")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Treatment of tardive dyskinesia with VMAT-2 inhibitors: a systematic review and meta-analysis of randomized controlled trials
Authors Solmi M , Pigato G, Kane JM , Correll CU
Received 31 January 2018
Accepted for publication 15 March 2018
Published 14 May 2018 Volume 2018:12 Pages 1215—1238
DOI https://doi.org/10.2147/DDDT.S133205
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Marco Solmi,1 Giorgio Pigato,2 John M Kane,3,4 Christoph U Correll3–5
1Neuroscience Department, Psychiatry Unit, University of Padua, Padua, Italy; 2Psychiatry Unit, Padua Hospital, Padua, Italy; 3Department of Psychiatry, Northwell Health, The Zucker Hillside Hospital, Glen Oaks, NY, USA; 4Department of Psychiatry and Molecular Medicine, Hofstra Northwell School of Medicine, Hempstead, NY, USA; 5Department of Child and Adolescent Psychiatry, Charité Universitätsmedizin, Berlin, Germany
Aim: The aim of this study was to summarize the characteristics, efficacy, and safety of vesicular monoamine transporter-2 (VMAT-2) inhibitors for treating tardive dyskinesia (TD).
Materials and methods: We conducted a literature search in PubMed, Cochrane Database, and ClinicalTrials.gov, screening for systematic reviews, meta-analyses or double-blind, randomized, placebo-controlled trials (DBRPCTs) reporting efficacy or safety data of VMAT-2 inhibitors (tetrabenazine, deutetrabenazine, and valbenazine) in patients with TD. A random effects meta-analysis of efficacy and safety data from DBRPCTs was performed.
Results: Two acute, 12-week DBRPCTs with deutetrabenazine 12–48 mg/day (n=413) and 4 acute, 4–6-week double-blind trials with valbenazine 12.5–100 mg/day (n=488) were meta-analyzable, without meta-analyzable, high-quality data for tetrabenazine. Regarding reduction in total Abnormal Involuntary Movement Scale (AIMS) scores (primary outcome), both deutetrabenazine (k=2, n=413, standardized mean difference [SMD] =-0.40, 95% confidence interval [CI] =-0.19, -0.62, p<0.001; weighted mean difference (WMD) =-1.44, 95% CI =-0.67, -2.19, p<0.001) and valbenazine (k=4, n=421, SMD =-0.58, 95% CI =-0.26, -0.91, p<0.001; WMD =-2.07, 95% CI =-1.08, -3.05, p<0.001) significantly outperformed placebo. Results were confirmed regarding responder rates (≥50% AIMS total score reduction; deutetrabenazine: risk ratio [RR] =2.13, 95% CI =1.10, 4.12, p=0.024, number-needed-to-treat [NNT] =7, 95% CI =3, 333, p=0.046; valbenazine: RR =3.05, 95% CI =1.81, 5.11, p<0.001, NNT =4, 95% CI =3, 6, p<0.001). Less consistent results emerged from patient-rated global impression-based response (p=0.15) and clinical global impression for deutetrabenazine (p=0.088), and for clinical global impression change for valbenazine (p=0.67). In an open-label extension (OLE) study of deutetrabenazine (≤54 weeks) and a dose-blinded valbenazine study (≤48 weeks), responder rates increased over time. With valbenazine, discontinuation effects were studied, showing TD symptom recurrence towards baseline severity levels within 4 weeks after valbenazine withdrawal. No increased cumulative or specific adverse (AEs) events versus placebo (acute trials) in extension versus acute trial data were observed.
Conclusion: The 2 VMAT-2 inhibitors, valbenazine and deutetrabenazine, are effective in treating TD, both acutely and long-term, without concerns about increased risk of depression or suicide in the TD population. No head-to-head comparison among VMAT-2 inhibitors and no high-quality, meta-analyzable data are available for tetrabenazine in patients with TD.
Keywords: tetrabenazine, deutetrabenazine, valbenazine, tardive dyskinesia, VMAT-2
Introduction
Tardive dyskinesia (TD) is a severe1 and potentially irreversible adverse effect of first- and second-generation antipsychotics (FGAs and SGAs), with a cumulative annual incidence of 5.4%2 to 7.7%3 with FGAs compared to 0.8%2 to 3.0%3 with SGAs in adults, and approximately a 3- to 5-fold higher incidence in the elderly, with both FGAs (25%–30%)4,5 and SGAs (5%–7%).6 A recent meta-analysis found across 41 studies and 11,493 patients (77% diagnosed with schizophrenia) pooled TD prevalence of 25.3% in patients treated with antipsychotics, with a lower frequency with current SGA treatment (20.7%) than FGA treatment (30.0%), and the lowest prevalence in SGA-treated patients without a documented lifetime history of FGA treatment (7.2%).7 Proposed risk factors for TD include both unmodifiable patient-related and illness-related risk factors (higher age, female sex, Caucasian race, African descent, longer illness duration, intellectual disability and brain damage, negative symptoms in schizophrenia, mood disorder diagnosis, cognitive symptoms in mood disorders, and gene polymorphisms involving antipsychotic metabolism and dopamine functioning) as well as modifiable comorbidity-related and treatment-related factors (diabetes, smoking, alcohol and substance abuse, FGA vs SGA treatment, higher cumulative or current antipsychotic dose or higher antipsychotic plasma levels, early neuromotor syndromes [parkinsonian side effects, acute dystonia], anticholinergic co-treatment, akathisia, and subtle/emergent dyskinesia).2,3,7–10
TD can be measured with Abnormal Involuntary Movement Scale (AIMS),11 a 12-item scale with items 1–7 assessing involuntary movements across body regions, with a score ranging from 0 (no dyskinesia) to 4 (severe, maximal amplitude, and persistence during observation of abnormal movements), or with other less frequently used instruments/scales, such as the Extrapyramidal Symptom Rating Scale.12
Although still not conclusively established, 3 main (non-mutually exclusive) pathogenetic hypotheses of TD have been proposed. These include oxidative stress,13 glutamate-mediated toxicity within striatopallidal or nigrostriatal GABA signaling pathways with decreased inhibitory activity in nigrostriatal circuits, and dopamine receptor hypersensitivity following receptor upregulation due to antidopaminergic treatment.14 The latter theory is supported by the worsening or un-masking of TD after abrupt cessation of dopamine D2 blocking agents.15
TD should ideally be prevented. Strategies include antipsychotic prescription only when indicated clinically, avoiding parkinsonian side effects, using conservative maintenance doses, and preferring SGAs over FGAs as first-line treatment.3,8,16 However, SGAs are not a homogeneous class with regard to parkinsonian adverse effect rates.17 Nevertheless, the differential TD risk among individual SGAs, except for a likely lowest risk with clozapine, has not been clear. In this regard, a recent meta-analysis of annualized incidence rate ratios comparing FGAs with SGAs and SGAs with each other in RCTs lasting ≥3 months (median: 1.0 [interquartile range =0.44–2.0] years) not only confirmed significantly lower TD annualized incidence rates with SGAs as a class vs FGAs (NNT =20) but also suggested that olanzapine and aripiprazole may have a small, yet statistically significant advantage over other non-clozapine SGAs (NNT =100 for olanzapine vs non-clozapine SGAs).18 Moreover, in 3 trials that compared clozapine to olanzapine or predominant olanzapine treatment, olanzapine did not differ from clozapine with regard to annualized TD risk. Finally, despite a relatively greater risk of parkinsonian side effect and anticholinergic co-treatment than many other SGAs,17 risperidone and paliperidone had a similar TD risk when compared head-to-head with other non-clozapine/non-olanzapine SGAs.18
When TD is present, a number of treatment approaches have been tested, including antipsychotic dose reduction, cessation, or switch to/prescription of specific antipsychotics; however, no convincing evidence supports such strategies,19 apart from a switch to clozapine when TD is diagnosed in patients with schizophrenia and ongoing antipsychotic treatment is needed.20,21 Nevertheless, clozapine is not easy to use,22 and when clozapine is withdrawn, TD seems to recur.23
Several additional treatment approaches for improving TD symptoms exist that target alternatively proposed pathogenetic pathways, such as oxidative stress and impaired clearance of phenylalanine, with in some cases very early, promising results, such as for branched-chain amino acids, ginkgo biloba, vitamin E or melatonin,24–26 but without definitive, large/high-quality trials to support their clinical use.
In addition, benzodiazepines, non-antipsychotic catecholaminergic agents, gamma-aminobutyric acid agonists, cholinergic or anticholinergic agents, or other miscellaneous treatments have been tested as augmentation strategies aimed at reducing TD severity, with almost no clinically relevant improvement27–32 if not detrimental effect, or with tolerability issues, such as sedation, ataxia, or risks correlated with insulin administration outweighing any potentially beneficial effect.29,30,33 Lastly, preliminary evidence suggested relevant efficacy and notable safety of deep brain stimulation for TD34 and of botulinum for focal dystonias.33 A recent comprehensive systematic review assessing the quality and level of evidence of pharmacological approaches for TD concluded that “underpowered trials of limited quality repeatedly fail to provide answers.”35
Beyond stopping or modifying antipsychotic treatment or the aforementioned off-label approaches based on preliminary evidence, reversible vesicular monoamine transporter-2 (VMAT-2) inhibitors, which block a transporter that packages neurotransmitters, particularly dopamine, but also noradrenalin, serotonin, and histamine, into presynaptic vesicles for release into the synaptic cleft, have been tested for TD. Tetrabenzaine was the first VMAT-2 inhibitor that was approved in the United States in 2008 for Huntington’s chorea and that has been used off-label for the treatment of TD,15,36 with promising results from observational studies.37 However, serum half-life of tetrabenazine is very short, requiring 3 times daily dosing and resulting in large peak-to-trough variations in plasma levels that have been associated with off-target adverse effects, such as akathisia, somnolence, and even depression and suicidality, for which a black box warning was required by the US Food and Drug Administration (FDA) in subjects suffering from Huntington’s chorea.38,39 More recently, 2 novel VMAT-2 inhibitors, deutetrabenazine and valbenazine,40 have been developed, aiming at pharmacokinetic improvements (deutetrabenazine) or pharmacokinetic and pharmacodynamic improvements (valbenazine) over tetrabenazine. These modifications have resulted in a prolonged half-life and reduced peak–trough variations compared to tetrabenazine, and, in the case of valbenazine, eliminated enantiomers that are responsible for the off-target receptor occupancy unrelated to VMAT-2.41,42 Since both deutetrabenazine and valbenazine have been tested in double-blind, randomized, placebo-controlled trials (DBRPCTs) and were FDA approved for the treatment of TD in 2017, and since to our knowledge no formal meta-analysis has compared data from all available RCTs of VMAT-2-inhibitors, we conducted a systematic review and meta-analysis aiming at reporting on the meta-analytically derived evidence for the efficacy and safety of VMAT-2 inhibitors for the treatment of patients with TD. We hypothesized that VMAT-2 inhibitors, pooled together and individually, would be superior to placebo in the reduction of TD.
Materials and methods
Search strategy and inclusion criteria
An electronic literature search from database inception and without language restriction was performed on November 8, 2017, in the Cochrane library (http://www.cochranelibrary.com/), PubMed, FDA website (https://www.fda.gov/), and ClinicalTrials.gov (https://ClinicalTrials.gov/ct2/home), plus relevant conference abstracts. The following search keywords were used in PubMed, with equivalent/appropriate syntax for other databases: “tardive dyskinesia”[All Fields] AND (“tetrabenazine”[All Fields] OR “deutetrabenazine”[All Fields] OR “valbenazine”[All Fields]) AND (Review[ptyp] OR Clinical Trial[ptyp]). In addition, bibliographies of included or relevant references were hand-searched to seek further potentially eligible papers.
