")
Back to Journals » OncoTargets and Therapy » Volume 11
Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial–mesenchymal transition
Authors Chen Y , Tan W, Wang C
Received 23 March 2018
Accepted for publication 12 May 2018
Published 4 July 2018 Volume 2018:11 Pages 3817—3826
DOI https://doi.org/10.2147/OTT.S168317
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yao Dai
Yongxu Chen,1,2,* Wei Tan,1 Changjun Wang1,2,*
1Guangdong General Hospital, Guangdong Academy of Medical Sciences, Guangdong Geriatric Institute, Guangzhou, Guangdong Province, People’s Republic of China; 2School of Medicine, South China University of Technology, Guangzhou, Guangdong Province, People’s Republic of China
*All authors contributed equally to this work
Abstract: Cancer stem cells are a small population of cells with the potential for self-renewal and multi-directional differentiation and are an important source of cancer initiation, treatment resistance, and recurrence. Epithelial–mesenchymal transition (EMT) is a process in which epithelial cells lose their epithelial phenotype and convert to mesenchymal cells. Recent studies have shown that cancer cells undergoing EMT can become stem-like cells. Many kinds of tumors are associated with chronic inflammation, which plays a role in tumor progression. Among the various immune cells mediating chronic inflammation, macrophages account for ~30%–50% of the tumor mass. Macrophages are highly infiltrative in the tumor microenvironment and secrete a series of inflammatory factors and cytokines, such as transforming growth factor (TGF)-β, IL-6, IL-10, and tumor necrosis factor (TNF)-α, which promote EMT and enhance the stemness of cancer cells. This review summarizes and discusses recent research findings on some specific mechanisms of tumor-associated macrophage-derived cytokines in EMT and cancer stemness transition, which are emerging targets of cancer treatment.
Keywords: macrophage, cancer stem cell, tumor immunology, inflammatory cytokine, tumor microenvironment
Introduction
Cancer is a malignant disease with a high mortality that causes a significant burden to the society. Data have shown that up to 14.1 million people are likely to develop cancer annually starting in 2014, which has increased from ~10 million in the year 2003. In both developed and developing countries, cancer ranks second in mortality behind cardiovascular diseases.1,2 Although substantial progress has been made in cancer treatments, major challenges remain, such as tumor recurrence, metastasis, and resistance after conventional treatment. Recent development of cancer stem cell (CSC) theory implies that CSCs within the tumor ultimately lead to cancer recurrence and metastasis causing patient mortality.3 Traditional therapies can only eliminate treatment-sensitive cancer cells; however, CSCs survive due to treatment resistance and divide into offspring cells, resulting in rapid cancer recurrence. It is known that cancer progression is involved with chronic inflammation, a complex process due to interactions between various immune cells and inflammatory factors. Macrophages are one of the main infiltrating immune cells in chronic inflammation, secreting inflammatory factors and cytokines and influencing tumor angiogenesis and metastasis, particularly in CSCs.
Tumor inflammatory microenvironment
Tumor cells and their peripheral microenvironment have been likened to the relationship between “seeds and soil”, a proposition first proposed by Stephen Paget in 1889.4 The components that constitute the “soil”, which in total are called the tumor microenvironment (TME), are extremely complex, and only some of them are understood, despite extensive research. The known elements include tumor cells, fibroblasts, inflammatory mediators, immune cells, reactive oxygen species, and tumor-associated cytokines, among others.5,6 In addition, most cancer patients have a history of specific virus or bacterial infection; therefore, TMEs contain microorganism-related proteins, such as HBX protein in hepatocellular carcinoma (HCC) and the highly abundant tumorigenic proteins cagA and VacA toxins expressed by Helicobacter pylori in gastric cancer.7,8
As previously demonstrated, gene mutations and epigenetic alterations fundamentally trigger tumor initiation and progression. However, scientists have found that the TME plays a non-negligible role in tumor invasion, angiogenesis, and epithelial–mesenchymal transition (EMT; Table 1).
 | Table 1 Influence and mechanisms of components in the TME of cancer cells |
Tumor-associated macrophages
The macrophages that infiltrate the TME are defined as tumor-associated macrophages (TAMs). TAMs are one of the most important immune cells in the TME, which act as a bridge connecting the inflammatory microenvironment and the malignant phenotype of tumor cells. Certain cytokines derived from tumor cells and the TME such as CSF-1, chemokine (C-X-C motif) ligand 12, and CCL2/MCP-1 recruit mononuclear cells into the TME and activate them to become TAMs.21,22
Macrophages are highly plastic and can be activated into two polarized states through two pathways stimulated by different cytokines and chemokines from the TME. The Th1 cytokines such as LPS, IFN-γ, and tumor necrosis factor (TNF)-α induce macrophages into the M1 polarized state, which plays a role in promoting inflammation and antitumor activity, while M2-TAMs polarized by IL-4 and IL-13 function opposite to the M1 type in immunosuppression and anti-inflammation response.23–26 Macrophages are abundant in multiple cancers compared with adjacent tissues, and their number positively correlates with cancer stage and poor prognosis, so M2-TAMs can be regarded as cancer-promoting cells.27–29 TAMs play an indispensable role in the TME by secreting inflammatory factors that mediate the inflammatory microenvironment to regulate proliferation, metastasis, angiogenesis, immunosuppression, and EMT of various cancers (Table 2).
 | Table 2 Specific mechanisms of tumor invasion and progression triggered by TAMs |
CSCs
CSCs are a small population of cells within tumors that were first found in human acute leukemia nearly 2 decades ago. They have the potential for self-renewal, differentiation, and unlimited proliferation and may divide into a series of heterogeneous cancer cell types resulting in cancer recurrence and treatment resistance.43 The CSC theory proposed that CSCs cause tumor treatment failure by acting as progenitor cells that survive conventional treatment, and thus, cancer can be completely cured only by eliminating CSCs.
The origin of CSCs has generated much controversy, and there is still no consensus. Some researchers have argued that CSCs may be derived from normal stem cells or highly differentiated progenitor cells that have dedifferentiated.44 Another view on the origin of CSCs is called the cell fusion theory. In this scenario, fusion genes such as CD74-NRG1, FOXF1, and SYT-SSX are generated after tumor cells fuse with bone marrow-derived progenitor cells, including hematopoietic stem cells and mesenchymal stem cells or mononuclear cells from the TME, transforming various kinds of tumor cells into CSCs.45–50 Cancer cells after cell fusion retain the ability of invasion and metastasis but have also acquired the potential for self-renewal and other stem-like characteristics. What has been confirmed is that CSCs are not like somatic stem cells or embryonic stem cells (ESCs), which exist in the body, but are acquired like tumor cells by tumorigenic factors, implying that the relationship between TME and CSCs is critical. What, if any, molecules from the TME promote the stemness transition?
