")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 7
Treatment of systemic-onset juvenile arthritis with canakinumab
Received 5 July 2014
Accepted for publication 9 September 2014
Published 18 February 2015 Volume 2015:7 Pages 23—31
DOI https://doi.org/10.2147/OARRR.S54215
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Chuan-Ju Liu
Joachim Peitz, Gerd Horneff
Pediatric Rheumatology Centre, Department of General Pediatrics and Neonatology, Asklepios Clinic, Sankt Augustin, Germany
Abstract: Treatment of systemic-onset juvenile idiopathic arthritis is challenging, but the availability of cytokine antagonists targeting interleukin-1 and interleukin-6 have markedly advanced the therapeutic options. In this review, we focus on the current experience with canakinumab, an interleukin-1 monoclonal human antibody for the treatment of systemic-onset juvenile idiopathic arthritis and describe its efficacy and safety. Canakinumab is an important, safe, and valid drug in the treatment of systemic-onset juvenile idiopathic arthritis.
Keywords: anakinra, canakinumab, interleukin-1, interleukin-6, systemic-onset juvenile idiopathic arthritis
Juvenile idiopathic arthritis and systemic-onset juvenile idiopathic arthritis
Juvenile idiopathic arthritis (JIA) includes a group of diseases with arthritis lasting more than 6 weeks, of no apparent cause, and an onset prior to the age of 16 years.1 It is the most common chronic rheumatic disease in children.2 Seven JIA categories are defined (Table 1), with differences in clinical presentation, course, prognosis, and response to treatment with disease-modifying drugs. Differences in the biology and pathogenesis of the disease may be responsible for the variability in responses to medical treatment.
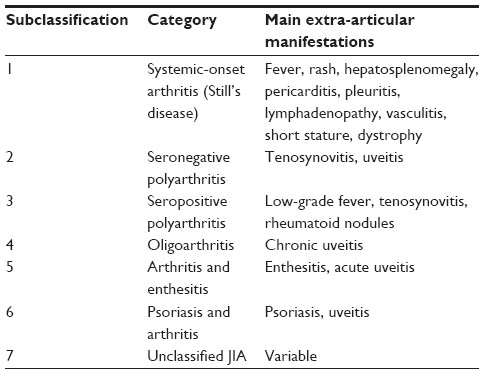 | Table 1 Classification of juvenile idiopathic arthritis |
Systemic-onset juvenile idiopathic arthritis (soJIA) is characterized by the variable occurrence of chronic aggressive arthritis, intermittently high spiking fever, maculopapular rash (often described as salmon-like colored) during fever episodes, hepatomegaly and splenomegaly, lymphadenopathy, serositis, and a marked increase in acute-phase reactant levels.3–5 These findings make it unique among the categories of JIA.
SoJIA, which is the most severe category of JIA, accounts for about 4%–17% of JIA cases.4 The diagnosis is applied when children up to 16 years of age present with arthritis and fever of at least 2 weeks’ duration, a spiking appearance and spontaneous disappearance of fever documented for at least 3 days, and the presence of at least one of following: erythematous rash, generalized lymphadenopathy, hepatomegaly, splenomegaly, or serositis. Furthermore, diagnosis of soJIA requires exclusion of other diseases possibly related to the clinical findings.