Inclusion criteria were: 1) most recent/comprehensive systematic review or meta-analysis on VMAT-2 inhibitors for TD, with included trials, or 2) individual, open, or randomized trials of VMAT-2 inhibitors for TD. For the formal meta-analysis, only data from DBRPCTs in patients with TD were considered, and only trials that reported results based on the AIMS scale were included.
In addition to the database search, data regarding the pharmacodynamic and pharmacokinetic properties of each drug were extracted from the official information provided by the manufacturers.
Two authors (MS and GP) independently conducted the search and selected the eligible papers. Any disagreement was resolved by consensus.
Data extraction
Relevant information about efficacy, safety, and clinically relevant pharmacokinetic and pharmacodynamic information was abstracted from systematic reviews/meta-analyses, individual trials, or manufacturer’s drug leaflets. Two authors (MS and GP) independently extracted the pharmacokinetic and pharmacodynamic properties of each VMAT-2 inhibitor, the number of studies (for reviews), study design, study duration, specific drug of interest, continuous and categorical efficacy data, and adverse effects frequencies. Any disagreement was resolved by consensus.
Quality assessment
Two authors (MS and GP) independently assessed the quality of the included studies with the Cochrane Collaboration’s tool for assessing the risk of bias.43
Meta-analysis
We performed a random effects meta-analysis44,45 by using comprehensive meta-analysis (CMA, version 2). For continuous variables, we calculated the standardized mean difference (SMD) as the primary outcome measure, complemented by the weighted mean difference (WMD) whenever the same rating scale was used across all trials. For categorical outcomes, we calculated the risk ratio (RR) and, whenever the RR result was statistically significant, the number-needed-to-treat or harm (NNT or NNH). Effect sizes were accompanied by their 95% confidence intervals (CIs). Heterogeneity was assessed with the Cochrane Q and I2 statistics for each analysis (with significant heterogeneity being indicated by a p<0.05 or I2≥50%, respectively).46 We meta-analyzed each VMAT-2 inhibitor separately and also calculated pooled effect sizes. Publication bias was assessed via visual inspection of funnel plots and with the Begg-Mazumdar Kendall’s tau47 and Egger bias test.48 In case that publication bias was suspected, we calculated the trim and fill adjusted analysis49 to remove the most extreme small studies from the positive side of the funnel plot and recalculated the effect size at each iteration until the funnel plot was symmetric around the (new/adjusted) effect size. In order to indirectly estimate any drug-related difference between VMAT-2 inhibitors, we also conducted sensitivity analyses based on the individual VMAT-2 inhibitor. In order to have a conservative approach and minimize biases, we used a “once randomized-analyzed,” or intent-to-treat (ITT) approach for all efficacy and safety analyses and ignored the results from the completer analyses. The exception was a prespecified modified ITT approach in the studies with deutetrabenazine where all eligible and consenting subjects were randomized, but a priori only those with AIMS scores ≥6 were analyzed.
Results
Search results
Figure 1 presents the flow diagram of the literature search. Our search yielded no article from the Cochrane database but 41 individual studies and 3 reviews from PubMed, of which 33 were excluded after title/abstract assessment with finally 8 published studies and 3 reviews being included after full-text assessment. In ClinicalTrials.gov, we identified 38 trials (DBRPCT and studies with a different design), of which 30 were excluded after full assessment of each trial results section on the website (with specific reasons for the exclusion of each trial reported in detail in Table S1). In addition, 2 relevant conference abstracts were included.
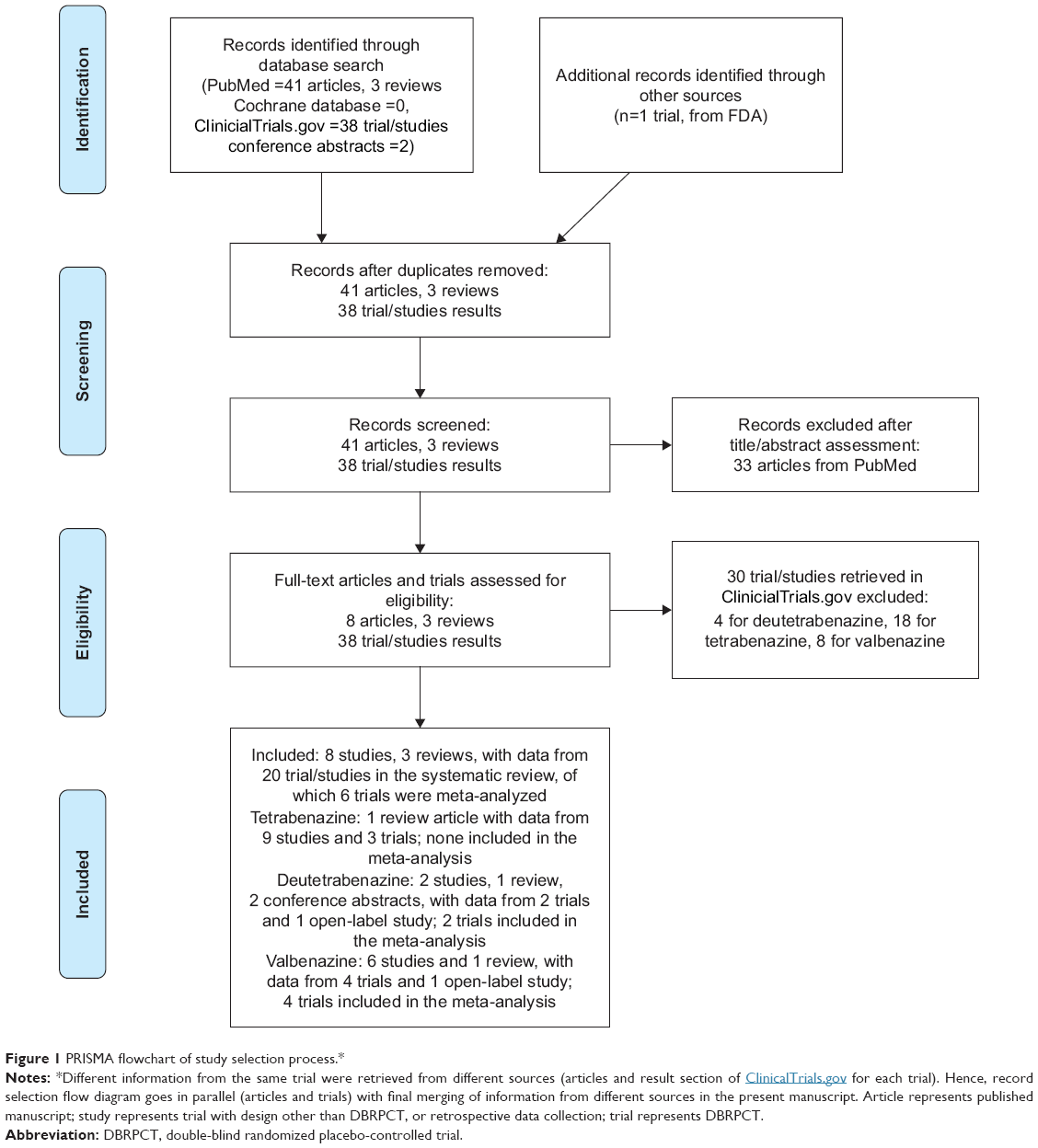 | Figure 1 PRISMA flowchart of study selection process.* |
Different articles reported results from the same trial, or from trials not registered in ClinicalTrials.gov (being too old), and some trials retrieved in ClinicalTrisls.gov did not have related publications. Hence, we merged the information gathered from all eligible studies and reviews from PubMed with data reported in ClinicalTrials.gov and in conference abstracts, and finally included information from 8 distinct DBRPCTs, 2 controlled single-blind studies, 7 distinct open-label studies, and 3 distinct studies with a retrospective or case-series design.
In particular, of these 20 included articles, tetrabenazine was evaluated in 2 DBPRCTs (n=10), 1 haloperidol-controlled RCT (n=13), 1 single-blind placebo-controlled study (n=12), 5 open-label studies, and 3 retrospective or case-series studies (total n=409). Deutetrabenazine was evaluated in 2 DBPRCTs (drug=281, placebo=132) and one open label study (n=304). Finally, valbenazine was investigated in 4 DBRPCs (drug=281, placebo=207) and 1 open label study (n=163). Results from all 20 trials and studies are summarized in our systematic review.
Finally, we formally meta-analyzed the 2 DBRPCTs for deutetrabenazine (n=413), and 4 DBRPCTs for valbenazine (n=488). In addition, the pharmacokinetic characteristics of each included VMAT-2 inhibitor were retrieved from the specific medication’s package insert sheet.
The main characteristics of the included studies are summarized in Table 1. Below, we descriptively summarize the pharmacological properties, clinical indications, efficacy, and safety of each drug from individual studies (reported in detail in Table 2), quality of included studies (Table 3), and a summary of results from meta-analysis (Table 4).