Markers of CSCs
CSCs share some common surface markers with normal stem cells, such as CD133, CD44, and CD99.51,52 ESC nuclear transcription factors such as SOX-2, Oct3/4, Klf-4, Nanog, and c-Myc are also regarded as CSC markers.53–55 One study showed that even Nestin, a specific marker of neural stem cells, can be used to identify CSCs.56 These markers can be utilized not only to identify and isolate CSCs but also to predict treatment efficacy in the clinic, shedding light on how CSCs contribute to poor survival and tumor progression.55 The markers shared between CSCs and normal stem cells imply that there are some similar biological characteristics between them, such as self-renewal and endless proliferation, under the suitable conditions.
TAM-induced EMT of cancers
EMT is a process by which epithelial cells lose the tight junctions between cells and gain an elongated, fibroblast-like morphology similar to mesenchymal cells, along with downregulation of epithelial markers (E-cadherin, occludins, and claudins) and upregulation of mesenchymal markers (vimentin, fibronectin, and N-cadherin).57,58 It is widely associated with human embryonic development,59 wound healing or tissue repair,60 and angiogenesis.61,62
Evidence shows the ability for metastasis and invasion of cancer cells after EMT is remarkably enhanced, and these mesenchymal-like cells are strongly resistant to targeted drugs or radio- or chemotherapy.63–65 Tumor cells after EMT express high levels of stem surface markers, indicating that these cells have become stem-like cells.66–68 One interesting study revealed that breast CSCs originate from the fusion of M2-TAMs and breast cancer cells; these hybrid cells overexpress mesenchymal-associated genes and stemness markers.48 Therefore, it can be said that tumor cells after EMT are likely becoming CSCs to some extent.
Factors that induce EMT come from the TME. These signals include aberrant expression of microRNAs, abnormal expression of hormone receptors, and factors secreted by cancer-associated stromal cells and fibroblasts, which are all involved with stem-like transition triggered by EMT.69–72 Macrophages secrete various soluble cytokines and inflammatory mediators that are not only involved in tumor angiogenesis, matrix degradation, and invasion but also promote conversion of cancer cells into stem-like cells, resulting in tumor recurrence and metastasis (Figure 1).12
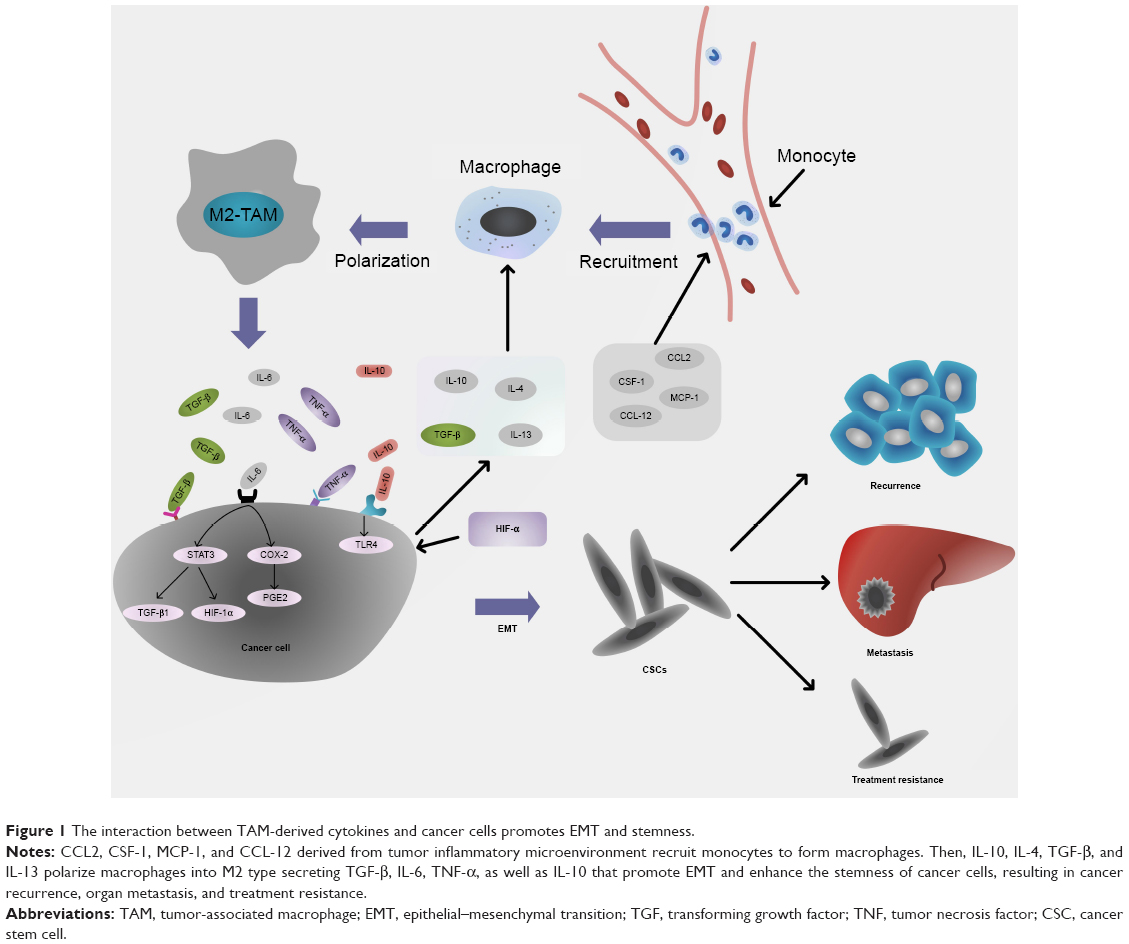 | Figure 1 The interaction between TAM-derived cytokines and cancer cells promotes EMT and stemness. |
Major cytokines derived from TAMs in EMT and CSCs
Transforming growth factor (TGF)-β
The TGF-β family is a group of extracellular growth factors that includes TGF-βs, activins, and bone morphogenetic proteins (BMPs) that regulate growth, migration, angiogenesis, and immune responses of cancer.73 TGF-β has a dual effect on tumor behavior. It plays an anticancer role by suppressing tumor proliferation, inducing apoptosis, and promoting cancer cell differentiation into normal cells but changes its role to become a “catalyst” of cancer progression in the later stages.74 However, the body produces compensatory TGF-β that stimulates angiogenesis and immunosuppression and enhances cell mobilization once cancer cell resistance to the suppressive effects of TGF-β occurs.