Given the as yet unknown pathogenesis and extreme heterogeneity of soJIA, the course of the disease varies between individuals, ranging from a single appearance of the disease, to recurrent predominantly systemic disease courses, to a progressive polyarthritis that frequently leads to severe and destructive joint disease. Complications of soJIA include growth impairment, osteoporosis, and the macrophage activation syndrome, which is potentially lethal (Table 2).6–8
 | Table 2 Criteria for macrophage activation syndrome in systemic-onset juvenile idiopathic arthritis |
The aim of treatment is to reach inactive disease or even remission as defined in Table 3. Current treatments for soJIA have proved largely unsatisfactory.9–12 Management of the disease relies on corticosteroids. Children with soJIA do not respond well to disease-modifying agents such as methotrexate, and poor responses have also been reported with newer agents, such as anti-tumor necrosis factor.13 Several reports have suggested a major role for cytokines in the disease, such as interleukin (IL)-6 and, more recently, IL-1β.14–21
 | Table 3 Criteria for inactive disease in juvenile idiopathic arthritis |
Role of interleukin-1
IL-1 is a proinflammatory cytokine that is produced by monocytes/macrophages and dendritic cells. Its stimulatory effects on T-cells were detected early on. IL-1 induces expression of numerous proinflammatory genes, including the cyclooxygenase type 2 gene, which is important for rheumatic inflammation. It enhances the expression of intercellular adhesion molecule-1 and other adhesion proteins, thereby enhancing intercellular adhesion and activation. Injection of recombinant IL-1 leads to fever, arterial hypotension, and severe flu-like symptoms, with myalgia, headache, body aches, and loss of appetite. IL-1 may therefore be responsible for many of the symptoms of inflammation, and by influencing the activity of osteoclasts and osteoblasts, even for the appearance of osteoporosis in systemic inflammation, as in soJIA.
The biologic effects of IL-1 can be neutralized as outlined in Figure 1. The first published report of successful treatment of systemic JIA with IL-1 inhibition occurred in 2004 with the report of a remarkable response in two patients whose severe disease manifestations were previously refractory to other therapies.22 At about the same time, other researchers found that serum from children with soJIA induced the transcription of IL-1β-related genes in the peripheral blood mononuclear cells of healthy controls. Based on this finding, these investigators treated patients with soJIA with anakinra, an IL-1 inhibitor, and achieved a dramatic clinical response. For example, seven of nine patients who were refractory to prior therapies achieved a remission. These data prompted clinicians to use anakinra for the treatment of soJIA in clinical practice, as reported in several case series. An early report showed a remarkable response to treatment with anakinra in ten of 21 patients. In these patients, it seemed that those with fewer involved joints might benefit more from anakinra therapy than those with arthritis in many joints.18 A larger retrospective case series of 46 patients with soJIA was limited to children who received anakinra as part of their initial glucocorticoid-sparing treatment regimen or even as first-line monotherapy. Fifty-nine percent of these patients showed a good clinical response. Of ten patients who received anakinra as first-line therapy without any glucocorticoids, eight achieved a complete remission.
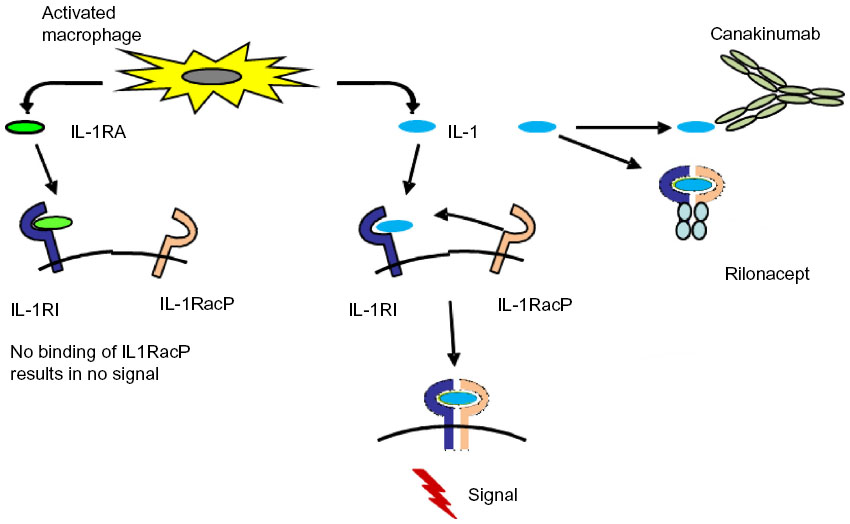 | Figure 1 Interaction of IL-1 and IL-1 receptor antagonist with the IL-1 receptor molecules. |
In addition to anakinra, other IL-1 inhibitors have been developed and subsequently studied for soJIA (Table 4). Currently, there are three different biological inhibitors of the IL-1 pathway available: anakinra, an IL-1 receptor antagonist with a relatively short half-life; canakinumab, a human IL-1β antibody with a long plasma half-life; and rilonacept, an IL-1 receptor fusion protein.