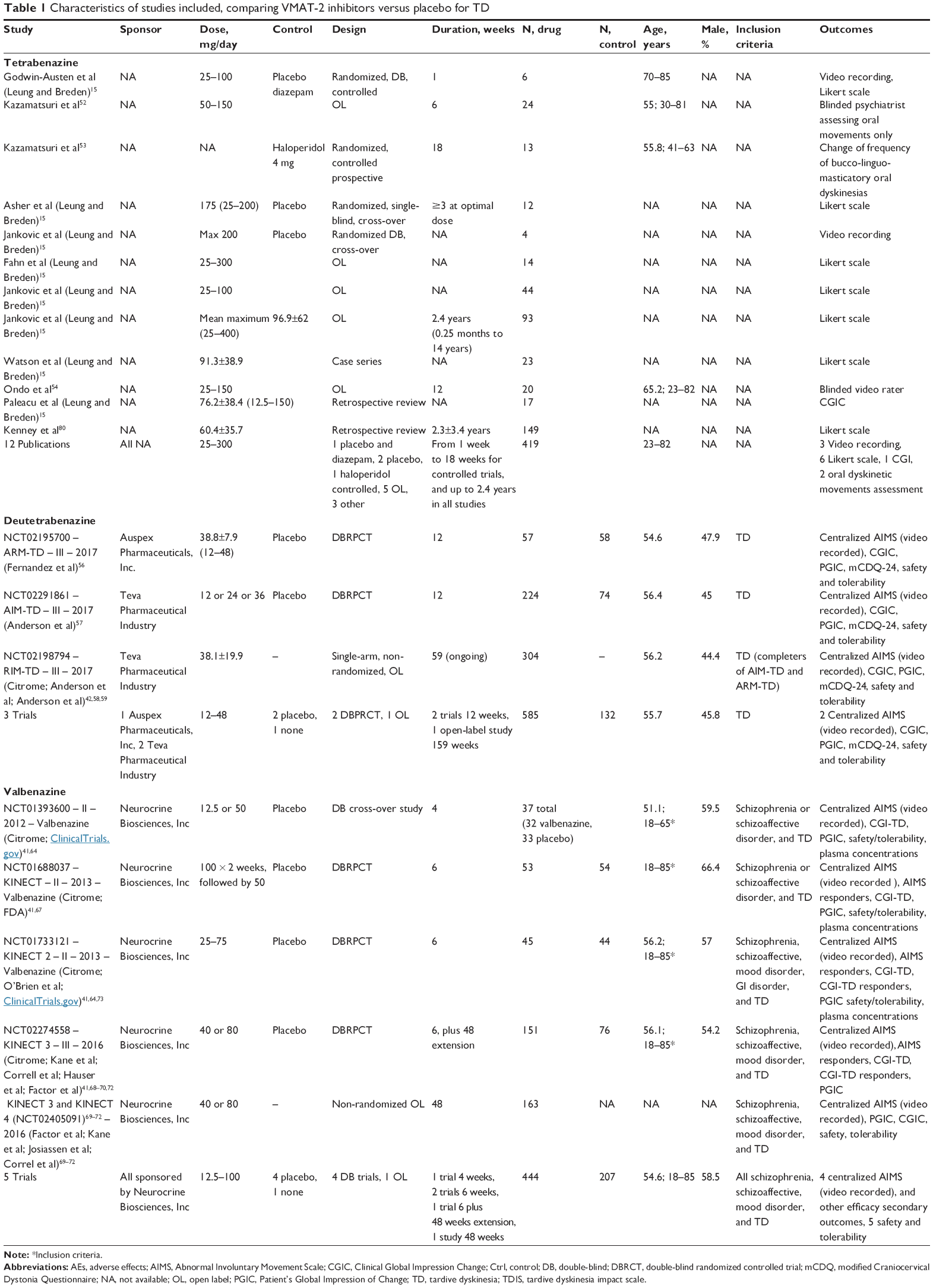 | Table 1 Characteristics of studies included, comparing VMAT-2 inhibitors versus placebo for TD |
 | Table 2 Efficacy and safety in trials of VMAT-2 inhibitors for TD* |
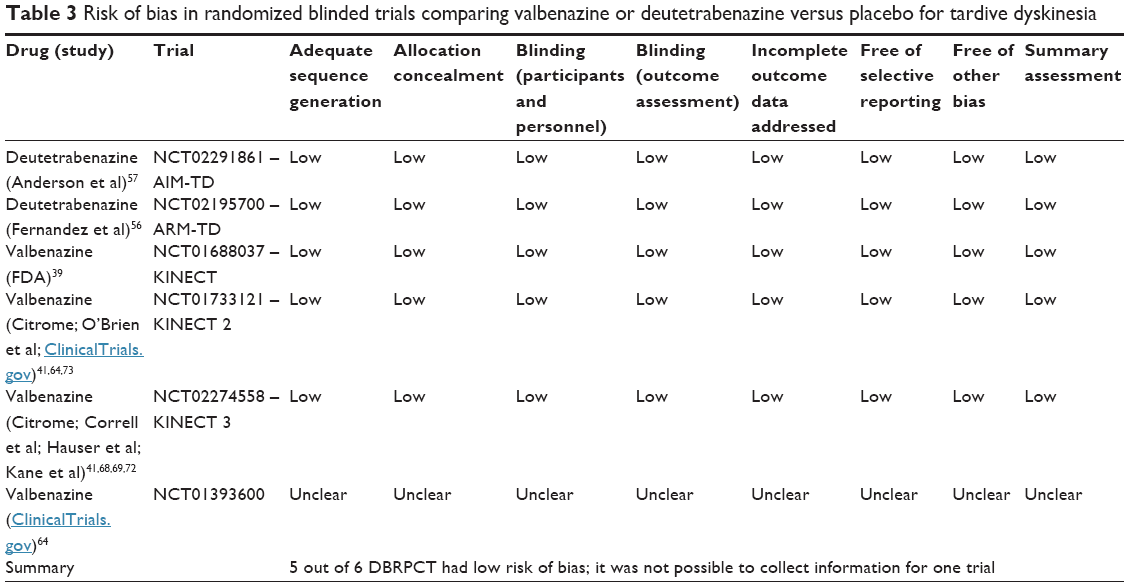 | Table 3 Risk of bias in randomized blinded trials comparing valbenazine or deutetrabenazine versus placebo for tardive dyskinesia |
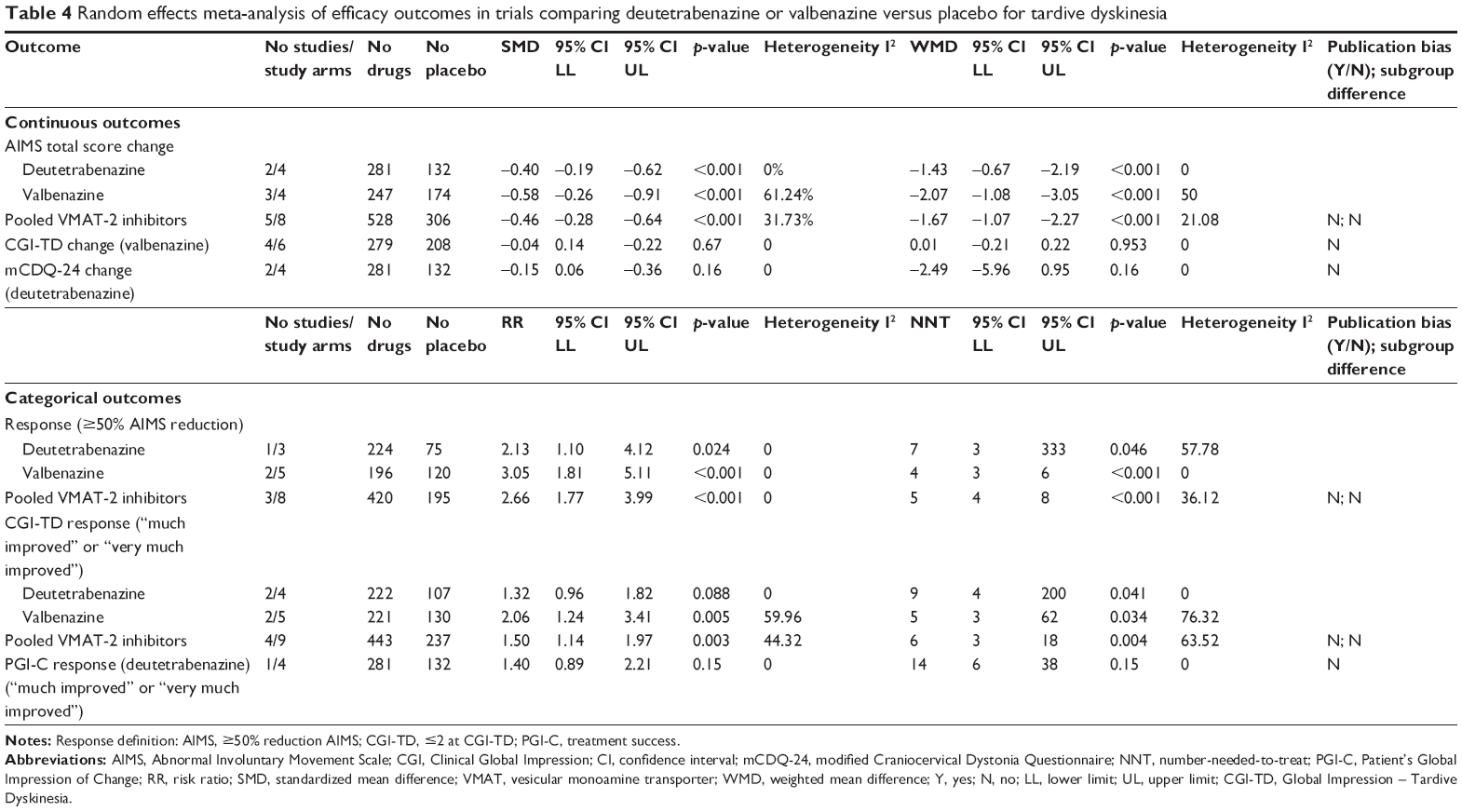 | Table 4 Random effects meta-analysis of efficacy outcomes in trials comparing deutetrabenazine or valbenazine versus placebo for tardive dyskinesia |
Tetrabenazine
Pharmacokinetics and pharmacodynamics
The FDA approved tetrabenazine for Huntington’s disease, not TD, in the United States, while the Agenzia Italiana del Farmaco approved tetrabenazine for TD in Italy along with several other countries including the United Kingdom and Canada. Tetrabenazine is composed of a one-to-one mixture of two enantiomers, alpha-tetrabenazine and beta-tetrabenazine. Each enantiomer is metabolized to 2 isomers. The (+)alpha and (−)alpha dihydrotetrabenazine (DHTBZ) isomers have VMAT-2 reversible inhibitory activity (affinity of tetrabenazine for VMAT-2 is Ki ≈100 nM), while the (+)beta and (−)beta DHTBZ isomers are weak dopamine D2 antagonists (Ki ≈2,100 nM). Tetrabenazine can be taken with or without food, and its bioavailability after oral administration is ≥75%, but its half-life is as short as 5 hours (thus 3 times daily dosing is required).50 The Cmax is reached within 1–1.5 hours, with blood plasma peaks50 and protein binding being close to 85%. Tetrabenazine is extensively metabolized by CYP2D6 and 75% of it being eliminated in urine and around 16% in feces.
Efficacy
One systematic review of the effects of tetrabenazine for TD15 reported results from 12 studies overall, with partially overlapping studies populations,51 with 2 DBRPCTs (n=10), 1 single-blind placebo-controlled study (n=12), and one haloperidol-controlled, randomized study (n=12). Each study had very small sample sizes, surprisingly with response rates up to 100% for tetrabenazine, and all used poor quality assessment tools and poor reporting of outcomes, precluding inclusion of any of these studies in our meta-analysis. Overall, considering also studies with open-label or retrospective design, 419 patients aged 20–82 years were studied. The study duration of the RCTs ranged from 1 to 20 weeks, and the tetrabenazine dose ranged between 50 and 150 mg/day. Notably, in 2 of the RCTs, which were conducted in the 1970s, patients had been on antipsychotic treatment (likely all FGAs) for >10 years and had been hospitalized for an average of 29 years.52,53 Moreover, in just 1 trial, tetrabenazine was compared with an active control intervention, namely, haloperidol, without showing a benefit in TD reduction versus haloperidol.53 In one prospective, single-blind trial,52 dyskinesia “disappeared” in 1 out of 3 patients, “improved” in 1 out of 4 patients, and the average oral movements per minute significantly decreased from baseline and then re-increased to baseline values after tetrabenazine cessation.52 In a more “recent,” single-blind, randomized trial (published in 1999),54 all patients reported at least mild improvement, and dyskinesia clearly improved from baseline.
Overall, there is a lack of high-quality double-blind randomized, placebo controlled RCTs, without convincing evidence of efficacy of tetrabenazine for TD. No “head to head” double-blind comparison with another VMAT-2 inhibitors has been performed.
