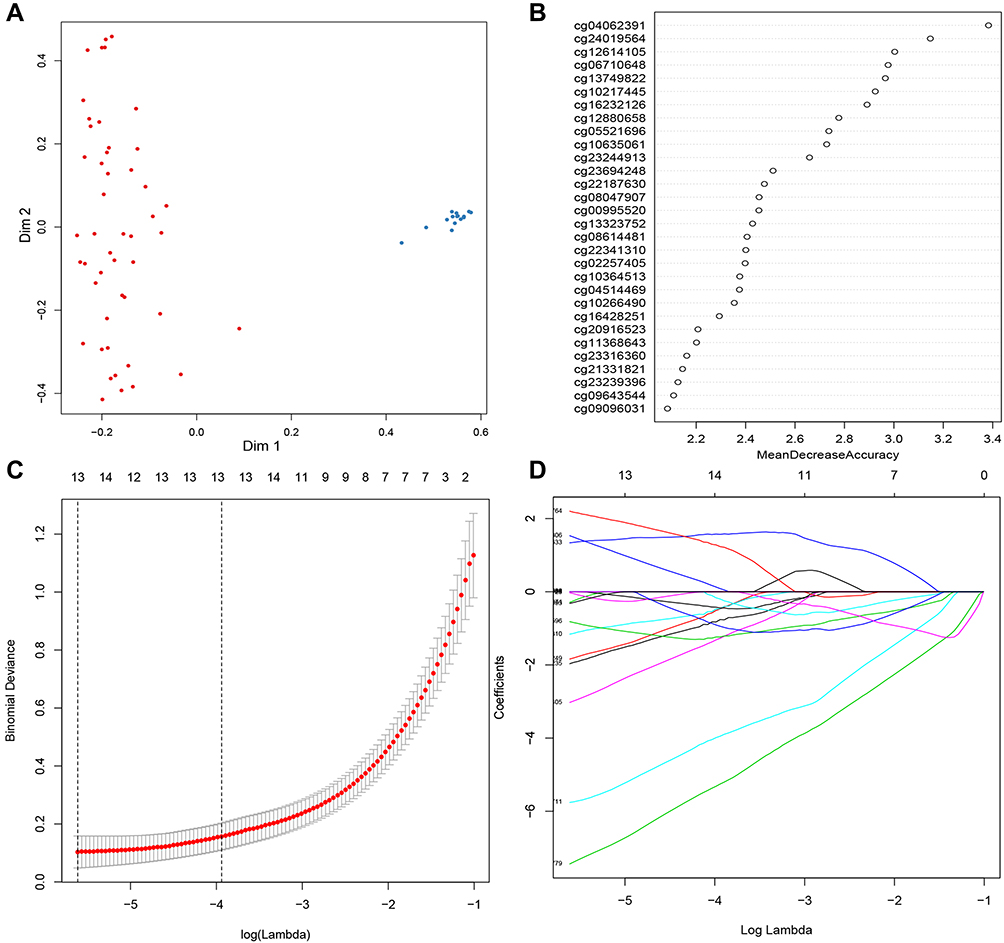 |
Figure 1 Screening candidate methylated markers. (A) Evaluation of random forest model with multidimensional scaling chart. (B) Variable importance for top 30 predictors. (C and D) Thirteen methylation markers identified by LASSO regression analysis and the cross-validation performed to prevent overfitting. |
The Identification of Methylation Marker Panels for Diagnostic Prediction
Five overlapping methylation markers including cg10635061, cg16428251, cg22341310, cg23316360, and cg24019564 were obtained between two methods of LASSO and random forest analysis (Figure 2A and B). We built a random forest classifier with the five markers to evaluate the accuracy of the model in the training and validation cohorts in GSE30759. The OOB error in two of all cohorts was 0; that means that the random forest classifier can predict the status of the all patients with 100% accuracy (Figure 3A and B). Therefore, we finally selected all five markers for internal and external verification. Wilcoxon test results in all datasets show that the methylation level of cg16428251, cg22341310, and cg23316360 is significantly different in the normal group and the cancer group. The cg10635061 and cg24019564 methylation level in the CC group is also significantly higher than in the normal group except in GSE41384 and GSE99511. The unsurprising thing is that in cohorts of GSE46306 and GSE99511, all five methylation sites in CIN3 is higher than in normal tissues, but lower than in tumor tissues except g10635061 and cg24019564 in GSE99511, which further confirms that CIN3 is early stage of carcinogenesis (Figure 4). Having explored the differences in methylation levels between HPV- and HPV + patients, we found that cg22341310 and cg24019564 were significantly hypermethylated in the HPV+ group (Figure S4), suggesting that the methylation levels of cg22341310 and cg24019564 may be related to HPV infection. Diagnosis efficiency of each of five methylation sites was also calculated in 6 datasets. cg16428251, cg22341310, and cg23316360 showed the best diagnostic performance yielding all AUCs >0.8 in all cohorts (Table S1–S6). The combination of five methylation sites was found to have a 100% correct rate in stratifying tumor tissue from all cohorts (Figure 5). Apart from this, the combination of five methylation sites was found to have a sensitivity of 76.47% and 69.44% and a specificity of 75.0% and 85.71% for the detection of CIN3 in GSE46306 and GSE99511, respectively (Figure 6).