TGF-β is the main inflammatory mediator in TME and participates in cell EMT and cancer stemness transition. Treatment of Hep3B or PLC/PRF/5 HCC cells by recombinant TGF-β promotes EMT along with expression of stemness-related markers CD44, EpCAM, and CD133.75–77 In HCC tumor specimens, the density of CD68+ TAMs is positively correlated with EpCAM+ tumor cell distribution; TGF-β1 secreted by M2-TAMs promotes EMT of Hepa1-6 cells to become stem-like cells.78 In brain tumors, heat shock protein-47 enhances high-level TGF-β expression, which induces the TGF-β pathway to promote EMT and stemness in glioblastoma.79 Ye et al80 found that the invasive front of glioma contains abundantly infiltrating TAMs and CD133+ glioma cells in both surgical resections and animal xenografts. These TAMs secrete immunosuppressive factors such as IL-10, and TGF-β1 in turn polarizes macrophages into the M2 type. The hypoxic microenvironment in the center of a tumor could induce stemness-associated transcription factors by enhancing TGF-β1 expression, resulting in regulatory T-cell and macrophage infiltration into the TME.81
IL-6
IL-6 is a multifunctional proinflammatory cytokine in serum and tissues and plays a key role in both acute and chronic inflammatory responses in autoimmune diseases,82 cardiovascular diseases,83 and cancers.84,85 IL-6 in the TME may originate from bone marrow-derived myofibroblasts,86 mesenchymal stem cells,87 mesenchymal stromal cells,88 and/or CD4+ T cells.89 However, the main source of IL-6 is TAMs, and it is closely connected with tumor progression and invasion by inducing lymphangiogenesis and EMT.90–92 The concentration of IL-6 in patient serum is related to advanced tumor stage and overall survival time, and it has potential as a biomarker to evaluate prognosis before surgery.93
IL-6 and CSCs mutually influence each other. Macrophage-derived IL-6 activates the STAT3 signaling pathway, increasing CD44+ HCC cells and enhancing sphere formation when HCC cells are cocultured with macrophages.94 In breast cancer, IL-6 activates the STAT3 pathway and its target genes, such as TGF-β1 and HIF-1α, to increase the proportion of CD44+/CD24− cancer stem-like cells during chemotherapy-induced apoptosis.95 There is a positive feedback loop between IL-6 and CSCs; CD133+ glioma stem cells but not bulk glioma cells induce microglial IL-6 secretion through MyD88-TLR4 signaling in macrophages, which in turn promotes glioma stem cell enlargement.96
Lung cell malignancies triggered by chronic inflammation caused by toxic cigarette extract have elevated IL-6 expression that promotes EMT and CSC formation through the STAT3 pathway.97 Moreover, increased IL-6 induces EMT through the COX-2/PGE2 pathway and promotes tumor cell invasion by activating β-catenin during interactions between macrophages and lung cancer cells.94
IL-10
IL-10 is derived from not only leukocytes but also normal and malignant epithelial cells in hypoxic conditions.98 However, concentration of macrophage-derived IL-10 is almost 10-fold greater than that from leukocytes within the tumor,99 playing a role in immunosuppression in tumors, which is related to tumor drug resistance,100 cellular growth, and proliferation.42,101 IL-10 inhibits both CC and CXC chemokines that are indispensable for activation or recruitment of monocytes, dendritic cells, and neutrophils. In addition, IL-10 directly inhibits cytokine production, CD4+ T-cell proliferation, and T-cell cloning.102
EMT is also triggered by IL-10. Tumor cells cocultured with macrophages go through EMT in an IL-10-dependent manner.103,104 IL-10 expression is elevated when M2-TAMs are cocultured with pancreatic cancer cell lines such as PANC-1 and BxPC-3, causing the EMT of cell lines through the TLR4/IL-10 signaling pathway combined with enhancing of CD133 and CD44, which suggests that IL-10 is the key element in these changes.105
In HCC, hypoxic stress induces the cells to release Netrin-1, resulting in EMT and a high-level of IL-10 expression,106 which may synergistically participate in the promotion of CSCs. HIF-1α in a hypoxic environment in the HCC mouse model drives hepatocytes to secrete IL-10, which activates tissue-resident macrophages to polarize toward the M2-TAM phenotype, a positive feedback enhancing tumor evolution.107 These studies together demonstrate that in an inflammatory microenvironment, IL-10 derived from TAMs or tumor cells plays a role in mediating EMT directly or indirectly, which may enhance tumor cell stemness transition.
TNF-α
TNF is a superfamily of proinflammatory cytokines involved in various inflammation responses, including rheumatoid arthritis and cardiovascular disease, in part through activation of the nuclear factor-kappa B (NF-κB) pathway.108 TNF-α is chiefly released by host innate immune cells, including activated macrophages, T lymphocytes, and natural killer (NK) cells in tumors. Other cells including fibroblasts, smooth muscle cells, and tumor cells also secrete a small quantity of TNF-α.109,110 TNF-α is a critical inflammation mediator in the TME and exerts its antitumor activity by promoting inflammation and immune response, host defense, tumor cell apoptosis, and tumor vasculature destruction.111,112 However, some recent studies have found that TNF-α plays a completely reversed role in tumor progression and is involved in tumor migration,113 metastasis,114 angiogenesis,115 and negative regulation of immune homeostasis.116 The mechanism of these double-edged effects has not been fully elucidated, and they depend on the distribution of TNF-α receptors, the tumor stage, and the tumor type.117–120
TNF-α in the microenvironment promotes EMT of various tumor cells and CSC transition in addition to tumorigenesis as discussed earlier.121–123 Mikami et al124 found that in clear cell renal cell carcinomas (ccRCC), TNF-α is significantly correlated with CD44+ cancer cells in late-stage patients, inducing ccRCC progression and sunitinib resistance through EMT. Murine mammary carcinoma cells show EMT along with a high proportion of CD24-/lowCD44+ phenotypes, when they are exposed to TGF-β/TNF-α in vitro.125 In that study, even the withdrawal of TGF-β/TNF-α from the conditioned medium did not completely reverse the mesenchymal phenotype, indicating that TNF-α promotes stemness at the gene level. This conclusion is in accordance with TNF-α triggering chromosomal instability in liver progenitor cells and contributes to their conversion to liver CSCs.126
Conclusion
EMT contributes to drug resistance, tumor invasion, and CSC transition, and thus, it is a potential target for inhibition to suppress CSC generation. However, critical regulatory pathways are still unknown due to the complicated mechanisms by which EMT promotes cancer cell stemness. Drugs and other methods to suppress this process have only recently begun to be investigated in vitro and have thus far achieved only modest results (Table 3).