Safety and tolerability
Most of the safety data come from trials targeting conditions other than TD (eg, Huntington’s disease). Generally, almost 9 out of 10 patients experienced ≥1 adverse effect during tetrabenazine treatment. In patients with TD, the most frequent adverse events (AEs) (>10% and >5% more frequent than placebo) were drowsiness (mean: 24.8%, range: 13%–36.5%), sedation/somnolence (mean: 18.5%, range: 6%–31%), parkinsonian side effects (mean: 11.9%, range: 4.3%–78.6%), insomnia (11%), anxiety (10.3%), depression (mean: 6.8%, range: 1.7%–11%), and akathisia (mean: 6.3%, range: 1.7%–9.5%). Sedation and acute motor syndromes (parkinsonian side effects, dystonic reaction, and akathisia), which can be due to D2 antagonism of the beta-isomers of tetrabenazine, have been reported in approximately 1 out of 4 patients.54 Also, based on data in Huntington’s disease, tetrabenazine has a black box warning for depression and suicidality,39 leading to the recommendation that attention should be paid to mood status or aberrant acting out behavior during tetrabenazine treatment. Somnolence and sedation may impair driving abilities. QT prolongation may occur, but does not seem to be clinically relevant. Dysphagia can also occur, increasing the risk of pneumonia. Hyperprolactinemia may occur as well. Notably, in the trials included in the abovementioned systematic review,15 specifically targeting TD, 1 patient out of 24 did not complete the study due to psychotic exacerbation.52
Poor metabolizers, special populations, and concomitant drugs
As per FDA labeling, tetrabenazine dose should not exceed 50 mg/day in poor CYP2D6 metabolizers, or 100 mg/day in extensive metabolizers, yet no clear correlation has been demonstrated between CYP2D6 status and clinical response.55 Guidance from labels in other countries may differ. Tetrabenazine is contraindicated in patients with any degree of hepatic impairment, and in patients with suicidal ideation, or untreated/inadequately treated depression. Pregnant or lactating women should be warned that data from animal studies suggest possible fetal harm but that no data are available from human studies. The tetrabenazine dose should be decreased in case of co-treatment with CYP2D6 inhibitors (eg, paroxetine and fluoxetine), and tetrabenazine must not be co-administered with monoamine oxidase inhibitors (MAOIs) or reserpine (an irreversible VMAT inhibitor), due to possible toxicity related to partially overlapping pharmacodynamic actions. No efficacy or safety data are available for pediatric or geriatric populations.
Deutetrabenazine
Pharmacokinetics and pharmacodynamics
Deutetrabenazine is a reversible VMAT-2 inhibitor. Similar to tetrabenazine, there are α and β enantiomers, and each gives rise to 2 isomers of a DHTBZ metabolite. However, the incorporation of deuterium, which is a stable, non-radioactive, non-toxic, and naturally occurring isotope of hydrogen, in place of hydrogen at the sites of primary metabolism results in slower metabolic clearance compared to tetrabenazine and lower Cmax values, despite similar effective doses as used in the tetrabenazine trials (36–48 mg). Deutetrabenazine should be taken with food and its bioavailability after oral administration is ≥80%. The half-life of deutetrabenazine is about 9–10 hours (thus twice-daily dosing is required),1 and its protein binding is close to 84%. Deutetrabenazine is extensively metabolized by CYP2D6, with minor contributions from CYP1A2 and CYP3A4/5, and 80% of it being eliminated in urine and around 10% in feces.
Efficacy
One systematic review reported results from 2 acute trials and 1 open-label, long-term study.42 In 2017, the US FDA approved deutetrabenazine for both Huntington’s disease and TD. One multicenter DBRPCT (“ARM-TD study”) included 117 (deutetrabenazine =58, placebo =59) patients with moderate to severe TD (82.9% of which with AIMS ≥6 based on blinded video assessment), and assessed AIMS, clinician’s and patient’s global impression (CGI and PGI) scores after 12 weeks. Patients were administered deutetrabenazine starting from 12 mg/day, with weekly increases of 6 mg up to 48 mg maximum (36 mg for patients taking strong CYP2D6 inhibitors), according to clinical response. Baseline AIMS values were 9.6 (4.1 SD) with deutetrabenazine and 9.6 (3.8 SD) with placebo. Although no significant difference was noted in CGI scores compared with placebo at study endpoint, or in modified Craniocervical Dystonia Questionnaire (mCDQ-24) (p not available for both comparisons), AIMS scores improved significantly more with deutetrabenazine than placebo (placebo-subtracted treatment difference −1.4, standard error [SE]: 0.6, p=0.019).56 Another multicenter DBRPCT (“AIM-TD study”) included 298 patients (deutetrabenazine =224 divided in 3 groups: 12, 24, and 36 mg/day; placebo =74) with TD (AIMS ≥6) duration ≥3 months, and assessed AIMS after 12 weeks (or at least at 1 follow-up observation).57 Baseline AIMS values were 9.6 (2.4 SD) with 12 mg/day deutetrabenazine, 9.4 (2.9 SD) with 24 mg/day, 10.1 (3.2 SD) with 36 mg/day, and 9.5 (2.7 SD) with placebo. Efficacy was demonstrated compared with placebo for deutetrabenazine 36 mg/day (placebo-subtracted treatment difference −1.9, SE: 0.58, p=0.001) and 24 mg/day (−1.8, SE: 0.60, p=0.003), but not for 12 mg/day (−0.7, SE: 0.57, p=0.217). Moreover, compared to placebo (12%), the proportion of patients with a ≥50% AIMS score reduction was significantly higher with deutetrabenazine 24 mg/day (35%, OR =3.96, 95% CI =1.46, 10.72, p=0.005) and 36 mg/day (33%, OR =3.80, 95% CI =1.40, 10.36, p=0.007). Conversely, in the ITT population, deutetrabenazine did not differ significantly from placebo regarding the Patient Global Impression of Change (PGIC) defined responder rate (12 mg/day: p=0.37, 24 mg/day: p=0.13, 36 mg/day: p=0.30) or mCDQ-24 scores (12 mg/day: p=0.66, 24 mg/day: p=0.24, 36 mg/day: p=0.12).57 In the OLE study58,59 (59 weeks, ongoing, n=304 from the ARM-TD and AIM-TD studies), all efficacy measures (continuous scores or responder rates) continued to improve from week 6 to week 54. No head to head comparison with other VMAT-2 inhibitors has been performed.
Safety and tolerability
Deutetrabenazine was generally well tolerated, with trial completion rate ranging from 89% to 95% in DBRPCTs,41 and the NNH for all-cause discontinuation was not significantly different from placebo.41 Moreover, deutetrabenazine had similar AEs rates in all dose arms compared with placebo (36 mg/day: 51.4%; 24 mg/day: 43.8%; 12 mg/day: 48.6%; placebo: 47.2%), with the 36 mg/day dose being associated with the highest frequency of AEs.57 Moreover, no worsening of EPS occurred,56 yet such a definition should be replaced with finer definitions of specific adverse motor events, with consistent reporting of rates of each of them in RCTs.60 Sedation/somnolence may occur with deutetrabenazine, but the manufacturer correctly advises that patients with Huntington’s disease should be advised about such a risk, while there is no such need in patients with TD, given the comparable rates of somnolence in the deutetrabenazine (2%–13.8%) and placebo group (4%–10.2%). Although deutetrabenazine, like tetrabenazine, has a black box warning for depression and suicidality in patients with Huntington’s disease (based on data from trials in this population), data from 2 trials in patients with TD did not show any increased risk versus placebo, which is why deutetrabenazine did not receive such a regulatory warning for the TD population.56 In the ARM-TD trial, depressed mood/depression was noted at the exact same frequency as placebo (1.7%), with no patients reporting suicidality.56 In the AIM-TD trial, suicidality was reported in the 24 and 36 mg/day arms (2.7% the highest),57 but without any difference from placebo. Also, in the long-term, OLE study, deutetrabenazine showed42,58,59 similar adjusted incidences of AEs, compared with both the acute-phase trials and the placebo. Moreover, >90% of psychiatric AEs were mild to moderate. Hence, the absence of any FDA warning about a putative increased risk of depression or suicide in patients with TD seems appropriate, at least in the included psychiatrically stable patients with TD. Deutetrabenazine has not been tested in the pediatric population.
Poor metabolizers, special populations, and concomitant drugs
Poor CYP2D6 should be prescribed lower deutetrabenazine doses. No data are available about deutetrabenazine in patients with renal impairment. Even if no data are available, based on data for tetrabenazine, lower doses should be considered when hepatic function is impaired. Deutetrabenazine dose should also be decreased in the presence of CYP2D6 inhibitors (eg, paroxetine and fluoxetine). However, since the slower metabolism and lower Cmax reduce the impact of slow CYP2D6 metabolizers, no genotyping has been recommended, unlike for tetrabenazine.
Valbenazine
Pharmacokinetics and pharmacodynamics
Valbenazine is a highly selective, reversible VMAT-2 inhibitor that consists of the (+)α-DHTBZ enantiomer, an oxidative metabolite of valbenazine, and its 2 isomers, with all 3 molecules having VMAT-2 binding only.61 It can be taken with or without food, although the Cmax is decreased by high-fat meals. Oral bioavailability of valbenazine is ~49%, its half-life is about 20 hours,62 enabling once daily dosing, tmax is between 0.5 and 1.0 hours,62 steady state is reached within 8 days,62 and protein binding is 99%. Area under the curve (AUC) analyses have shown a dose-proportional increase from 50 to 150 mg/day and more than dose-proportion from 1 to 50 mg/day.62
The affinity of valbenazine for VMAT-2 is Ki ≈150 nM, with virtually no affinity for VMAT-1 (Ki >10,000 nM). Valbenazine and its metabolites do not have any affinity for other than VMAT-2 targets, namely, serotonin, dopamine, adrenergic, histaminergic, or muscarinic receptors, thus minimizing untoward effects beyond those specifically linked to its specific binding site. It is extensively metabolized by hydrolysis and oxidative metabolism (CYP3A4/5); (+)-α-HTBZ seems to be further metabolized in part by CYP2D6. Approximately 60% of elimination takes place in urine and 30% in feces.
Efficacy
The US FDA approved valbenazine for TD in 2017.63 One systematic review41 has identified 6 completed trials (2 long-term extension studies and 4 DBRPCTs), including a total of 660 subjects, aged 18–85 years, with a study duration ranging from 12 days to 48 weeks, and with the valbenazine dose ranging from 12.5 to 100 mg/day. Four double-blind trials (NCT01393600,64 KINECT, KINECT 2, and KINECT 3)41,65–73 and 1 dose-blind extension study (KINECT 4)41,69–72 reported on the efficacy and safety of valbenazine,41,68–73 in patients who developed TD during antipsychotic treatment for schizophrenia, schizoaffective disorder, or mood disorders, or during metoclopramide treatment for a gastrointestinal conditions.
Baseline AIMS scores in KINECT 2 were 8.0 (3.5 SD) in the valbenazine and 7.9 (4.5 SD) in the placebo group, being 10.4 (3.6) for 80 mg/day, 9.7 (4.1 SD) for 40 mg/day, and 9.9 (4.3 SD) for placebo in the KINECT 3 study.
Although valbenazine did not separate from placebo in the small dose-finding studies NCT01393600 (4 weeks, 37 patients in a blinded cross-over study, analyzed: drug =32, placebo =33) and KINECT (6 weeks, drug =50, placebo =54),41,64–67 it showed significantly greater improvement in the AIMS total score compared to placebo at week 6 in 2 larger, acute-phase KINECT 2 (6 weeks, valbenazine: n=45, placebo n=44), and KINECT 3 trials (6 weeks, valbenazine: n=151, placebo: n=76, with extension to 48 weeks: n=198).68,70,73 In KINECT 2, improvement in total AIMS scores were consistently higher with valbenazine than placebo (treatment difference −2.4, SE: 0.7, p<0.001), as well as CGI-TD score (treatment difference −0.8, SE: 0.2, p<0.001). The same was true in KINECT 3 with regard to the AIMS score change (80 mg/day: −3.2, SE: 0.4, p<0.001; 40 mg/day: −1.9, SE: 0.4, p<0.001, vs placebo −0.1, SE: 0.4).