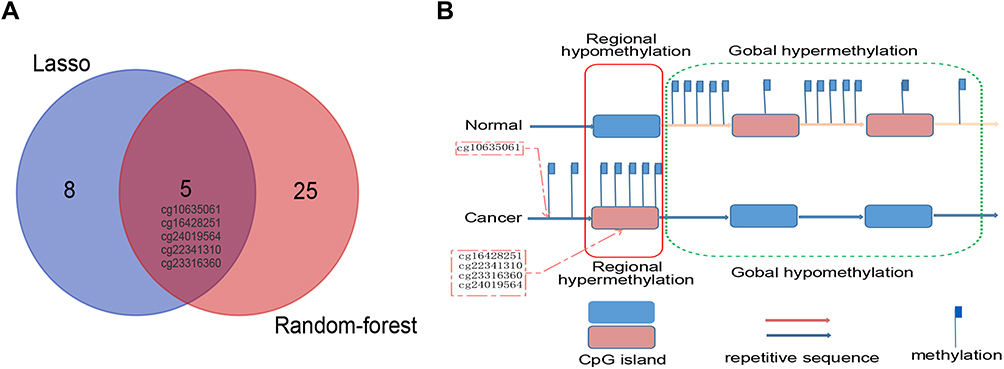 |
Figure 2 Alterations of DNA methylation in CC. (A) Five overlapping aberrant methylation markers selected by LASSO-based and random forest-based analysis. (B) Regional hypermethylation and global hypomethylation are present in cancer cells. Regional hypermethylation (aberrant DNA methylation) indicates increased DNA methylation of CpG islands unmethylated in normal cells (demonstrated in red dotted boxes). |
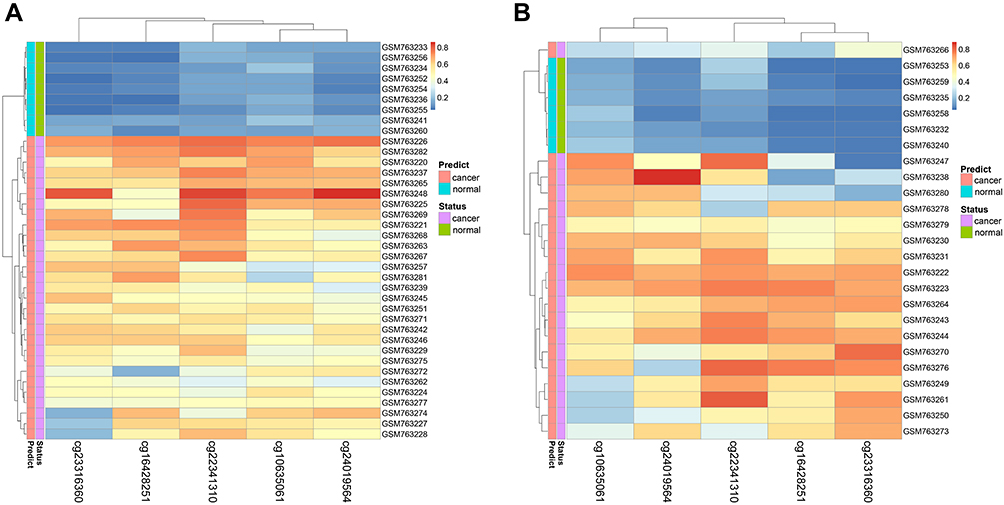 |
Figure 3 Performance of the DNA methylation diagnostic based on random forest (RF) classifier. (A) Unsupervised hierarchical clustering of five methylation markers selected for predicting the status of training dataset. (B) Predicting the status of testing dataset. |
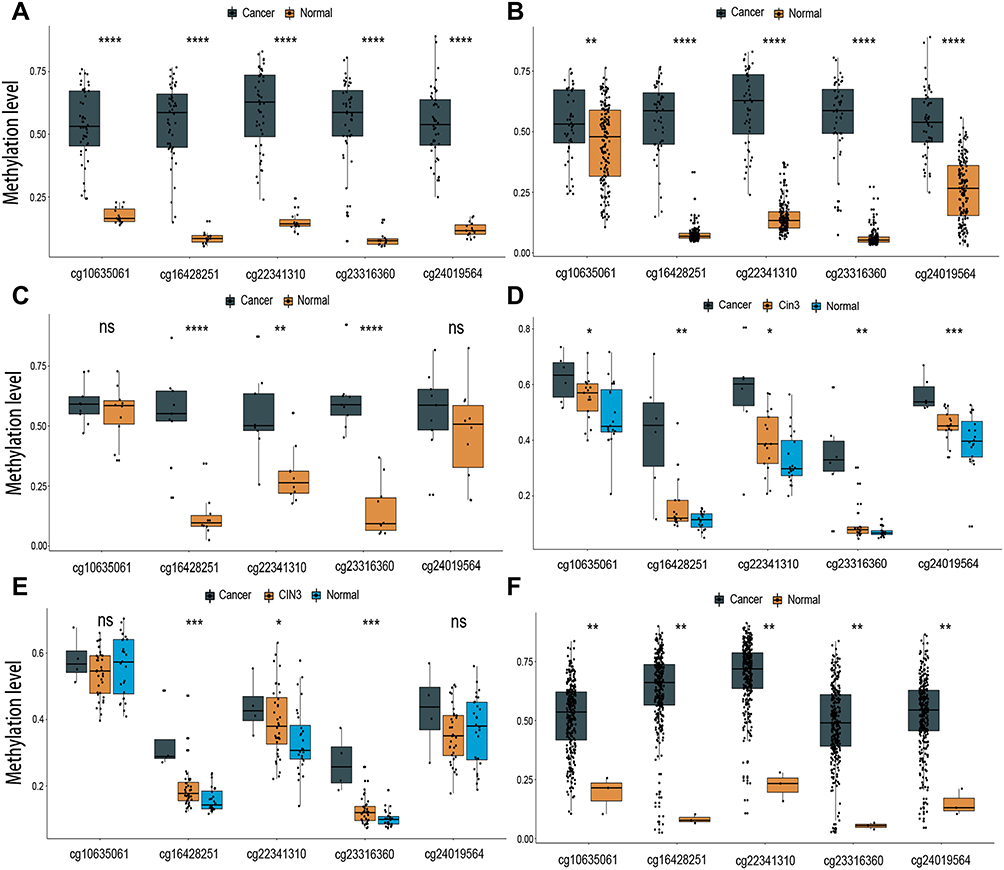 |
Figure 4 Boxplot presenting the beta value distribution of the five markers in six independent datasets. (A) GSE30759, (B) GSE30760, (C) GSE41384, (D) GSE46306, (E) GSE99511, (F) TCGA. (ns not significant, *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). |
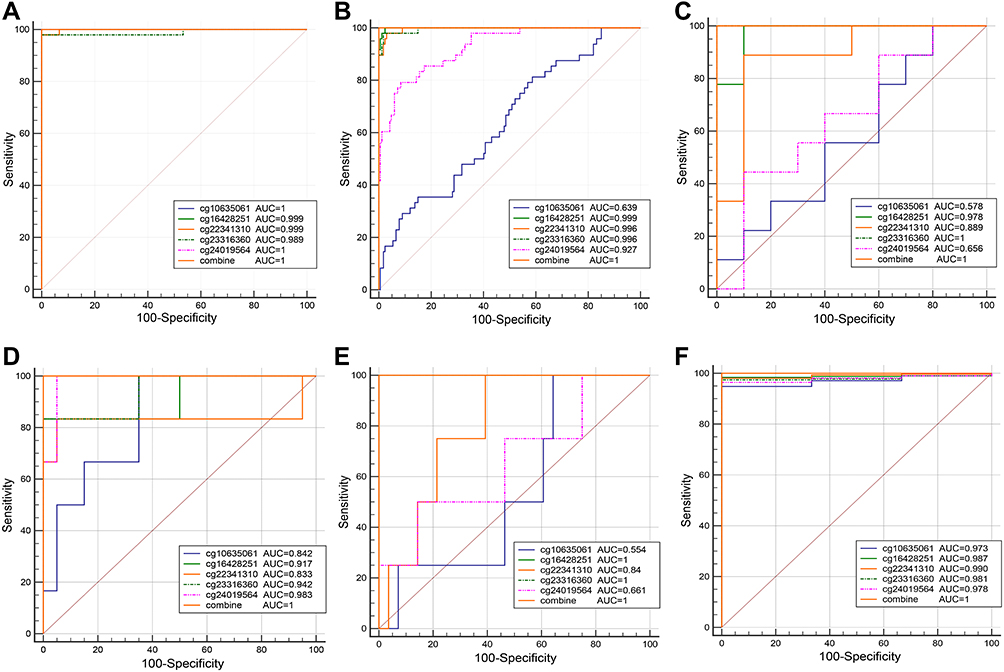 |
Figure 5 Performance of the DNA methylation diagnostic markers in CC prediction evaluated by ROC. (A) GSE30759, (B) GSE30760, (C) GSE41384, (D) GSE46306, (E) GSE99511, (F) TCGA. |
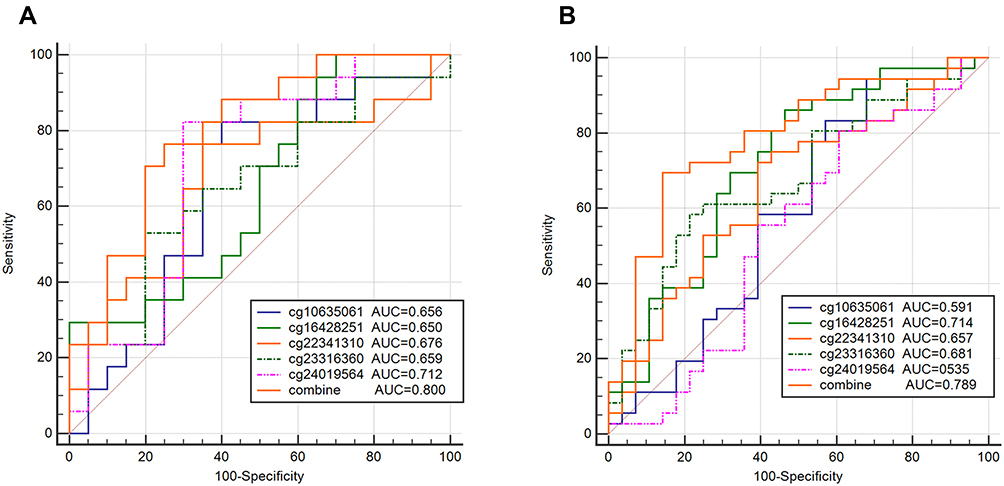 |
Figure 6 The diagnosis efficiency of DNA methylation diagnostic markers in CIN3 prediction. (A) GSE46306, (B) GSE99511. |
DNA-Methylation-Based Prognostic Prediction Model for CC
Eighty-one candidate methylation sites were identified by univariate Cox survival analysis in GSE30759; dimensionality reduction was implemented by LASSO Cox and multivariate Cox analysis (Figure S5), and a prognostic prediction model was constructed by multivariate Cox analysis to predict prognosis with the methylation sites of cg08849574 and cg13084335 in GSE30759 and TCGA. The risk score for predicting survival time of CC was generated with the following formula based on the two methylation markers: risk score = (−2.52×cg08849574 methy) + (−16.7×cg13084335 methy). We divided all patients from the training cohort of GSE30759 and test cohort of TCGA into high- and low-risk groups according to the median value of the risk score. Furthermore, the predictive model has shown excellent stratified ability and can be a useful tool to predict the outcomes of CC patients, which was confirmed and depicted by Kaplan–Meier curves. The 5-year and 10-year survival rates of the low-risk group (training cohort: 70.8% and 45.8%, validation cohort: 21.4% and 7.1%) were significantly better than those of the high-risk group (training cohort: 37.5% and 25%, validation cohort: 10% and 4.2%, Figure 7A and B). Kaplan–Meier curves also showed that CC patients with different staging have significantly different prognosis (P<0.005, Figure 8A and B).