 | Table 3 Recent studies on drugs or chemical substances that suppress EMT-induced cancer stem-like cell initiation |
Eliminating CSCs is a critical approach to suppress tumor recurrence and increase treatment sensitivity. However, targeting CSCs has some difficulties due to the limitations of culturing, identifying, and isolating them, which indicates that more attention should be paid to the TME, especially regarding protumor factors. It is reasonable to regard tumor mass and CSCs as parts of a whole, with the physiological functioning of the integrated parts requiring support from the TME. Therefore, it is desirable to destruct the “soil” that is favorable for tumor growth to slow down progression.
In this review, we have summarized TAM-derived cytokines in CSC transition. In addition to secretory pathways and direct contact by cell fusion, some recent studies have found that exosomes may serve as a delivery vehicle and mediate communication between TAMs and cancer cells,136,137 which emphasizes that the cardinal work is to figure out interplays between TAMs in TME and CSCs.
Targeting TAMs is a promising direction, but additional factors need to be considered. First, TAMs infiltrate the TME in abundance, but there are no highly specific markers of TAMs. One study has shown that cancer cells after coculture with TAMs can even express the macrophage-specific marker CD163.103 Second, macrophages are the most important immune cells, and they exert their antitumor role by discovering and eliminating mutated cells in the early stages of tumor initiation. So which macrophage phenotype should be targeted and when is the best time to target it? TAMs can paradoxically promote tumor metastasis and angiogenesis once anti-TAM therapy has ceased.138 Third, some CSCs are in a dormant state where they are arrested in G0/S and have no physiological activity. These CSCs are not regulated by TAMs;139 whether TAMs break the dormant state of CSCs is unknown. These issues and others should be thoroughly studied before targeting of TAMs can become a clinical reality.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (Nos 81774261 and 81373582), the Natural Science Foundation of Guangdong Province (No S2013010012636), the Administration of Traditional Chinese Medicine of Guangdong (Nos 20171001, 20181008 and 20181002.) and the Foundation of GuangZhou Science Technology and Innovation Commission (Nos 201803010088, 201804010118).
Author contributions
Yongxu Chen and Wei Tan contributed to searching literature, drafting, and editing of the manuscript. Changjun Wang participated in the conception of the idea. All authors contributed toward data analysis, drafting, and revising the paper and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Mcguire S. World cancer report 2014. Geneva, Switzerland: world health organization, international agency for research on cancer, WHO press, 2015. Adv Nutr. 2016;7(2):418–419. | ||
Townsend N, Wilson L, Bhatnagar P, Wickramasinghe K, Rayner M, Nichols M. Cardiovascular disease in Europe: epidemiological update 2016. Eur Heart J. 2013;34(39):3028–3034. | ||
Shimokawa M, Ohta Y, Nishikori S, et al. Visualization and targeting of LGR5+ human colon cancer stem cells. Nature. 2017;545(7653):187–192. | ||
Mathot L, Stenninger J. Behavior of seeds and soil in the mechanism of metastasis: a deeper understanding. Cancer Sci. 2012;103(4):626–631. | ||
Chiantore MV, Mangino G, Zangrillo MS, et al. Role of the microenvironment in tumourigenesis: focus on virus-induced tumors. Curr Med Chem. 2015;22(8):958–974. | ||
Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol. 2016;27(8):1482–1492. | ||
Jia Q, Dong Q, Qin L. CCN: core regulatory proteins in the microenvironment that affect the metastasis of hepatocellular carcinoma. Oncotarget. 2016;7(2):1203–1214. | ||
Hardbower DM, Peek RM Jr, Wilson KT. At the bench: Helicobacter pylori, dysregulated host responses, DNA damage, and gastric cancer. J Leukoc Biol. 2014;96(2):201–212. | ||
Ji X, Zhu X, Lu X. Effect of cancer-associated fibroblasts on radiosensitivity of cancer cells. Future Oncol. 2017;13(17):1537–1550. | ||
Zhu Y, Zhang L, Zha H, et al. Stroma-derived fibrinogen-like protein 2 activates cancer-associated fibroblasts to promote tumor growth in lung cancer. Int J Biol Sci. 2017;13(6):804–814. | ||
Zhao XL, Lin Y, Jiang J, et al. HMGB1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J Pathol. 2017;243(3):376–389. | ||
Landskron G, Fuente MDL, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014;2014(2):149185. | ||
Tzanakakis G, Kavasi RM, Voudouri K, et al. The role of the extracellular matrix in cancer-associated epithelial to mesenchymal transition phenomenon. Dev Dyn. 2017;23(6):881–889. | ||
Brooks M, Mo Q, Krasnow R, et al. Positive association of collagen type I with non-muscle invasive bladder cancer progression. Oncotarget. 2016;7(50):82609–82619. | ||
Insua-Rodrã-Guez J, Oskarsson T. The extracellular matrix in breast cancer. Adv Drug Deliv Rev. 2016;97:41–55. | ||
Ren L, Yu Y, Wang L, Zhu Z, Lu R, Yao Z. Hypoxia-induced CCL28 promotes recruitment of regulatory T cells and tumor growth in liver cancer. Oncotarget. 2016;7(46):75763–75773. | ||
Qi Z, Yu L, Zhang J, et al. Hypoxia-inducible factor-2α promotes tumor progression and has crosstalk with Wnt/β-catenin signaling in pancreatic cancer. Mol Cancer. 2017;16(1):119. | ||
Thompson ED, Taube JM, Aschkendrick RJ, et al. PD-L1 expression and the immune microenvironment in primary invasive lobular carcinomas of the breast. Mod Pathol. 2017;30(11):1551–1560. | ||