Valbenazine also outperformed placebo in response rates, regardless of the definition of response. For example, in KINECT 2, a ≥50% AIMS total score reduction was observed in 48.9% with valbenazine versus 18.2% with placebo (p<0.001), with rates of patients achieving a CGI-TD ≤2 (ie, “much or very much improved”) being 67% with valbenazine and 16% with placebo (p<0.001).41 Similarly, in KINECT 3, a ≥50% AIMS total score reduction was observed in 40.0% with valbenazine 80 mg/day (p<0.001), 23.8% with 40 mg/day (p=0.02), and 8.7% with placebo, with the 80 mg/day dose consistently showing the highest response rates.41,68–73 Notably, the same efficacy and dose-related profile was confirmed in both the schizophrenia and schizoaffective disorder subgroup,69 as well as the mood disorder subgroup.72
The NNT for treatment response was consistently around 4 for valbenazine 80 mg daily, ranging from 2 to 9, with doses from 25 to 75 mg/day. In KINECT 3, the NNT for the AIMS-based response in all patients was 4 for 80 mg/day and 7 for 40 mg/day, being consistent in patients with mood disorders and schizophrenia/schizoaffective disorder (mood disorder subgroup: 80 mg/day: NNT =4 for AIMS-based definition and NNT =6 for CGI-TD, 40 mg/day: NNT =9 for AIMS and NNT =8 for CGI-TD; schizophrenia subgroup: 80 mg/day: NNT =4 for AIMS-based definition and NNT =17 for CGI-TD, 40 mg/day: NNT =6 for AIMS and NNT =10 for CGI-TD).41,61,68,69,72
In exploratory analyses, statistical separation of valbenazine from placebo occurred as early as week 2 at 80 mg/day and did not differ according to sex, age, ethnicity, underlying main diagnosis, or TD baseline severity.68,73
However, in KINECT 2, the PGIC-based (treatment success) response rate statistics were not reported, only showing numerically higher rates with valbenazine versus placebo (57.8% vs 31.8%, p=NA),73 and in KINECT 3, the endpoint CGI-TD score differences missed statistical significance (treatment difference −0.3, SE: 0.1, p=0.074).68
In the OLE study (48 weeks, plus 4 weeks after valbenazine withdrawal, n=164), all efficacy measures (continuous scores or responder rates) continued to improve from week 6 to week 48, but then worsened until week 52 after valbenazine was discontinued, trending back to the acute baseline values, supporting the need for continuation treatment.69,70,72
No “head to head” comparison with other VMAT-2 inhibitors or other agents has been performed.
Safety and tolerability
Valbenazine was generally well tolerated,41 with comparable study completion rates on valbenazine and placebo (KINECT 2: valbenazine: 76%, placebo: 80%; KINECT 3: valbenazine 80 mg/day: 88.8%, valbenazine 40 mg/day: 82.9%, placebo: 91%). Furthermore, no detrimental effects were noted on core psychopathologic symptoms of the underlying mental condition,41 in both the acute and the long-term extension trials.71 In addition, there was no increase in suicidal behavior or depression noted, at least in the included psychiatrically stable patients. Calgary Depression Scale for Schizophrenia and Montgomery and Asberg Depression Rating Scale scores remained stable after treatment with valbenazine 40 and 80 mg/day in both the 6-week KINECT 3 trial68 and the KINECT 4 long-term tolerability trial.71
Two potential tolerability concerns have been reported,41 namely, QTc prolongation and somnolence. However, QTc prolongation, which was modeled by the FDA, was not clinically relevant at regular doses74 and should be monitored particularly in those with QTc prolongation or other arrhythmias associated with QT prolongation. Nevertheless, no clinically relevant ECG alteration was noted in the acute 6-week and 48-week long-term trials.41,71 Moreover, although patients with TD experienced numerically higher rates of somnolence compared with placebo (KINECT 2: valbenazine: 5.9%, placebo: 2%, p=NA; KINECT 3: valbenazine: 5.3%, placebo: 3.9%, p=NA), the rates were very low. Interestingly, in all patients, decreased appetite was reported more frequently with valbenazine versus placebo (KINECT 2: valbenazine: 7.8%, placebo: 0%).73
The most frequent long-term AEs in both schizophrenia/schizoaffective and mood disorder subsamples in the 48-week extension study were somnolence, urinary tract infections, and headache.71 Finally, a potential risk of dose-related prolactin, bilirubin, and alkaline phosphatase increase was noted as per the FDA evaluation, but was not observed in the acute trials, and no clinically relevant laboratory alteration was noted in the 48-week, long-term tolerability trial.71
Poor metabolizers, special populations, and concomitant drugs
Lower doses (ie, 40 mg/day) should be prescribed in patients with known slow CYP2D6 metabolizer status or intolerability to 80 mg/day.41 Patients with moderate to severe hepatic impairment should take the lower dose of valbenazine, and pregnant or lactating women should be advised of the potential risk for the fetus or newborn, even if no data about such risks in humans are available to date. As for renal impairment, no dose adjustment is required for mild to moderate renal impairment, while valbenazine should be avoided in case of severe renal failure since no data are available.41
No dose adjustment is required in the elderly population, and no efficacy or safety data are currently available for pediatric populations.41 Valbenazine should not be prescribed with MAOIs, or strong CYP3A4 inducers (eg, carbamazepine, St John’s wort, phenytoin, and rifampin) and should be prescribed at a reduced dose when given together with strong inhibitors of CYP2D6 (eg, paroxetine, fluoxetine, and quinidine) or CYP3A4 (eg, itraconazole, ketoconazole, and clarithromycin), and digoxin concentrations should be monitored when valbenazine is administered with digoxin, due to possible increase of blood levels of digoxin with concomitant administration.41 However, no genotyping has been recommended unlike for tetrabenazine. In the 48-week, long-term tolerability trial, no relevant interaction with ongoing concomitant medications was reported.71
Quality of included studies
Table 3 reports the quality of included studies. According to the Cochrane Risk of Bias Tool, 5 out of 6 blinded trials had low risk of bias, while for 1 (NCT01393600), no detailed methodological information was available (unclear risk of bias).
Meta-analysis
Meta-analyses were performed for AIMS change (primary outcome), CGI-TD change, mCDQ-24 change, and treatment response based on AIMS (≥50% total score reduction), CGI-TD (score ≤2, “much improved” or “very much improved”), or PGI-C (“treatment success”). Table 4 summarizes the results of random effects meta-analysis for VMAT-2 inhibitors pooled together and for deutetrabenazine and valbenazine individually, and Figure 2 presents the forest plot of AIMS change. All other forest plots and funnel plots of meta-analyses are available upon request.
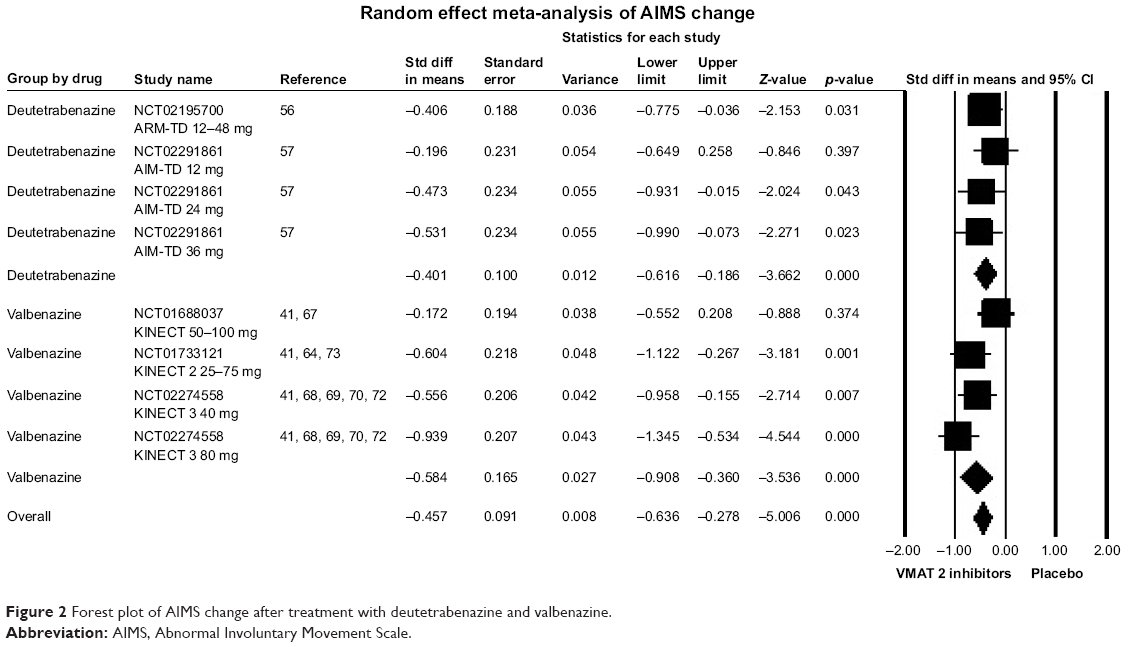 | Figure 2 Forest plot of AIMS change after treatment with deutetrabenazine and valbenazine. |
Results confirmed the efficacy of VMAT-2 inhibitors for TD. AIMS scores reduced significantly more with VMAT-2 inhibitors as a class than placebo (trial arms k=8; SMD =−0.46, 95% CI =−0.28, −0.64, p<0.001; WMD =−1.67, 95% CI =−1.07, −2.27; p<0.001; I2=21%) and with both deutetrabenazine (k=4; SMD =−0.40, 95% CI =−0.19, −0.62, p<0.001; WMD =−1.43, 95% CI =−0.67, −2,19; p<0.001; I2=0%) and valbenazine (k=4; SMD =−0.58, 95% CI =−0.26, −0.91, p<0.001; WMD =−2.07; 95% CI =−1.08, −3.05; p<0.001; I2=50%) individually. VMAT-2 inhibitors as a class were superior for treatment response, defined according to AIMS (≥50% of AIMS score improvement from baseline) (k=8; RR =2.66; 95% CI =1.77, 3.99, p<0.001; I2=0%; NNT =5, 95% CI =4, 8, p<0.001). The same was true for deutetrabenazine (k=3; RR =2.13; 95% CI =1.10, 4.12; p=0.024; I2=0%; NNT =7, 95% CI =3, 333, p=0.046), and valbenazine (k=5; RR =3.05; 95% CI =1.81, 5.11; p<0.001; I2=0%; NNT =4, 95% CI =3, 6, p<0.001) individually. Similar results were found for VMAT-2 inhibitors as a class pertaining to response defined according to CGI criteria (k=9; RR =1.50; 95% CI =1.14, 1.97; p=0.003; I2=44.32%; NNT =6, 95% CI =3, 18, p=0.004), with consistent results for valbenazine (k=5; RR =2.06, 95% CI =1.24, 3.41; p=0.005; I2=59.96%; NNT =5, 95% CI =3, 62, p=0.034), while deutetrabenazine showed only a trend toward significance (k=4; RR =1.32; 95% CI =0.96,1.82; p=0.088; I2=0%; NNT =9, 95% CI =4, 200, p=0.041).