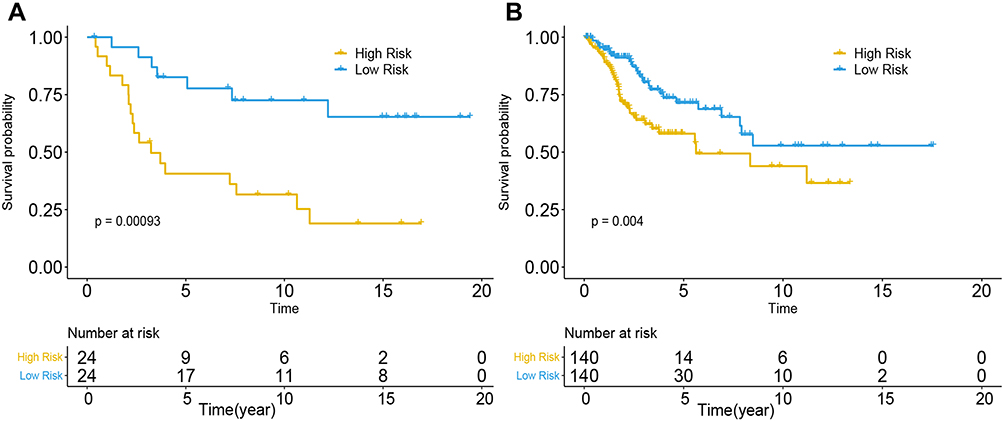 |
Figure 7 Performance of the DNA methylation prognostic model in the OS prediction of patients with CC. (A) GSE30759, (B) TCGA. |
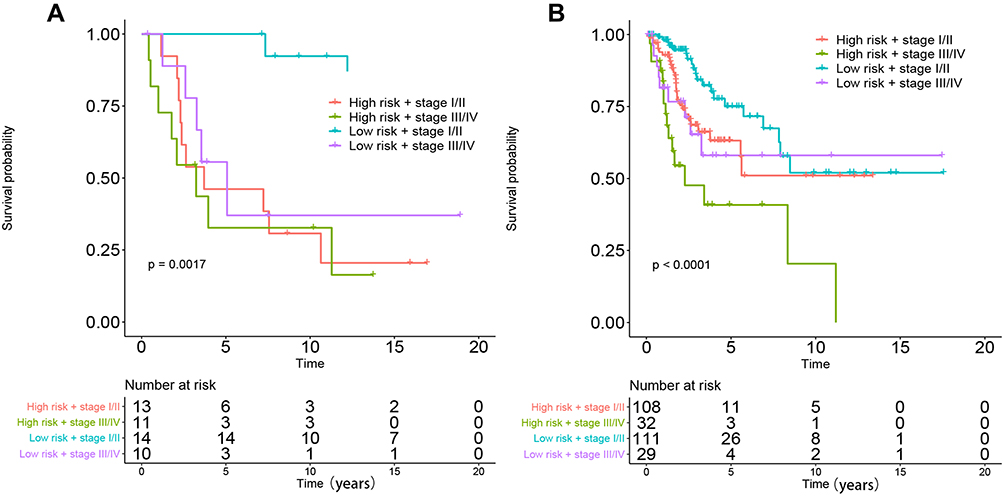 |
Figure 8 Performance of combinations of the DNA methylation prognostic model and stage in the OS prediction of patients with CC. (A) GSE30759, (B) TCGA. |
Independent Prognostic Role of the Prognostic Prediction Model
To explore clinical relevance between prognostic model and clinical features, univariate and multivariate Cox regression analyses were performed by including age, stage, histology, grade, surgery, radiation, chemotherapy, risk, pharmaceutical, and T, M, N as explanatory variables in GSE30759 and TCGA. In univariate Cox regression, stage, age, and risk were significantly related to OS in GSE30759; age, M, N, and risk were associated with OS in TCGA (P < 0.01, Table 1). After multivariate adjustment, the prognostic prediction model remained a powerful and independent prognostic factor for patients with CC (training cohort: HR = 3.09, 95% CI = 1.24–7.70, P = 0.016, validation cohort: HR = 1.825, 95% CI = 1.13–2.95, P = 0.014, Table 1), presenting the potential to act as an independent factor for OS prediction. To develop a clinically feasible and convenient method that could predict the survival probability of individual patients, we constructed a prognostic nomogram, which included clinical variables and risk to predict the 3- and 5-year OS of CC patients (Figure S6A and B). The calibration curve also shows satisfactory agreement between predictive values and observation values for the probabilities of 3- and 5-year OS in the cohort, with the Harrell’s concordance index for predicting OS of CC being 0.736 and 0.678, respectively (Figure S6C and D), indicating that the nomogram could be helpful for predicting the survival of CC patients.