Chung W, Eum HH, Lee HO, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. 2017;8:15081. | ||
Lundgren S, Karnevi E, Elebro J, et al. The clinical importance of tumour-infiltrating macrophages and dendritic cells in periampullary adenocarcinoma differs by morphological subtype. J Transl Med. 2017;15(1):152. | ||
Morita Y, Zhang R, Leslie M, et al. Pathologic evaluation of tumor-associated macrophage density and vessel inflammation in invasive breast carcinomas. Oncol Lett. 2017;14(2):2111–2118. | ||
Karagiannis SN, Josephs DH, Bax HJ, Spicer JF. Therapeutic IgE antibodies: harnessing a macrophage-mediated immune surveillance mechanism against cancer. Cancer Res. 2017;77(11):2779–2783. | ||
Franklin RA, Liao W, Sarkar A, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–925. | ||
Zheng X, Turkowski K, Mora J, et al. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget. 2017;8(29):48436–48452. | ||
Yin S, Huang J, Li Z, et al. The prognostic and clinicopathological significance of tumor-associated macrophages in patients with gastric cancer: a meta-analysis. PLoS One. 2017;12(1):e0170042. | ||
Lu G, Zhu Q, Jing H, et al. Cyclooxygenase-2 in tumor-associated macrophages promotes metastatic potential of breast cancer cells through Akt pathway. Int J Biol Sci. 2016;12(12):1533–1543. | ||
Cassetta L, Pollard JW. Repolarizing macrophages improves breast cancer therapy. Cell Res. 2017;27(8):963–964. | ||
Huang YJ, Yang CK, Wei PL, et al. Ovatodiolide suppresses colon tumorigenesis and prevents polarization of M2 tumor-associated macrophages through YAP oncogenic pathways. J Hematol Oncol. 2017;10(1):60. | ||
Piaggio F, Kondylis V, Pastorino F, et al. A novel liposomal Clodronate depletes tumor-associated macrophages in primary and metastatic melanoma: anti-angiogenic and anti-tumor effects. J Controlled Release. 2016;223:165–177. | ||
Storr SJ, Safuan S, Ahmad N, El-Refaee M, Jackson AM, Martin SG. Macrophage-derived interleukin-1beta promotes human breast cancer cell migration and lymphatic adhesion in vitro. Cancer Immunol Immunother. 2017;66(10):1287–1294. | ||
Ling L, Chen YS, Yao YD, et al. CCL18 from tumor-associated macrophages promotes angiogenesis in breast cancer. Oncotarget. 2015;6(33):34758–34773. | ||
Kong L, Zhou Y, Hong B, Lv T, Shi Y, Yang J. Deletion of interleukin-6 in monocytes/macrophages suppresses the initiation of hepatocellular carcinoma in mice. J Exp Clin Cancer Res. 2016;35(1):131. | ||
Fu XT, Dai Z, Song K, et al. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/snail pathway. Int J Oncol. 2015;46(2):587–596. | ||
Fujita N, Nishie A, Aishima S, et al. Role of tumor-associated macrophages in the angiogenesis of well-differentiated hepatocellular carcinoma: pathological-radiological correlation. Oncol Rep. 2014;31(6):2499–2505. | ||
Peng LS, Zhang JY, Teng YS, et al. Tumor-associated monocytes/macrophages impair NK-cell function via TGFβ1 in human gastric cancer. Cancer Immunol Res. 2017;5(3):248–256. | ||
Ma HY, Liu XZ, Liang CM. Inflammatory microenvironment contributes to epithelial-mesenchymal transition in gastric cancer. World J Gastroenterol. 2016;22(29):6619–6628. | ||
Mu L, Wang J, Chen Y, et al. Hypoxia-inducible factor-1α and semaphorin4D genes involved with tumor-associated macrophage-induced metastatic behavior and clinical significance in colon cancer. Chin Med J (Engl). 2014;127(20):3568–3575. | ||
Wang X, Zhao X, Wang K, Wu L, Duan T. Interaction of monocytes/macrophages with ovarian cancer cells promotes angiogenesis in vitro. Cancer Sci. 2013;104(4):516–523. | ||
Shieh YS, Hung YJ, Hsieh CB, Chen JS, Chou KC, Liu SY. Tumor-associated macrophage correlated with angiogenesis and progression of mucoepidermoid carcinoma of salivary glands. Ann Surg Oncol. 2009;16(3):751–760. | ||
Tjiu JW, Chen JS, Shun CT, et al. Tumor-associated macrophage-induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase-2 induction. J Invest Dermatol. 2009;129(4):1016–1025. | ||
Zhang X, Zeng Y, Qu Q, et al. PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol. 2017;22(6):1026–1033. | ||
Rui W, Meng L, Chen H, et al. Increased IL-10 mRNA expression in tumor-associated macrophage correlated with late stage of lung cancer. J Exp Clin Cancer Res. 2011;30(1):62. | ||
Azam B, Mozafar K, Rasool KM. New findings on breast cancer stem cells: a review. J Breast Cancer. 2015;18(4):303–312. | ||
Wang B, Jacob ST. Role of cancer stem cells in hepatocarcinogenesis. Genome Med. 2011;3(2):1–6. | ||
Murayama T, Nakaoku T, Enari M, et al. Oncogenic fusion gene CD74-NRG1 confers cancer stem cell-like properties in lung cancer through a IGF2 autocrine/paracrine circuit. Cancer Res. 2016;76(4):974–983. | ||
Wei HJ, Nickoloff JA, Chen WH, et al. FOXF1 mediates mesenchymal stem cell fusion-induced reprogramming of lung cancer cells. Oncotarget. 2014;5(19):9514–9529. | ||
Ding J, Jin W, Chen C, Shao Z, Wu J. Tumor associated macrophage × – cancer cell hybrids may acquire cancer stem cell properties in breast cancer. PLoS One. 2012;7(7):e41942. | ||
Zhou Y, Chen D, Qi Y, et al. Evaluation of expression of cancer stem cell markers and fusion gene in synovial sarcoma: insights into histogenesis and pathogenesis. Oncol Rep. 2017;37(6):3351–3360. | ||
Fei L, Tong L, Wang J, et al. Bone marrow mesenchymal stem cells participate in prostate carcinogenesis and promote growth of prostate cancer by cell fusion in vivo. Oncotarget. 2016;7(21):30924–30934. | ||