Deutetrabenazine and valbenazine were not significantly superior to placebo regarding the following secondary outcomes for which results were only available for one or the other VMAT-2 inhibitor. Deutetrabenazine: mCDQ-24 (k=4; p=0.157) and PGI-C response (k=4; p=0.149); valbenazine: CGI-TD scores change (k=6; p=0.953). Notably, when 12 mg group in AIM-TD trial was removed from analyses, a trend toward a significant improvement in mCDQ-24 appeared (p=0.079). Also, when deutetrabenazine 12 mg arm was removed from analyses, heterogeneity significantly dropped across the above-described analyses.
With regard to safety, a meta-analysis was performed for all adverse effects reported in the included data sources, and all results are reported in Table 5. No increased risk of AEs versus placebo emerged with VMAT-2 inhibitors as a class or with deutetrabenazine or valbenazine individually. This non-differential adverse effect risk included no increased risk of depression, suicidal ideation, sedation, or somnolence, which had been a concern with tetrabenazine (but whose data could not be meta-analyzed due to insufficient trial design). In fact, VMAT-2 inhibitors were associated with a significantly lower risk of nausea compared with placebo (k=4, RR =0.31, 95% CI =0.10, 0.95, p=0.04).
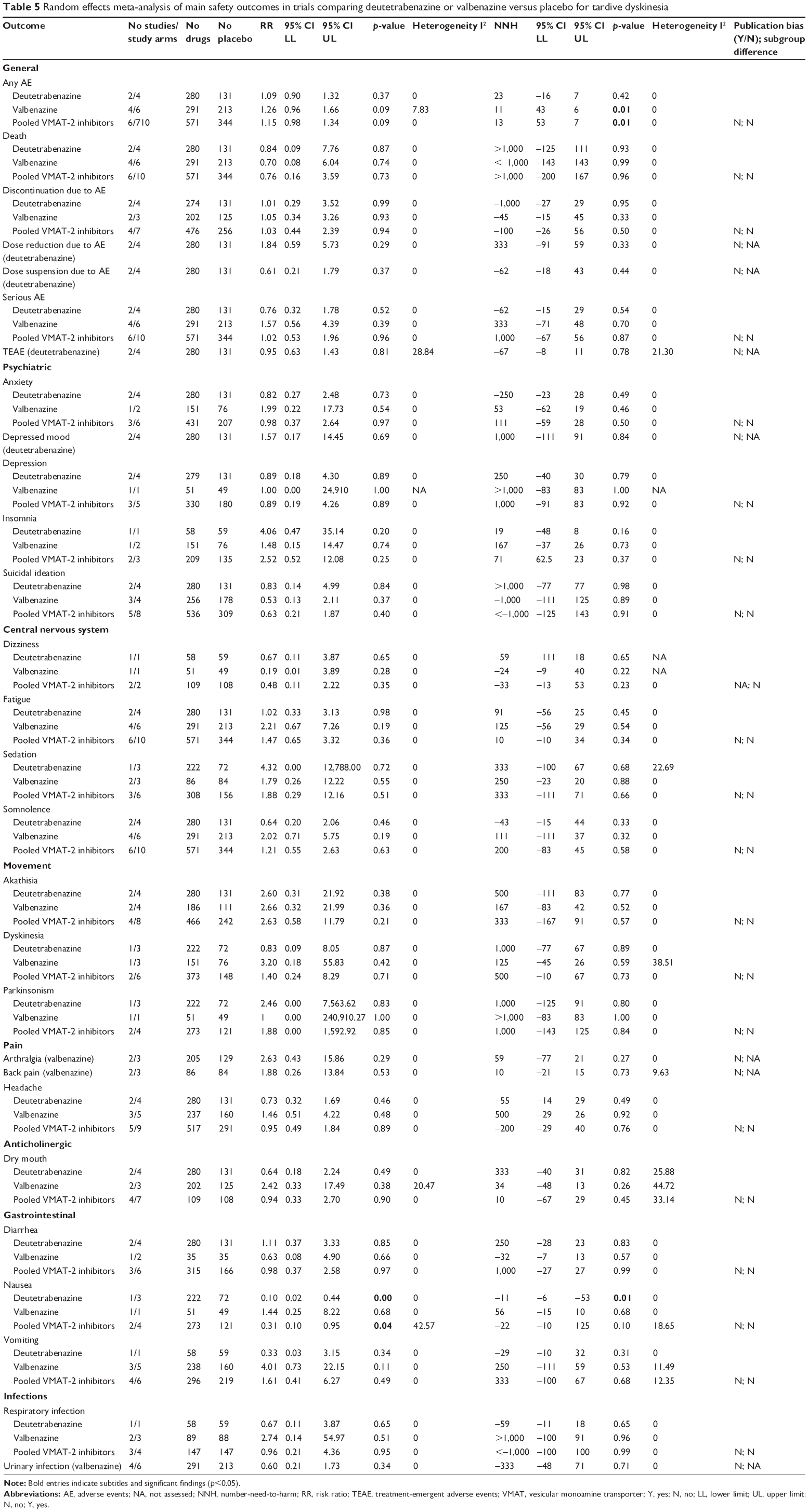 | Table 5 Random effects meta-analysis of main safety outcomes in trials comparing deutetrabenazine or valbenazine versus placebo for tardive dyskinesia |
Discussion
Tetrabenazine, deutetrabenazine, and valbenazine inhibit VMAT-2 action, resulting in less dopamine being transported from the cytoplasm into presynaptic vesicles, which leads to less dopamine release from the pre-synaptic neurons into the synaptic cleft.36 This reduced dopamine release results in less stimulation of post-synaptic dopamine receptors in the nigro-striatal pathway, which is thought to subsequently decrease dyskinetic movements. Despite this common mechanism of action, pharmacological factors, efficacy, and safety should be considered when selecting a VMAT-2 inhibitor to treat TD, since any preference of one VMAT-2 inhibitor over another is currently based on individual medication properties, rather than on head-to-head comparison trials, which are still lacking.
Results of this systematic review and meta-analysis indicate that high-quality evidence for the efficacy and safety in patients with TD exists for the 2 FDA-approved VMAT-2 inhibitors, deutetrabenazine and valbenazine, but not for tetrabenazine. Furthermore, pharmacokinetic and also pharmacodynamic properties of VMAT-2 inhibitors differ. Tetrabenazine has a short half-life, needs to be given 3 times daily, leading to high peak levels and high peak–trough variations that seem to be responsible for off-target adverse effects, such as sedation/somnolence, acute motor syndromes, including parkinsonism and akathisia, QTc prolongation, and, possibly, depression and suicidality.15 In contrast, deutetrabenazine has a longer half-life, can be given twice daily and has less peak levels and peak–trough variation, which reduces its adverse effect risk, although it still gives rise to 4 active metabolites, 2 of which have weak dopamine receptor blocking properties.42 Valbenazine, on the other hand, has an even longer half-life, can be given once daily and is metabolized to +-(α)HTBZ with its 2 isomers, which each have highly selective VMAT-2 activity.41
Regarding efficacy, both deutetrabenazine and valbenazine have demonstrated robust efficacy for the reduction of AIMS scores and, regarding treatment response,41,42,56–59,64,68–73 generally with higher doses producing greater efficacy and with medium effect sizes and clinically relevant NNTs of 2–9 for variously defined treatment response. In contrast to clinician-rated outcomes, patient-rated outcomes were less robust or nonsignificant. However, this reduced the ability to demonstrate superiority compared to placebo on patient-rated outcomes may have to do with the fact that not all patients are fully aware of the presence or impact of the TD.75–79 In this context, data are missing that put the efficacy results into the context of patient awareness and burden caused by the TD, such as in subgroup analyses. To comprehensively assess the impact of TD and of treatments for TD, the field will have to define TD-specific rating scales that tap into the subjective and functional relevance of TD that may not necessarily be directly related to total TD severity, but also (rather) to local body area distribution and the functional impact of the abnormal involuntary movements.
Moreover, since, like with clozapine,23 discontinuation of valbenazine was associated with a general recurrence of TD, even after 48 weeks of treatment,59,71 it will be important to clarify if there is a subgroup of patients in whom TD does not recur after remission during VMAT-2 inhibitor treatment, be it those in whom dopamine blocking agents could be stopped, or in those with a short duration or milder or locally more limited forms of TD. Notably, effects of discontinuation have not yet been studied with deutetrabenazine, while observational studies have shown TD recurrence after withdrawal of tetrabenazine.37,80 Moreover, data are missing that could help to determine if starting a VMAT-2 inhibitor in patients with only subtle dyskinetic movements that are still below the threshold of Schooler–Kane criteria81 can effectively prevent progression to TD.
Different from tetrabenazine, the safety and tolerability of deutetrabenazine and valbenazine was very good. There was no increased treatment discontinuation versus placebo and no decrease in efficacy of the antipsychotics for the underlying psychiatric condition. Moreover, the QTc prolonging effects of VMAT-2 inhibitors seemed modest.68,71 Similarly, in patients with TD, parkinsonian side effects, akathisia, and somnolence/sedation were far less common with either deutetrabenazine (parkinsonian side effects: 0%–1%; akathisia: 0%–5.2%; sedation/somnolence: 0%–13.8%) or valbenazine (parkinsonian side effects: 0%–3.8%; akathisia: 0%–5.3%; sedation/somnolence: 0%–5.9%) than with tetrabenazine (parkinsonian side effects: 4.3%–78.6%; akathisia: 1.7%–9.5%; sedation/somnolence: 6%–31%), despite ongoing treatment with antipsychotics.
Importantly, due to results from studies in patients with, tetrabenazine has a black box warning for depression and suicidality. Based on the limited data in TD patients, tetrabenazine was still associated with depression in 6.8% (range 1.7%–11%) of patients. Conversely, neither depression nor suicidality emerged as a concern with deutetrabenazine or valbenazine, with placebo-level rates in the acute trials (deutetrabenazine: depression: 1%–4%; suicidality: 0%–3%; valbenazine: depression: 0%–1%; suicidality: 0%–4.2%) and illness base-rate frequencies82–85 in the long-term studies lasting up to 1 year (deutetrabenazine: depression: 6%–20%; suicidality: 0%–13%; valbenazine: depression: 2.6%–3.2%; suicidality: 4.5%–4.5%). These reassuring results are reflected by an absence of a black box labeling by the FDA for both deutetrabenazine and valbenazine in the patient population with TD with regard to depression and suicidality. However, results are currently based on selected patient populations agreeing to participate in research studies. Since the vast majority of patients with TD (apart from those with gastrointestinal conditions and metoclopramide therapy) have an underlying psychiatric condition, mainly schizophrenia or schizoaffective disorder and mood disorders (roughly two-thirds and one-third in the valbenazine or deutetrabenazine trials, respectively), they have an illness-inherent risk for the emergence or worsening of depression and suicidality.82–85 Therefore, despite the current absence of a signal for depression and suicidal behavior in the deutetrabenazine and valbenazine studies, clinicians need to monitor mood status and suicidality in patients on VMAT-2 inhibitors, be it illness related or as part of an adverse effect. Moreover, since patients with active major depression or suicidality were excluded from the regulatory trials with VMAT-2 inhibitors, their safety in these populations is currently unclear.