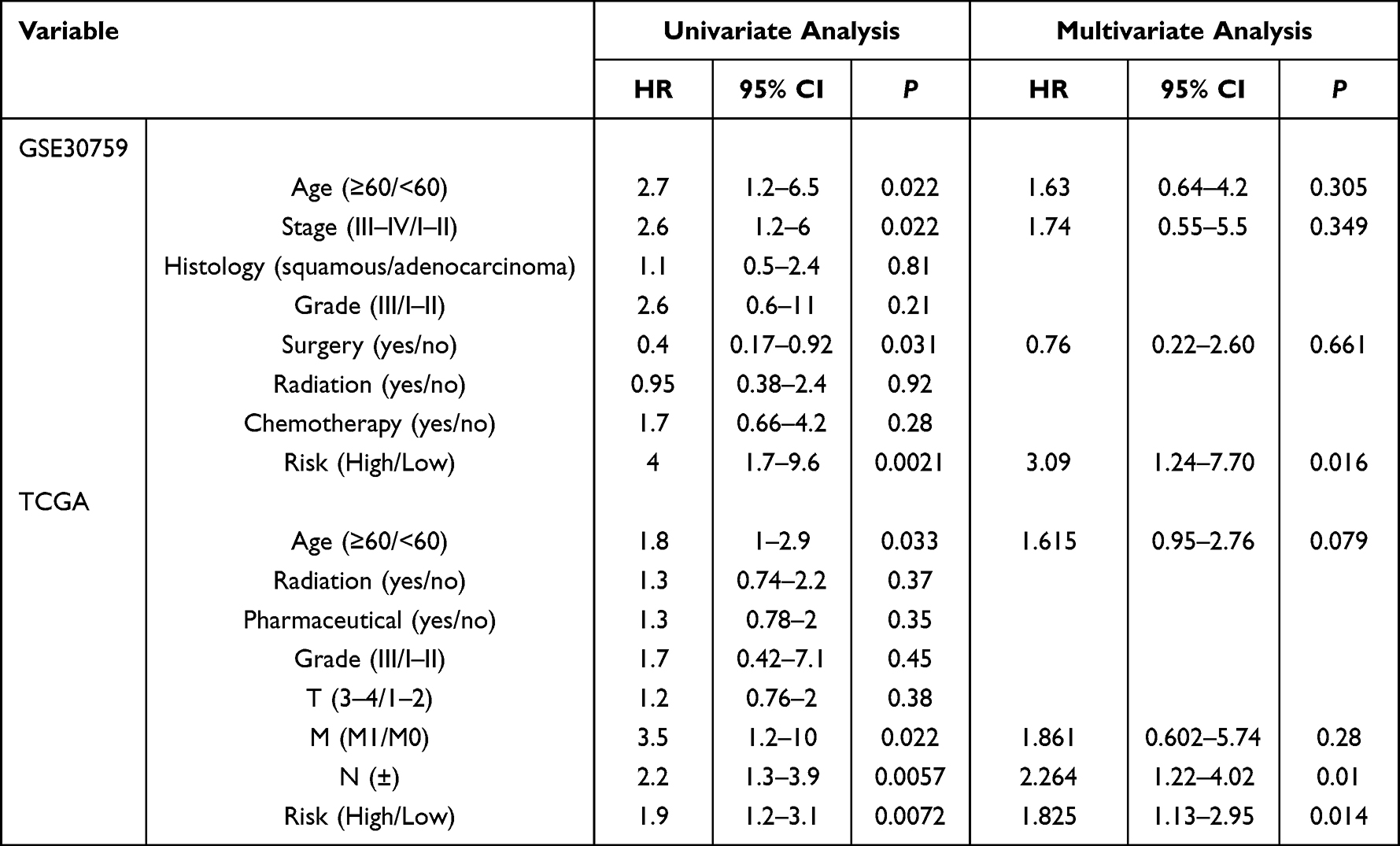 |
Table 1 Univariate and Multivariate Cox Regression Analyses of the Prognostic Prediction Model in CC Patients |
Discussion
The diagnosis of CC mainly depends on the results of colposcopy. However, colposcopy is not suitable for CC screening due to its invasive and the expensive features, and abuse of colposcopy will lead to waste of medical resources.19 On the other hand, HPV detection has higher sensitivity than cytology, but its lower specificity leads to some unnecessary referral and treatment.20 Recent developments in high-throughput approaches are considered the main elements of the most important revolution of the modern medicine and offering opportunities in the field of cancer methylation research, and DNA methylation markers could potentially provide better sensitivity and specificity for early detection of CC and provide opportunities for clinicians to implement intervention treatment before disease progression.20 DNA methylation is not as molecularly constrained and can be detected in almost any tissue types such as biopsied tissues, blood, urine, stools, or other body fluids.10,21,22 The most important advantage of DNA methylation assays is that it can detect early-stage cancers, and several reasons may contribute to this fascinating ability: (i) aberrant DNA methylation occurs in the early phase of tumorigenesis and can be specific for cancer-type or tissue-type;23 (ii) the patterns of DNA methylation are widespread across the same tumor types and tumor tissue, whereas somatic mutations are usually limited to subpopulations of tumor cells.24 Thus, DNA methylation profiles can be used to develop novel markers or classifiers that can be applied to non-invasive and specific detection of early cancer.