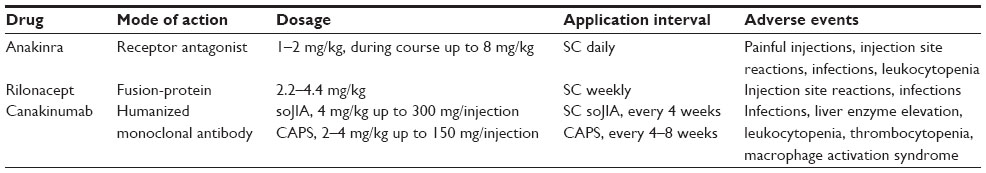 | Table 4 Differences in inhibition of interleukin-1 |
Canakinumab in soJIA
Canakinumab is a human monoclonal antibody (Table 4). Its half-life in plasma is 23–26 days, which is much longer than that of IL-1β. It binds Il-1β selectively and prevents binding of IL-1β to the IL-1 receptor. Furthermore, canakinumab does not bind IL-1α or the IL-1 receptor antagonist, and does not interact with the IL-1 receptor.
Canakinumab is given as a subcutaneous injection once a month. It has proven efficacy in the treatment of genetic fever syndromes and autoinflammatory diseases, including cryopyrin-associated periodic syndrome,23 familial Mediterranean fever.24 More recently, positive case reports and small case series in hyper-IgD syndrome,25 tumor necrosis factor receptor-associated periodic fever syndrome,26 Schnitzler’s syndrome,27,28 Behçet’s disease,29 and other immune-mediated diseases,30–33 make canakinumab an interesting option for use in systemic arthritis,23 which has found its way into recent recommendations.34 Not surprisingly, it has also been shown to be effective in adult Still’s disease.35
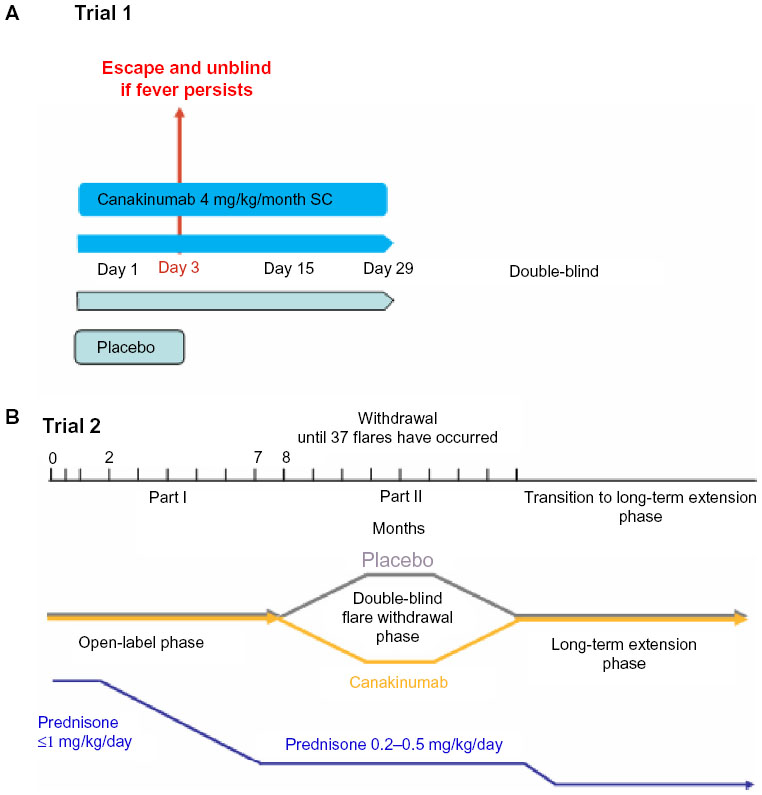
In a Phase II trial that included children with soJIA, 23 children and adolescents aged 4–19 years received a single injection of canakinumab with doses from 0.5 to 9 mg/kg. That trial was an open-label study of dose escalation.36 Canakinumab was readministered at the time of recurrence of disease. Thirteen of 22 (59%) patients showed an immediate response, with an improvement of at least a PedACR50 (American College of Rheumatology Pediatric Criteria) by day 15, which defines an improvement of at least 50% in at least three of six parameters without worsening of more than one of the remaining parameters. These six parameters include global disease activity by physician’s and parent’s evaluation, functional disabilities by the Childhood Health Assessment Questionnaire, systemic inflammation as determined by erythrocyte sedimentation rate, number of joints with active arthritis, and number of joints with limited range of motion. Inactive disease, defined as the absence of any disease activity indicator, was achieved in four patients (18%). Seventeen of the 23 patients were previously treated with anakinra. Six of eleven patients who had not responded to anakinra before achieved at least a PedACR50 response by day 15 after a single dose of canakinumab. This shows that failure of previous anakinra therapy does not seem to be a negative predictor of response to treatment with canakinumab. The best predictor of improvement from baseline was a low number of active joints, an observation that seems similar to that seen with anakinra. The median time to recurrence of disease activity correlated to the dosage as well as the probability of recurrence within 1 month. With 4 mg/kg of canakinumab given subcutaneously once per month the rate of relapse in between 1 month was estimated to be 6%. The injections were well tolerated. Side effects were mild to moderate, mainly comprising infections and gastrointestinal disturbances. Three serious adverse events were observed.