The results of this meta-analysis need to be interpreted within its strengths and limitations. This meta-analysis has the following strengths. First, to our knowledge, this is the first comprehensive meta-analysis assessing the efficacy and safety of deutetrabenazine and valbenazine in TD. Moreover, given the conservative choice not to consider post hoc analyses from individual publications (which yield significant results in single trials as opposed to some nonsignificant findings from ITT or prespecified mITT data56,57), our significant results can be considered solid and unlikely be the result of a bias (Table 4). Moreover, results were confirmed in diagnostic subgroups. Finally, central ratings by blinded evaluators, which was applied in all DBRPCTs included in the meta-analysis and in both extension studies with deutetrabenazine and valbenazine, provides an unbiased and methodologically superior outcome assessment.86
However, some limitations should also be considered that are mainly due to the available evidence. Tetrabenazine essentially lacks sufficiently large and methodologically sound data from sufficiently large DBRPCTs to evaluate its utility in patients with TD. No RCT that compares different VMAT-2 inhibitors head-to-head currently exists. Moreover, all included trials evaluating the efficacy and safety of deutetrabenazine and valbenazine were industry-funded. However, results from this meta-analysis did not suggest the presence of any publication bias. Furthermore, results from patient-rated outcomes were inconsistent, and data on the functional impact of TD reduction with VMAT-2 inhibitors and information provided by family members/caregivers are lacking. Moreover, data regarding whether valbenazine or deutetrabenazine is effective in milder forms of TD are missing. Finally, patients included in the registration trials were psychiatrically stable and did not have active major depressive disorder or active suicidality (although patients with a history of suicidality were eligible for the studies). Thus, generalizability of the results to a more severely psychiatrically ill population with TD is currently not clear. Nevertheless, results from this meta-analysis confirm statistically and clinically relevant efficacy of deutetrabenazine and valbenazine in patients with TD, indicating their safety and tolerability in this important and previously underserved patient population.
Conclusion
Deutetrabenazine and valbenazine are FDA approved for TD and have at present the best high-quality evidence supporting their efficacy for TD, at the dose of 24–36 mg/day for deutetrabenazine and 40–80 mg/day for valbenazine, likely with a positive dose–response relationship. Moreover, the efficacy seems to progress further beyond the acute 6–12 weeks, as treatment continues for up to 1 year, both in patients with schizophrenia or schizoaffective disorder and in patients with mood disorders. Both deutetrabenazine and valbenazine are safe, at least in the studied, psychiatrically stable patient populations, and according to meta-analytic results, neither of the 2 agents seems to have an increased risk for depression or suicidality in stable patients with TD.86
Tetrabenazine has no high-quality evidence for its efficacy or safety in patients with TD and should, at best, be considered a third-line, off-label treatment for TD, unless until methodologically sound trials, ideally comparing tetrabenazine with other VMAT-2 inhibitors, show its evental non-inferiority regarding effiacy and tolerability compared with deutetrabenazine and valbenazine.
Additional RCTs with VMAT-2 inhibitors in patients with TD are needed, ideally targeting long-term safety in patients who are representative of clinical samples as well as targeting functionality and quality of life, rated by clinicians, patients, and family members/caregivers. Furthermore, head-to-head comparisons of the 2 FDA-approved agents should be conducted. In addition, studies should look to identify predictors of response and remission of TD as well as of subgroups in whom the VMAT-2 inhibitor may successfully be withdrawn after remission of TD.50 Finally, strategies for patients not responding sufficiently to the currently available VMAT-2 inhibitors also require further study.
Author contributions
Marco Solmi and Christoph U Correll designed the study. Marco Solmi and Giorgio Pigato run the literature search, extracted evidence. Marco Solmi, Giorgio Pigato, and Christoph U Correll drafted the manuscript. Marco Solmi, Christoph U Correll, and John M Kane interpreted the evidence and revised and refined the manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work. All the authors approved the final version of the manuscript.
Disclosure
Christoph U Correll has been a consultant and/or advisor to or has received honoraria from Alkermes, Allergan, Bristol-Myers Squibb, Gerson Lehrman Group, IntraCellular Therapies, Janssen/J&J, LB Pharma, Lundbeck, Medavante, Medscape, Neurocrine, Otsuka, Pfizer, ROVI, Sunovion, Takeda, and Teva. He has provided expert testimony for Bristol-Myers Squibb, Janssen, and Otsuka. He served on a Data Safety Monitoring Board for Lundbeck and Pfizer. He received grant support from Janssen and Takeda. He is a shareholder of LB Pharma. John M Kane has been a consultant and/or advisor to or has received honoraria from Alkermes, Allergan, IntraCellular Therapies, Janssen/J&J, LB Pharma, Lundbeck, Medscape, Neurocrine, Otsuka, Pfizer, Pierre Fabre, Roche, Sunovion, Takeda, and Teva. He has received grant support from Janssen, Lundbeck, and Otsuka. He is a shareholder of The Vanguard Research Group and LB Pharma. The other authors report no other conflicts of interest in this work.
References
Cummings MA, Proctor GJ, Stahl SM. Deuterium tetrabenazine for tardive dyskinesia. Clin Schizophr Relat Psychoses. 2018;11(4):214–220. | ||
Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414–425. | ||
Correll CU, Schenk EM. Tardive dyskinesia and new antipsychotics. Curr Opin Psychiatry. 2008;21(2):151–156. | ||
Woerner MG, Alvir JM, Saltz BL, Lieberman JA, Kane JM. Prospective study of tardive dyskinesia in the elderly: rates and risk factors. Am J Psychiatry. 1998;155(11):1521–1528. | ||
Jeste DV, Caligiuri MP, Paulsen JS, et al. Risk of tardive dyskinesia in older patients. A prospective longitudinal study of 266 outpatients. Arch Gen Psychiatry. 1995;52(9):756–765. | ||
Woerner MG, Correll CU, Alvir JM, Greenwald B, Delman H, Kane JM. Incidence of tardive dyskinesia with risperidone or olanzapine in the elderly: results from a 2-year, prospective study in antipsychotic-naive patients. Neuropsychopharmacology. 2011;36(8):1738–1746. | ||
Carbon M, Hsieh CH, Kane JM, Correll CU. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J Clin Psychiatry. 2017;78(3):e264–e278. | ||
Solmi M, Pigato G, Kane JM, Correll CU. Clinical risk factors for the development of tardive dyskinesia. J Neurol Sci. 2018;389:21–27. | ||
Jankelowitz SK. Treatment of neurolept-induced tardive dyskinesia. Neuropsychiatr Dis Treat. 2013;9:1371–1380. | ||
Kane JM, Correll CU. Pharmacologic treatment of schizophrenia. Dialogues Clin Neurosci. 2010;12(3):345–357. | ||
Guy W. Abnormal Involuntary Movement Scale (117-AIMS), in ECDEU Assessment Manual for Psychopharmacology. 1976. Available from: https://mmcp.health.maryland.gov/pap/docs/Abnormal%20Involuntary%20Movement%20Scale.pdf. Accessed May 2, 2018. | ||
Chouinard G, Margolese HC. Manual for the extrapyramidal symptom rating scale (ESRS). Schizophr Res. 2005;76(2–3):247–265. | ||
Cho CH, Lee HJ. Oxidative stress and tardive dyskinesia: pharmacogenetic evidence. Prog Neuropsychopharmacol Biol Psychiatry. 2013;46:207–213. | ||
Margolese HC, Chouinard G, Kolivakis TT, Beauclair L, Miller R. Tardive dyskinesia in the era of typical and atypical antipsychotics. Part 1: pathophysiology and mechanisms of induction. Can J Psychiatry. 2005;50(9):541–547. | ||
Leung JG, Breden EL. Tetrabenazine for the treatment of tardive dyskinesia. Ann Pharmacother. 2011;45(4):525–531. | ||
Solmi M, Murru A, Pacchiarotti I, et al. Safety, tolerability, and risks associated with first- and second-generation antipsychotics: a state-of-the-art clinical review. Ther Clin Risk Manag. 2017;13:757–777. | ||
Leucht S, Cipriani A, Spineli L, et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382(9896):951–962. | ||
Carbon M, Kane J, Leucht S, Correll C. Tardive dyskinesia risk with first-generation and second-generation antipsychotics in comparative randomized controlled trials: a meta-analysis. World Psychiatry. In press 2018. | ||
Soares-Weiser K, Rathbone J. Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for tardive dyskinesia. Cochrane Database Syst Rev. 2006;1:CD000459. | ||
Mentzel T, Snoek R, vander Oorschot M, et al. Clozapine as a treatment for tardive dyskinesia: a meta-analysis. Schizophrenia International Research Society; April, 2–6, 2016; Florence. | ||
Spivak B, Mester R, Abesgaus J, et al. Clozapine treatment for neuroleptic-induced tardive dyskinesia, parkinsonism, and chronic akathisia in schizophrenic patients. J Clin Psychiatry. 1997;58(7):318–322. | ||
Nielsen J, Correll CU, Manu P, Kane JM. Termination of clozapine treatment due to medical reasons: when is it warranted and how can it be avoided? J Clin Psychiatry. 2013;74(6):603–613; quiz 613. | ||
Small JG, Milstein V, Marhenke JD, Hall DD, Kellams JJ. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment-resistant psychosis. J Clin Psychiatry. 1987;48(7):263–267. | ||
Zheng W, Xiang YQ, Ng CH, Ungvari GS, Chiu HF, Xiang YT. Extract of ginkgo biloba for tardive dyskinesia: meta-analysis of randomized controlled trials. Pharmacopsychiatry. 2016;49(3):107–111. | ||
Soares-Weiser K, Maayan N, McGrath J. Vitamin E for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev. 2011;2:CD000209. | ||