In this study, we developed a five methylation marker panel (FHL2/SOX14/ZNF541/EDNRB/RUNX3) corresponding to five CpGs (cg10635061, cg16428251, cg22341310, cg23316360, and cg24019564) to distinguish CC tissues from normal tissues. It is widely documented that cancer-related changes in DNA methylation of cytosine usually occur in areas called CpG islands, and with a relatively aggregated representation of CpGs,25–27 our results show that cg16428251, cg22341310, cg23316360, and cg24019564 are located on CpG island except cg10635061, which is consistent with the theoretical basis. In the subsequent verification process, to ensure the independence of each dataset, we did not perform batch correction and merge into a dataset. As we know, hypermethylation levels vary between different studies which may not only have to do with storage, platform, and test characteristics, but may also represent differences between heterogeneity and populations of CC and other unknown factors.28 cg16428251, cg22341310, and cg23316360 show excellent performance in stratifying tumor tissue and normal tissue in six different cohorts (AUC > 0.8), but cg10635061 and cg24019564 sometimes exhibit unsatisfactory efficacy except in some datasets. The combination of five methylation sites was found to have a correct rate of 79.8–80.0% for the detection of CIN3, with performance comparable to that of another methylation panel (JAM3, EPB41L3, and TERT).29 Apart from this, the combined diagnostic classification of five methylation sites in five independent cohorts not only were significantly better than the single marker (sensitivity 100%; specificity 100%) in discriminating CC and normal samples, but also showed significantly better performance in distinguishing CIN3 from mixed samples, similar to previously demonstrated results on methylation markers to distinguish tumor tissue from mixed tissues.30–32 This result verifies that the increasing degree of methylation proposed by Valentina Fiano is related to the severity of cervical carcinogenesis.33 There is no discrepancy of cg10635061, cg16428251, and cg24019564 in methylation levels between HPV-negative and HPV-positive patients, supporting the theory that HPV infection is necessary but not the only prerequisite for cervical carcinogenesis,34 and DNA aberrant methylation in CC patients may be responsible for this phenomenon.16 The results indicate that combined diagnostic classification of five methylation sites is suitable for all HPV-positive and -negative CC patients.
Several downstream genes of the five CpG markers have previously been proposed as candidate DNA methylation markers for the early detection of malignancies, such as SOX14, EDNRB, and RUNX3. SOX14 is a member of the SOX family of transcription factors and is reported to promote proliferation of CC cells by mediating the Wnt/β-catenin pathway.35 However, a recently published study has also confirmed that SOX14 plays a tumor suppressor role in CC cells by upregulating the p53 signaling pathway.36 The specific mechanism of SOX14 in the carcinogenesis of CC needs further exploration. Furthermore, Rong Wang et al revealed that SOX14 is significantly hypermethylated in CC and achieved 96% accuracy in CC diagnosis,37 which results are extremely consistent with our study. Aberrant hypermethylation and expression inhibition of EDNRB had been found in multitype tumor tissues compared to normal groups in a series of studies,38–41 and EDNRB methylation status was shown to have sensitivity and specificity of 38% and 78% for detection of oral cancer.42 In our study, we focused on evaluating the diagnostic performance of a specific DNA methylation site, which is upstream of EDNRB, with AUC values for discrimination between the CC and the control groups of 0.989, 0.996, 1,0.942,1 and 0.981, respectively. Another candidate methylation site corresponding gene, RUNX3, was also previously shown to increase methylation in various types of malignancies, including gliomas and gastric, breast, colorectal, liver, lung, and bladder cancers.43,44 Meanwhile, RUNX3 methylation was illustrated to be useful in detecting early gastric cancer.45 All of the results concerning the above CpG markers methylation status in cancer are in agreement with our study performed in CC and suggesting that the CpG markers can serve as an alternative method to HPV test or as a supplementary tool for early detection of CC.
There is an increasing application of prognostic prediction models with multi-CpG markers or multi-genes that is used to predict the outcome of cancer patients,15,46,47 due to the carcinogenesis and development of neoplasms usually resulting from the interaction of multiple molecular elements. Therefore, a prognostic risk model was then constructed by using two methylation site markers of cg08849574 and cg13084335. By utilizing this prognostic prediction model, significant statistical difference was seen in the survival curve between the high- and low-risk groups in the training dataset, and the discriminative performance was validated in an external dataset. The prognostic prediction model not only can help clinicians distinguish low-risk patients to prevent overtreatment, but also can be helpful for oncologists to conduct more aggressive treatment and surveillance for those high-risk patients. A further study of the correlation between the prognostic prediction model and clinical characteristics showed that our model has an independent prognostic factor by performing the univariate and multivariate Cox analysis. Moreover, a nomogram comprising clinical features and the risk model was constructed to facilitate prediction of each individual patient prognosis. In our study, the c-index for the nomogram in the training cohort and the testing cohort of CC patients was 0.736 and 0.678, respectively (3-, 5-year OS), significantly higher than previous research reported by Jin Yang et al and comparable to the research conducted by Polterauer et al in predicting the outcome of CC patients.48,49 Therefore, the nomogram based on the prognostic prediction model can potentially serve as a useful tool for predicting the survival time of CC patients and helping oncologists to individualize treatment for patients.
Limitations of the present study also should be acknowledged. Although we searched all available methylation data in the public database, the sample size might not be adequate and may lead to selection bias. More external independent datasets and larger samples are needed for further validation. Second, additional experimental researches are required to elucidate the specific mechanism and the function of these methylation markers that are included in the diagnosis panel and prognostic prediction model in the carcinogenesis and progression of CC.
Conclusion
Collectively, the newly discovered DNA methylation markers in our study may enable better discrimination between CC and normal control, providing the promise of novel biomarkers for early cancer prediction and diagnosis. In addition, the prognostic prediction model which uses two methylation sites demonstrated the excellent value of risk stratification and could help oncologists to individualize treatment for patients. Most importantly, pioneering diagnostic methodologies and risk stratification based on the field of methylation research may be integrated into the oncologist’s practice in the future.
Data Sharing Statement
The datasets analyzed for this study can be found in the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/), Xena website (https://xenabrowser.net/datapages/), and TCGA database (GDC, https://portal.gdc.cancer.gov/).
Ethics Approval and Consent to Participate
The need for ethics approval was waived by the Department of Scientific Research Management, Nanping First Hospital Affiliated to Fujian Medical University.