Melzer C, Von der Ohe J, Lehnert H, Ungefroren H, Hass R. Cancer stem cell niche models and contribution by mesenchymal stroma/stem cells. Mol Cancer. 2017;16(1):28. | ||
Sun JH, Luo Q, Liu LL, Song GB. Liver cancer stem cell markers: progression and therapeutic implications. World J Gastroenterol. 2016;22(13):3547–3557. | ||
Bartakova A, Michalova K, Presl J, Vlasak P, Kostun J, Bouda J. CD44 as a cancer stem cell marker and its prognostic value in patients with ovarian carcinoma. J Obstet Gynaecol. 2017;38(1):110–114. | ||
Zhu F, Qian W, Zhang H, et al. SOX2 is a marker for stem-like tumor cells in bladder cancer. Stem Cell Reports. 2017;9(2):429–437. | ||
Gwak JM, Kim M, Kim HJ, Jang MH, Park SY. Expression of embryonal stem cell transcription factors in breast cancer: Oct4 as an indicator for poor clinical outcome and tamoxifen resistance. Oncotarget. 2017;8(22):36305–36318. | ||
De LAA, Ferrari F, Xi R, et al. Hallmarks of pluripotency. Nature. 2015;525(7570):469–478. | ||
Ruggieri S, Tamma R, Resta N, et al. Stat3-positive tumor cells contribute to vessels neoformation in primary central nervous system lymphoma. Oncotarget. 2017;8(19):31254–31269. | ||
Chen D, Zhou H, Liu G, Zhao Y, Cao G, Liu Q. SPOCK1 promotes the invasion and metastasis of gastric cancer through slug-induced epithelial-mesenchymal transition. J Cell Mol Med. 2017;22(2):797–807. | ||
Xie K, Ye Y, Zeng Y, Gu J, Yang H, Wu X. Polymorphisms in genes related to epithelial-mesenchymal transition and risk of non-small cell lung cancer. Carcinogenesis. 2017;38(10):1029–1035. | ||
Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014;7(344):re8. | ||
Arthurfarraj PJ, Morgan CC, Adamowicz M, et al. Changes in the coding and non-coding transcriptome and DNA methylome that define the Schwann cell repair phenotype after nerve injury. Cell Rep. 2017;20(11):2719–2734. | ||
Ribatti D. Epithelial-mesenchymal transition in morphogenesis, cancer progression and angiogenesis. Exp Cell Res. 2017;353(1):1–5. | ||
Li C, Li Q, Cai Y, et al. Overexpression of angiopoietin 2 promotes the formation of oral squamous cell carcinoma by increasing epithelial–mesenchymal transition-induced angiogenesis. Cancer Gene Ther. 2016;23(9):295–302. | ||
Wang W, Zhao Y, Yao S, et al. Nigericin inhibits epithelial ovarian cancer metastasis by suppressing the cell cycle and epithelial-mesenchymal transition. Biochemistry (Mosc). 2017;82(8):933–941. | ||
Liu Z, Gao W. Leptomycin B reduces primary and acquired resistance of gefitinib in lung cancer cells. Toxicol Appl Pharmacol. 2017;335:16–27. | ||
Zhang R, Sun S, Ji F, et al. CNTN-1 enhances chemoresistance in human lung adenocarcinoma through induction of epithelial-mesenchymal transition by targeting the PI3K/Akt pathway. Cell Physiol Biochem. 2017;43(2):465–480. | ||
Wei H, Liang F, Cheng W, et al. The mechanisms for lung cancer risk of PM2.5: induction of epithelial-mesenchymal transition and cancer stem cell properties in human non-small cell lung cancer cells. Environ Toxicol. 2017;32(11):2341–2351. | ||
Li B, Lu Y, Yu L, et al. miR-221/222 promote cancer stem-like cell properties and tumor growth of breast cancer via targeting PTEN and sustained Akt/NF-κB/COX-2 activation. Chem Biol Interact. 2017;277:33–42. | ||
Silva ILD, Monteromontero L, Martínvillar E, et al. Reduced expression of the murine HLA-G homolog Qa-2 is associated with malignancy, epithelial-mesenchymal transition and stemness in breast cancer cells. Sci Rep. 2017;7(1):6276. | ||
Findlay VJ, Wang C, Watson DK, Camp ER. Epithelial-to-mesenchymal transition and the cancer stem cell phenotype: insights from cancer biology with therapeutic implications for colorectal cancer. Cancer Gene Ther. 2014;21(5):181–187. | ||
Lam SS, Mak AS, Yam JW, Cheung AN, Ngan HY, Wong AS. Targeting estrogen-related receptor alpha inhibits epithelial-to-mesenchymal transition and stem cell properties of ovarian cancer cells. Mol Ther. 2014;22(4):743–751. | ||
Kong L, Guo S, Liu C, et al. Overexpression of SDF-1 activates the NF-κB pathway to induce epithelial to mesenchymal transition and cancer stem cell-like phenotypes of breast cancer cells. Int J Oncol. 2016;48(3):1085–1094. | ||
Pistore C, Giannoni E, Colangelo T, et al. DNA methylation variations are required for epithelial-to-mesenchymal transition induced by cancer-associated fibroblasts in prostate cancer cells. Oncogene. 2017;36(40):5551–5566. | ||
Yan Y, Zhang J, Li JH, et al. High tumor-associated macrophages infiltration is associated with poor prognosis and may contribute to the phenomenon of epithelial-to-mesenchymal transition in gastric cancer. Oncotargets Ther. 2016;9(Issue 1):3975–3983. | ||
Shen W, Tao GQ, Zhang Y, Cai B, Sun J, Tian ZQ. TGF-β in pancreatic cancer initiation and progression: two sides of the same coin. Cell Biosci. 2017;7:39. | ||
Malfettone A, Soukupova J, Bertran E, et al. Transforming growth factor-β-induced plasticity causes a migratory stemness phenotype in hepatocellular carcinoma. Cancer Lett. 2017;392:39–50. | ||
Bae WJ, Lee SH, Rho YS, Koo BS, Lim YC. Transforming growth factor β1 enhances stemness of head and neck squamous cell carcinoma cells through activation of Wnt signaling. Oncol Lett. 2016;12(6):5315–5320. | ||
Chen W, Zhou S, Mao L, et al. Crosstalk between TGF-β signaling and miRNAs in breast cancer metastasis. Tumor Biol. 2016;37(8):10011–10019. | ||
Fan QM, Jing YY, Yu GF, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014;352(2):160–168. | ||