Richardson MA, Small AM, Read LL, Chao HM, Clelland JD. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92–96. | ||
Bhoopathi PS, Soares-Weiser K. Benzodiazepines for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev. 2006;3:CD000205. | ||
Soares KV, McGrath JJ. Anticholinergic medication for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev. 2000;2:CD000204. | ||
McGrath JJ, Soares KV. Miscellaneous treatments for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev. 2000;2:CD000208. | ||
Alabed S, Latifeh Y, Mohammad HA, Rifai A. Gamma-aminobutyric acid agonists for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev. 2011;4:CD000203. | ||
El-Sayeh HG, Lyra da Silva JP, Rathbone J, Soares-Weiser K. Non-neuroleptic catecholaminergic drugs for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev. 2006;1:CD000458. | ||
Tammenmaa IA, McGrath JJ, Sailas E, Soares-Weiser K. Cholinergic medication for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev. 2002;3:CD000207. | ||
Vijayakumar D, Jankovic J. Drug-induced dyskinesia, part 2: treatment of tardive dyskinesia. Drugs. 2016;76(7):779–787. | ||
Mentzel CL, Tenback DE, Tijssen MA, Visser-Vandewalle VE, van Harten PN. Efficacy and safety of deep brain stimulation in patients with medication-induced tardive dyskinesia and/or dystonia: a systematic review. J Clin Psychiatry. 2012;73(11):1434–1438. | ||
Bergman H, Walker DM, Nikolakopoulou A, Soares-Weiser K, Adams CE. Systematic review of interventions for treating or preventing antipsychotic-induced tardive dyskinesia. Health Technol Assess. 2017;21(43):1–218. | ||
Bernstein AI, Stout KA, Miller GW. The vesicular monoamine transporter 2: an underexplored pharmacological target. Neurochem Int. 2014;73:89–97. | ||
Miguel R, Mendonca MD, Barbosa R, et al. Tetrabenazine in treatment of hyperkinetic movement disorders: an observational study. Ther Adv Neurol Disord. 2017;10(2):81–90. | ||
Muller T. Valbenazine granted breakthrough drug status for treating tardive dyskinesia. Expert Opin Investig Drugs. 2015;24(6):737–742. | ||
Shen V, Clarence-Smith K, Hunter C, Jankovic J. Safety and efficacy of tetrabenazine and use of concomitant medications during long-term, open-label treatment of chorea associated with Huntington’s and other diseases. Tremor Other Hyperkinet Mov (N Y). 2013;3:tre-03-191-4337-1. | ||
Jankovic J. Dopamine depleters in the treatment of hyperkinetic movement disorders. Expert Opin Pharmacother. 2016;17(18):2461–2470. | ||
Citrome L. Valbenazine for tardive dyskinesia: a systematic review of the efficacy and safety profile for this newly approved novel medication – what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2017;71(7):1–14. | ||
Citrome L. Deutetrabenazine for tardive dyskinesia: a systematic review of the efficacy and safety profile for this newly approved novel medication – what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2017;71(11):1–17. | ||
Higgins JP, Altman DG, Gotzsche PC, et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928. | ||
DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7(3):177–188. | ||
DerSimonian R, Kacker R. Random-effects model for meta-analysis of clinical trials: an update. Contemp Clin Trials. 2007;28(2):105–114. | ||
Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327(7414):557–560. | ||
Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. 1994;50(4):1088–1101. | ||
Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629–634. | ||
Duval S, Tweedie R. Trim and fill: a simple funnel-plot-based method of testing and adjusting for publication bias in meta-analysis. Biometrics. 2000;56(2):455–463. | ||
Correll CU, Carbon M. A new class of VMAT-2 inhibitors for tardive dyskinesia. Lancet Psychiatry. 2017;4(8):574–575. | ||
Caroff SN, Aggarwal S, Yonan C. Treatment of tardive dyskinesia with tetrabenazine or valbenazine: a systematic review. J Comp Eff Res. 2018;7(2):135–148. | ||
Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95–99. | ||
Kazamatsuri H, Chien CP, Cole JO. Long-term treatment of tardive dyskinesia with haloperidol and tetrabenazine. Am J Psychiatry. 1973;130(4):479–483. | ||
Ondo WG, Hanna PA, Jankovic J. Tetrabenazine treatment for tardive dyskinesia: assessment by randomized videotape protocol. Am J Psychiatry. 1999;156(8):1279–1281. | ||
Mehanna R, Hunter C, Davidson A, Jimenez-Shahed J, Jankovic J. Analysis of CYP2D6 genotype and response to tetrabenazine. Mov Disord. 2013;28(2):210–215. | ||
Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003–2010. | ||
Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595–604. | ||
Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for the treatment of tardive dyskinesia: results from an open-label, long-term study. P7-009. The American Psychiatric Association 2017 Annual Meeting; May 20–24, 2017; San Diego, CA. | ||
Anderson KE, Stamler D, Davis MD, et al. Long-term safety of deutetrabenazine for the treatment of tardive dyskinesia: results from an open-label, long-term study. P07-010. The American Psychiatric Association 2017 Annual Meeting; May 20–24, 2017; San Diego, CA. | ||
Obeso JA. 2018 message from the editor in chief-Movement Disorders journal at cruise speed. Mov Disord. 2018;33(1):1–2. | ||
Freudenreich O, Remington G. Valbenazine for tardive dyskinesia. Clin Schizophr Relat Psychoses. 2017;11(2):113–119. | ||
Luo R, Bozigian H, Jimenez R, Loewen G, O’Brien CF. Single dose and repeat once-daily dose safety, tolerability and pharmacokinetics of valbenazine in healthy male subjects. Psychopharmacol Bull. 2017;47(3):44–52. | ||
Sarva H, Henchcliffe C. Valbenazine as the first and only approved treatment for adults with tardive dyskinesia. Expert Rev Clin Pharmacol. 2018;11(3):209–217. | ||
Clinicaltrials.gov. National Institute of Health. National Library of Medicine. Available from: https://clinicaltrials.gov/ct2/home. Accessed May 2, 2018. | ||
Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–389. | ||
Ostergaard SD, Bech P, Miskowiak KW. Fewer study participants needed to demonstrate superior antidepressant efficacy when using the Hamilton melancholia subscale (HAM-D) as outcome measure. J Affect Disord. 2016;190:842–845. | ||
Food and Drug Administration. US Department of Health and Human Services. Available from: https://www.fda.gov/. Accessed May 2, 2018. | ||
Hauser RA, Factor SA, Marder SR, et al. KINECT 3: a phase 3 randomized, double-blind, placebo-controlled trial of valbenazine for tardive dyskinesia. Am J Psychiatry. 2017;174(5):476–484. | ||
Kane JM, Correll CU, Liang GS, Burke J, O’Brien CF. Efficacy of valbenazine (NBI-98854) in treating subjects with tardive dyskinesia and schizophrenia or schizoaffective disorder. Psychopharmacol Bull. 2017;47(3):69–76. | ||
Factor SA, Remington G, Comella CL, et al. The effects of valbenazine in participants with tardive dyskinesia: results of the 1-year KINECT 3 extension study. J Clin Psychiatry. 2017;78(9):1344–1350. | ||
Josiassen RC, Kane JM, Liang GS, Burke J, O’Brien CF. Long-term safety and tolerability of valbenazine (NBI-98854) in subjects with tardive dyskinesia and a diagnosis of schizophrenia or mood disorder. Psychopharmacol Bull. 2017;47(3):61–68. | ||
Correll CU, Josiassen RC, Liang GS, Burke J, O’Brien CF. Efficacy of valbenazine (NBI-98854) in treating subjects with tardive dyskinesia and mood disorder. Psychopharmacol Bull. 2017;47(3):53–60. | ||
O’Brien CF, Jimenez R, Hauser RA, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Mov Disord. 2015;30(12):1681–1687. | ||
Thai-Cuarto D, O’Brien CF, Jimenez R, Liang GS, Burke J. Cardiovascular profile of valbenazine: analysis of pooled data from three randomized, double-blind, placebo-controlled trials. Drug Saf. 2017 Epub Dec 7. | ||
Alexopoulos GS. Lack of complaints in schizophrenics with tardive dyskinesia. J Nerv Ment Dis. 1979;167(2):125–127. | ||
Arango C, Adami H, Sherr JD, Thaker GK, Carpenter WT Jr. Relationship of awareness of dyskinesia in schizophrenia to insight into mental illness. Am J Psychiatry. 1999;156(7):1097–1099. | ||
Chong SA, Remington G, Mahendran R, Chua HC. Awareness of tardive dyskinesia in Asian patients with schizophrenia. J Clin Psychopharmacol. 2001;21(2):235–237. | ||
Emsley R, Niehaus DJ, Oosthuizen PP, Koen L, Chiliza B, Fincham D. Subjective awareness of tardive dyskinesia and insight in schizophrenia. Eur Psychiatry. 2011;26(5):293–296. | ||
Macpherson R, Collis R. Tardive dyskinesia. Patients’ lack of awareness of movement disorder. Br J Psychiatry. 1992;160:110–112. | ||
Kenney C, Hunter C, Jankovic J. Long-term tolerability of tetrabenazine in the treatment of hyperkinetic movement disorders. Mov Disord. 2007;22(2):193–197. | ||
Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486–487. | ||
Hawton K, Sutton L, Haw C, Sinclair J, Harriss L. Suicide and attempted suicide in bipolar disorder: a systematic review of risk factors. J Clin Psychiatry. 2005;66(6):693–704. | ||
Valtonen H, Suominen K, Mantere O, Leppamaki S, Arvilommi P, Isometsa ET. Suicidal ideation and attempts in bipolar I and II disorders. J Clin Psychiatry. 2005;66(11):1456–1462. | ||
Sokero TP, Melartin TK, Rytsala HJ, Leskela US, Lestela-Mielonen PS, Isometsa ET. Suicidal ideation and attempts among psychiatric patients with major depressive disorder. J Clin Psychiatry. 2003;64(9):1094–1100. | ||
Hor K, Taylor M. Suicide and schizophrenia: a systematic review of rates and risk factors. J Psychopharmacol. 2010;24(4 Suppl):81–90. | ||
Davis MC, Miller BJ, Kalsi JK, Birkner T, Mathis MV. Efficient trial design – FDA approval of valbenazine for tardive dyskinesia. N Engl J Med. 2017;376(26):2503–2506. |
Supplementary material
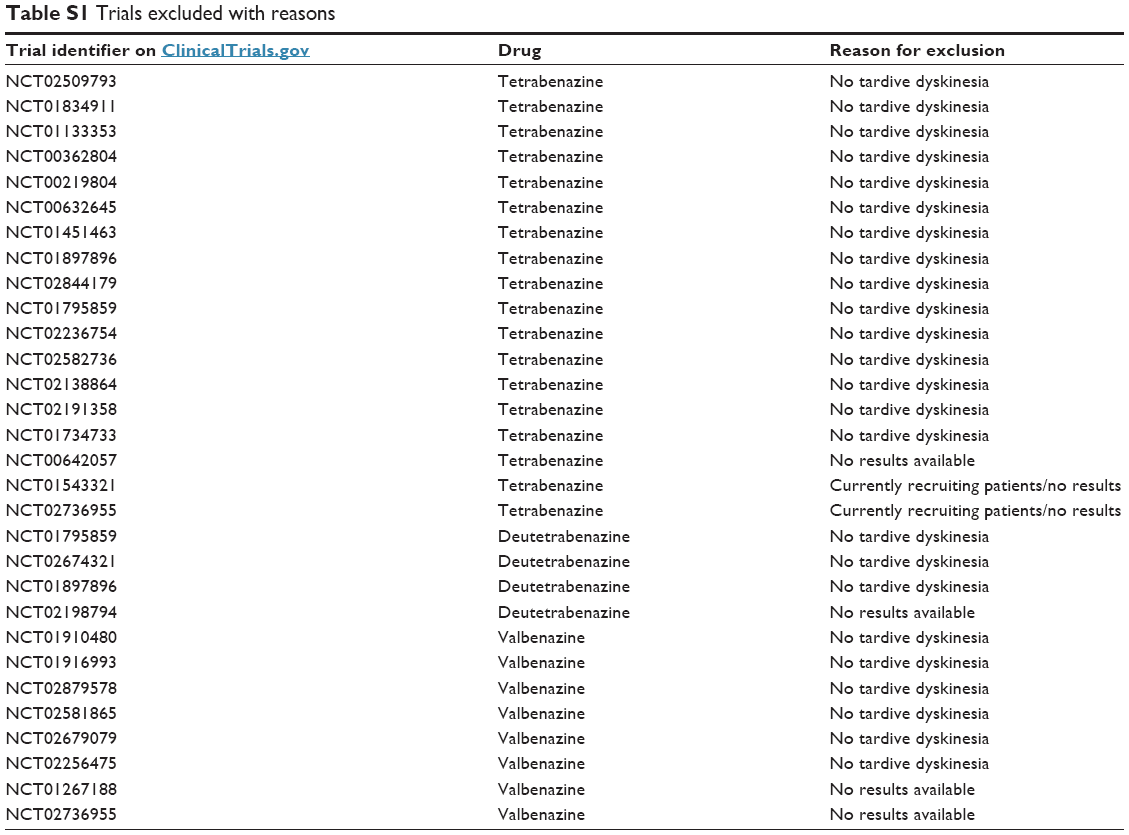 | Table S1 Trials excluded with reasons |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.