Acknowledgments
We would like to acknowledge the TCGA and the GEO database for providing data.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no conflicts of interest in relation to this work.
References
1. Cancer Genome Atlas Research N, Albert Einstein College of M, Analytical Biological S, Barretos Cancer H, Baylor College of M, Beckman Research Institute of City of H, Buck Institute for Research on A, Canada’s Michael Smith Genome Sciences C, Harvard Medical S, Helen FGCC. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543(7645):378–384. doi:10.1038/nature21386
2. Arbyn M, Weiderpass E, Bruni L, et al. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Glob Health. 2020;8(2):e191–e203. doi:10.1016/S2214-109X(19)30482-6
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
4. Kagabu M, Nagasawa T, Fukagawa D, et al. Immunotherapy for uterine cervical cancer. Healthcare. 2019;7:108.
5. Marret G, Borcoman E, Le Tourneau C. Pembrolizumab for the treatment of cervical cancer. Expert Opin Biol Ther. 2019;19(9):871–877. doi:10.1080/14712598.2019.1646721
6. Pimple SA, Mishra GA. Global strategies for cervical cancer prevention and screening. Minerva Ginecol. 2019;71(4):313–320. doi:10.23736/S0026-4784.19.04397-1
7. Ronco G, Dillner J, Elfstrom KM, et al. Efficacy of HPV-based screening for prevention of invasive cervical cancer: follow-up of four European randomised controlled trials. Lancet. 2014;383(9916):524–532. doi:10.1016/S0140-6736(13)62218-7
8. d’Errico M, Alwers E, Zhang Y, Edelmann D, Brenner H, Hoffmeister M. Identification of prognostic DNA methylation biomarkers in patients with gastrointestinal adenocarcinomas: a systematic review of epigenome-wide studies. Cancer Treat Rev. 2020;82:101933. doi:10.1016/j.ctrv.2019.101933
9. Li C, Ke J, Liu J, Su J. DNA methylation data-based molecular subtype classification related to the prognosis of patients with cervical cancer. J Cell Biochem. 2020;121(3):2713–2724. doi:10.1002/jcb.29491
10. Wong CC, Li W, Chan B, Yu J. Epigenomic biomarkers for prognostication and diagnosis of gastrointestinal cancers. Semin Cancer Biol. 2019;55:90–105. doi:10.1016/j.semcancer.2018.04.002
11. Bhat S, Kabekkodu SP, Noronha A, Satyamoorthy K. Biological implications and therapeutic significance of DNA methylation regulated genes in cervical cancer. Biochimie. 2016;121:298–311. doi:10.1016/j.biochi.2015.12.018
12. Urbano A, Smith J, Weeks RJ, Chatterjee A. Gene-specific targeting of DNA methylation in the mammalian genome. Cancers. 2019;11(10):1515. doi:10.3390/cancers11101515
13. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–220. doi:10.1038/nrg3354
14. Koch A, Joosten SC, Feng Z, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol. 2018;15(7):459–466. doi:10.1038/s41571-018-0004-4
15. Luo H, Zhao Q, Wei W, et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci Transl Med. 2020;12(524). doi:10.1126/scitranslmed.aax7533.
16. Lu Q, Ma D, Zhao S. DNA methylation changes in cervical cancers. Methods Mol Biol. 2012;863:155–176.
17. Xu RH, Wei W, Krawczyk M, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater. 2017;16(11):1155–1161. doi:10.1038/nmat4997
18. Schoonjans F, Zalata A, Depuydt CE, Comhaire FH. MedCalc: a new computer program for medical statistics. Comput Methods Programs Biomed. 1995;48(3):257–262. doi:10.1016/0169-2607(95)01703-8
19. Lu J, Song E, Ghoneim A, Alrashoud M. Machine learning for assisting cervical cancer diagnosis: an ensemble approach. Future Gener Comput Syst. 2020;106:199–205. doi:10.1016/j.future.2019.12.033
20. van Leeuwen RW, Ostrbenk A, Poljak M, van der Zee AGJ, Schuuring E, Wisman GBA. DNA methylation markers as a triage test for identification of cervical lesions in a high risk human papillomavirus positive screening cohort. Int J Cancer. 2019;144(4):746–754. doi:10.1002/ijc.31897
21. Liu B, Ricarte-Filho J, Mallisetty A, et al. Detection of promoter DNA methylation in urine and plasma aids the detection of non-small cell lung cancer. Clin Cancer Res. 2020;26(16):4339–4348.
22. Liu X, Wen J, Li C, Wang H, Wang J, Zou H. High-Yield methylation markers for stool-based detection of colorectal cancer. Dig Dis Sci. 2020;65(6):1710–1719. doi:10.1007/s10620-019-05908-9
23. Rosenquist R, Esteller M, Plass C. Introduction: epigenetics in cancer. Semin Cancer Biol. 2018;51:iv–v. doi:10.1016/j.semcancer.2018.07.002
24. Roy D, Tiirikainen M. Diagnostic power of DNA methylation classifiers for early detection of cancer. Trends Cancer. 2020;6(2):78–81. doi:10.1016/j.trecan.2019.12.006
25. Lorincz AT. Cancer diagnostic classifiers based on quantitative DNA methylation. Expert Rev Mol Diagn. 2014;14(3):293–305. doi:10.1586/14737159.2014.897610
26. Farkas SA, Milutin-Gasperov N, Grce M, Nilsson TK. Genome-wide DNA methylation assay reveals novel candidate biomarker genes in cervical cancer. Epigenetics. 2013;8(11):1213–1225. doi:10.4161/epi.26346
27. Vrba L, Futscher BW. A suite of DNA methylation markers that can detect most common human cancers. Epigenetics. 2018;13(1):61–72. doi:10.1080/15592294.2017.1412907
28. von Knebel Doeberitz M, Meijer CJLM, Lorincz A, Doorbar J, Leeman A: Infection to Cancer—Finding useful biomarkers for predicting risk of progression to cancer. 2020:269–282.