Jiang X, Zhou T, Wang Z, Qi B, Xia H. HSP47 promotes glioblastoma stem-like cell survival by modulating tumor microenvironment extracellular matrix through TGF-β pathway. ACS Chem Neurosci. 2016;8(1):128–134. | ||
Ye X, Xu S, Xin Y, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J Immunol. 2012;189(1):444–453. | ||
Hasmim M, Noman MZ, Messai Y, et al. Cutting edge: hypoxia-induced nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J Immunol. 2013;191(12):5802–5806. | ||
Yao X, Huang J, Zhong H, et al. Targeting interleukin-6 in inflammatory autoimmune diseases and cancers. Pharmacol Ther. 2014;141(2):125–139. | ||
Hashimotokataoka T, Hosen N, Sonobe T, et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc Natl Acad Sci U S A. 2015;112(20):E2677–E2686. | ||
Xia Y, Khoi PN, Yoon HJ, et al. Piperine inhibits IL-1β-induced IL-6 expression by suppressing p38 MAPK and STAT3 activation in gastric cancer cells. Mol Cell Biochem. 2015;398(1–2):147–156. | ||
Waldner MJ, Neurath MF. Master regulator of intestinal disease: IL-6 in chronic inflammation and cancer development. Semin Immunol. 2014;26(1):75–79. | ||
Zhu L, Cheng X, Shi J, et al. Crosstalk between bone marrow-derived myofibroblasts and gastric cancer cells regulates cancer stemness and promotes tumorigenesis. Oncogene. 2016;35(41):5388–5399. | ||
So KA, Min KJ, Hong JH, Lee JK. Interleukin-6 expression by interactions between gynecologic cancer cells and human mesenchymal stem cells promotes epithelial-mesenchymal transition. Int J Oncol. 2015;47(4):1451–1459. | ||
Jiang C, Zhang Q, Shanti RM, et al. Mesenchymal stromal cell-derived IL-6 promotes epithelial-mesenchymal transition and acquisition of epithelial stem-like cell properties in ameloblastoma epithelial cells. Stem Cells. 2017;35(9):2083–2094. | ||
Chen Q, Yang D, Zong H, et al. Growth-induced stress enhances epithelial-mesenchymal transition induced by IL-6 in clear cell renal cell carcinoma via the Akt/GSK-3β/β-catenin signaling pathway. Oncogenesis. 2017;6(8):e375. | ||
Zhao G, Zhu G, Huang Y, et al. IL-6 mediates the signal pathway of JAK-STAT3-VEGF-C promoting growth, invasion and lymphangiogenesis in gastric cancer. Oncol Rep. 2016;35(3):1787–1795. | ||
Dehai C, Bo P, Qiang T, et al. Enhanced invasion of lung adenocarcinoma cells after co-culture with THP-1-derived macrophages via the induction of EMT by IL-6. Immunol Lett. 2014;160(1):1–10. | ||
Che D, Zhang S, Jing Z, et al. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE2/β-catenin signalling pathway. Mol Immunol. 2017;90:197–210. | ||
Shimazaki J, Goto Y, Nishida K, et al. In patients with colorectal cancer, preoperative serum interleukin-6 level and granulocyte/lymphocyte ratio are clinically relevant biomarkers of long-term cancer progression. Oncology. 2013;84(6):356–361. | ||
Wan S, Zhao E, Kryczek I, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147(6):1393–1404. | ||
Zhou N, Zhang Y, Zhang X, et al. Exposure of tumor-associated macrophages to apoptotic MCF-7 cells promotes breast cancer growth and metastasis. Gastroenterology. 2015;16(6):11966–11982. | ||
Dzaye OD, Hu F, Derkow K, et al. Glioma stem cells but not bulk glioma cells upregulate IL-6 secretion in microglia/brain macrophages via toll-like receptor 4 signaling. J Neuropathol Exp Neurol. 2016;75(5):429–440. | ||
Liu Y, Luo F, Xu Y, et al. Epithelial-mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract. Toxicol Appl Pharmacol. 2015;282(1):9–19. | ||
O’Garra A, Barrat FJ, Castro AG, Vicari A, Hawrylowicz C. Strategies for use of IL-10 or its antagonists in human disease. Immunol Rev. 2008;223(1):114–131. | ||
Ruffell B, Chang-Strachan D, Chan V, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623–637. | ||
Yang C, He L, He P, et al. Increased drug resistance in breast cancer by tumor-associated macrophages through IL-10/STAT3/bcl-2 signaling pathway. Med Oncol. 2015;32(2):352. | ||
Jung M, Weigert A, Tausendschön M, et al. Interleukin-10-induced neutrophil gelatinase-associated lipocalin production in macrophages with consequences for tumor growth. Mol Cell Biol. 2012;32(19):3938–3948. | ||
Moore KW, De WMR, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19(1):683–765. | ||
Maniecki MB, Etzerodt A, Ulhøi BP, et al. Tumor-promoting macrophages induce the expression of the macrophage-specific receptor CD163 in malignant cells. Int J Cancer. 2012;131(10):2320–2331. | ||
Liu CY, Xu JY, Shi XY, et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Invest. 2013;93(7):844–854. | ||
Meng F, Li C, Li W, Gao Z, Guo K, Song S. Interaction between pancreatic cancer cells and tumor-associated macrophages promotes the invasion of pancreatic cancer cells and the differentiation and migration of macrophages. IUBMB Life. 2014;66(12):835–846. | ||
Yan W, Han P, Zhou Z, et al. Netrin-1 induces epithelial-mesenchymal transition and promotes hepatocellular carcinoma invasiveness. Dig Dis Sci. 2014;59(6):1213–1221. | ||
Ambade A, Satishchandran A, Saha B, et al. Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1αinduced IL-10. Oncoimmunology. 2016;5(10):e1221557. | ||
Gupta SC, Tyagi AK, Deshmukh-Taskar P, Hinojosa M, Prasad S, Aggarwal BB. Downregulation of tumor necrosis factor and other proinflammatory biomarkers by polyphenols. Arch Biochem Biophys. 2014;559(5):91–99. | ||
Horssen RV, Hagen TLMT, Eggermont AMM. TNF-α in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006;11(4):397–408. | ||