29. Eijsink JJ, Lendvai A, Deregowski V, et al. A four-gene methylation marker panel as triage test in high-risk human papillomavirus positive patients. Int J Cancer. 2012;130(8):1861–1869. doi:10.1002/ijc.26326
30. Hao X, Luo H, Krawczyk M, et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proc Natl Acad Sci U S A. 2017;114(28):7414–7419. doi:10.1073/pnas.1703577114
31. Jiang H, Ou Z, He Y, et al. DNA methylation markers in the diagnosis and prognosis of common leukemias. Signal Transduct Target Ther. 2020;5(1). doi:10.1038/s41392-019-0090-5.
32. Ying J, Xu T, Wang Q, Ye J, Lyu J. Exploration of DNA methylation markers for diagnosis and prognosis of patients with endometrial cancer. Epigenetics. 2018;13(5):490–504. doi:10.1080/15592294.2018.1474071
33. Fiano V, Trevisan M, Fasanelli F, et al. Methylation in host and viral genes as marker of aggressiveness in cervical lesions: analysis in 543 unscreened women. Gynecol Oncol. 2018;151(2):319–326. doi:10.1016/j.ygyno.2018.08.031
34. Yang HJ. Aberrant DNA methylation in cervical carcinogenesis. Chin J Cancer. 2013;32(1):42–48. doi:10.5732/cjc.012.10033
35. Li F, Wang T, Tang S. SOX14 promotes proliferation and invasion of cervical cancer cells through Wnt/beta-catenin pathway. Int J Clin Exp Pathol. 2015;8(2):1698–1704.
36. Stanisavljevic D, Petrovic I, Vukovic V, et al. SOX14 activates the p53 signaling pathway and induces apoptosis in a cervical carcinoma cell line. PLoS One. 2017;12(9):e0184686. doi:10.1371/journal.pone.0184686
37. Wang R, van Leeuwen RW, Boers A, et al. Genome-wide methylome analysis using MethylCap-seq uncovers 4 hypermethylated markers with high sensitivity for both adeno- and squamous-cell cervical carcinoma. Oncotarget. 2016;7(49):80735–80750. doi:10.18632/oncotarget.12598
38. Mousavi Ardehaie R, Hashemzadeh S, Behrouz Sharif S, Ghojazadeh M, Teimoori-Toolabi L, Sakhinia E. Aberrant methylated EDNRB can act as a potential diagnostic biomarker in sporadic colorectal cancer while KISS1 is controversial. Bioengineered. 2017;8(5):555–564. doi:10.1080/21655979.2017.1283458
39. Yuan Y, Du Y, Wang L, Liu X. The value of endothelin receptor type B promoter methylation as a biomarker for the risk assessment and diagnosis of prostate cancer: a meta-analysis. Pathol Res Pract. 2020;216(2):152796. doi:10.1016/j.prp.2019.152796
40. Chen C, Wang L, Liao Q, et al. Hypermethylation of EDNRB promoter contributes to the risk of colorectal cancer. Diagn Pathol. 2013;8(1):199. doi:10.1186/1746-1596-8-199
41. Tao K, Wu C, Wu K, et al. Quantitative analysis of promoter methylation of the EDNRB gene in gastric cancer. Med Oncol. 2012;29(1):107–112. doi:10.1007/s12032-010-9805-8
42. Schussel J, Zhou XC, Zhang Z, et al. EDNRB and DCC salivary rinse hypermethylation has a similar performance as expert clinical examination in discrimination of oral cancer/dysplasia versus benign lesions. Clin Cancer Res. 2013;19:3268–3275. doi:10.1158/1078-0432.CCR-12-3496
43. Liang Y, He L, Yuan H, Jin Y, Yao Y. Association between RUNX3 promoter methylation and non-small cell lung cancer: a meta-analysis. J Thorac Dis. 2014;6(6):694–705. doi:10.3978/j.issn.2072-1439.2014.04.09
44. Steponaitis G, Kazlauskas A, Vaitkiene P, Deltuva VP, Mikuciunas M, Skiriute D. Oncosuppressive Role of RUNX3 in Human Astrocytomas. J Oncol. 2019;2019:1232434. doi:10.1155/2019/1232434
45. Hideura E, Suehiro Y, Nishikawa J, et al. Blood free-circulating DNA testing of methylated RUNX3 is useful for diagnosing early gastric cancer. Cancers. 2020;12(4):789. doi:10.3390/cancers12040789
46. Xu L, He J, Cai Q, Li M, Pu X, Guo Y. An effective seven-CpG-based signature to predict survival in renal clear cell carcinoma by integrating DNA methylation and gene expression. Life Sci. 2020;243:117289. doi:10.1016/j.lfs.2020.117289
47. Peng Y, Wu Q, Wang L, Wang H, Yin F. A DNA methylation signature to improve survival prediction of gastric cancer. Clin Epigenetics. 2020;12(1):15. doi:10.1186/s13148-020-0807-x
48. Yang J, Tian G, Pan Z, et al. Nomograms for predicting the survival rate for cervical cancer patients who undergo radiation therapy: a SEER analysis. Future Oncol. 2019;15:3033–3045.
49. Polterauer S, Grimm C, Hofstetter G, et al. Nomogram prediction for overall survival of patients diagnosed with cervical cancer. Br J Cancer. 2012;107(6):918–924. doi:10.1038/bjc.2012.340
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.