Vinnakota K, Zhang Y, Selvanesan BC, et al. M2-like macrophages induce colon cancer cell invasion via matrix metalloproteinases. J Cell Physiol. 2017;232(12):3468–3480. | ||
Lu L, Li ZJ, Li LF, et al. Vascular-targeted TNFα improves tumor blood vessel function and enhances antitumor immunity and chemotherapy in colorectal cancer. J Control Release. 2015;210:134–146. | ||
Jeon H, Kim D, Choi M, et al. Targeted cancer therapy using a fusion protein of TNFα and a tumor-associated fibronectin-specific aptide. Mol Pharm. 2017;14(11):3772–3779. | ||
Xia P, Zhang R, Ge G. C/EBPβ Mediates TNF-α-induced cancer cell migration by inducing MMP expression dependent on p38 MAPK. J Cell Biochem. 2015;116(12):2766–2777. | ||
Ji H, Cao R, Yang Y, et al. TNFR1 mediates TNF-α-induced tumour lymphangiogenesis and metastasis by modulating VEGF-C-VEGFR3 signalling. Nat Commun. 2014;5:4944. | ||
Zhou B, Zhuang XM, Wang YY, et al. Tumor necrosis factor α induces myofibroblast differentiation in human tongue cancer and promotes invasiveness and angiogenesis via secretion of stromal cell-derived factor-1. Oral Oncol. 2015;51(12):1095–1102. | ||
Zhang YH, Yan HQ, Wang F, et al. TIPE2 inhibits TNF-α-induced hepatocellular carcinoma cell metastasis via Erk1/2 downregulation and NF-κB activation. Int J Oncol. 2015;46(1):254–264. | ||
Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015;15(6):362–374. | ||
Obeed OAA, Alkhayal KA, Sheikh AA, et al. Increased expression of tumor necrosis factor-α is associated with advanced colorectal cancer stages. World J Gastroenterol. 2014;20(48):18390–18396. | ||
Lu X, Cheng C, Wang G, Shu X, Ma J, Tong Q. Synergistic enhancement of cancer therapy using a combination of fusion protein MG7-scFv/SEB and tumor necrosis factor alpha. Protein Pept Lett. 2013;20(4):467–472. | ||
Ham B, Fernandez MC, D’Costa Z, Brodt P. The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 2016;11(1):1–27. | ||
Singh R, Shankar BS, Sainis KB. TGF-β1–ROS–ATM–CREB signaling axis in macrophage mediated migration of human breast cancer MCF7 cells. Cell Signal. 2014;26(7):1604–1615. | ||
Storci G, Sansone P, Mari S, et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J Cell Physiol. 2010;225(3):682–691. | ||
Dong W, Sun S, Cao X, et al. Exposure to TNF-α combined with TGF-β induces carcinogenesis in vitro via NF-κB/Twist axis. Oncol Rep. 2017;37(3):1873–1882. | ||
Mikami S, Mizuno R, Kosaka T, Saya H, Oya M, Okada Y. Expression of TNF-α and CD44 is implicated in poor prognosis, cancer cell invasion, metastasis and resistance to the sunitinib treatment in clear cell renal cell carcinomas. Int J Cancer. 2015;136(7):1504–1514. | ||
Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL. TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011;71(13):4707–4719. | ||
Li XF, Chen C, Xiang DM, et al. Chronic inflammation-elicited liver progenitor cell conversion to liver cancer stem cell with clinical significance. Hepatology. 2017;66(6):1934–1951. | ||
Li W, Wang Q, Su Q, et al. Honokiol suppresses renal cancer cells’ metastasis via dual-blocking epithelial-mesenchymal transition and cancer stem cell properties through modulating miR-141/ZEB2 signaling. Mol Cells. 2014;37(5):383–388. | ||
Cao X, Ren K, Song Z, et al. 7-Difluoromethoxyl-5,4′-di-n-octyl genistein inhibits the stem-like characteristics of gastric cancer stem-like cells and reverses the phenotype of epithelial-mesenchymal transition in gastric cancer cells. Oncol Rep. 2016;36(2):1157–1165. | ||
Fumiyuki S, Yoshimasa K, Mitsuteru N, et al. EGFR inhibitors prevent induction of cancer stem-like cells in esophageal squamous cell carcinoma by suppressing epithelial-mesenchymal transition. Cancer Biol Ther. 2015;16(6):933–940. | ||
Wu K, Ning Z, Zeng J, et al. Silibinin inhibits β-catenin/ZEB1 signaling and suppresses bladder cancer metastasis via dual-blocking epithelial–mesenchymal transition and stemness. Cell Signal. 2013;25(12):2625–2633. | ||
Palagani V, El KM, Kossatz U, et al. Epithelial mesenchymal transition and pancreatic tumor initiating CD44+/EpCAM+ cells are inhibited by γ-secretase inhibitor IX. PLoS One. 2012;7(10):e46514. | ||
Kanamoto A, Ninomiya I, Harada S, et al. Valproic acid inhibits irradiation-induced epithelial-mesenchymal transition and stem cell-like characteristics in esophageal squamous cell carcinoma. Int J Oncol. 2016;49(5):1859–1869. | ||
Nallanthighal S, Elmaliki KM, Reliene R. Pomegranate extract alters breast cancer stem cell properties in association with inhibition of epithelial-to-mesenchymal transition. Nutr Cancer. 2017;69(7):1088–1098. | ||
Bosukonda A, Carlson WD. Harnessing the BMP signaling pathway to control the formation of cancer stem cells by effects on epithelial-to-mesenchymal transition. Biochem Soc Trans. 2017;45(1):223–228. | ||
Kou Y, Ji L, Wang H, et al. Connexin 43 upregulation by dioscin inhibits melanoma progression via suppressing malignancy and inducing M1 polarization. Int J Cancer. 2017;141(8):1690–1703. | ||
Zheng P, Lei C, Yuan X, et al. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J Exp Clin Cancer Res. 2017;36(1):53. | ||
Chen X, Ying X, Wang X, Wu X, Zhu Q, Wang X. Exosomes derived from hypoxic epithelial ovarian cancer deliver microRNA-940 to induce macrophage M2 polarization. Oncol Rep. 2017;38(1):522–528. | ||
Bonapace L, Coissieux MM, Wyckoff J, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515(7525):130–133. | ||
Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 2013;14(6):329–340. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.