In a unique investigational plan, two randomized placebo-controlled trials were performed, including a total of 177 patients. In the first placebo-controlled, double-blind study of soJIA, 84 patients with systemically active JIA (fever was obligatory) received a single subcutaneous injection of canakinumab 4 mg/kg (maximum dose 300 mg) or placebo.37 Eligible children were aged 2–19 years with soJIA and had active systemic features and arthritis (two active joints, intermittent spiking fever >38°C, and a C-reactive protein level >30 mg/L [normal range <10 mg/L]). Background therapy with a prednisone equivalent of 1.0 mg/kg/day and stable doses of nonsteroidal anti-inflammatory drugs and methotrexate (up to a maximum of 20 mg/m2/week) were permitted. Major exclusion criteria were concomitant treatment with another biologic or disease-modifying drug, diagnosis of macrophage activation syndrome within the previous 6 months, active tuberculosis, and a live vaccination within 3 months prior to enrollment.
The primary outcome criterion was achievement of at least an adapted PedACR30 response and no fever. Patients with continuing fever could be unblinded from day 3, and if they had received placebo, they were offered canakinumab in an open trial arm. Six patients (14.0%) on canakinumab and 37 (90.2%) on placebo discontinued prematurely, all due to an unsatisfactory response. On day 15, 36/43 (83.7%) patients in the canakinumab group and 4/41 (9.8%) of patients in the placebo group met the primary endpoint of a modified PedACR30 with simultaneous defervescence. The difference was highly significant (Figure 2). Furthermore, at day 15, 14 (32.6%) patients on canakinumab versus none on placebo reached inactive disease status. One patient in each group developed a macrophage activation syndrome, and also one patient in each group got a serious infection.
 | Figure 2 Flow diagram of the study for the open-label and withdrawal design. |
Patients in the above-mentioned first controlled trial could change to a second study with a withdrawal design. Seventy-one patients from this trial, ten from the above-mentioned Phase II trial, and 96 newly enrolled patients contributed to the total of 177 patients who received canakinumab 4 mg/kg, with a maximum of 300 mg every 4 weeks. In the open-label phase of the study, all patients received canakinumab. In this first part of the study, the primary outcome criterion was a reduction in the steroid dose taken by at least 25% of patients at entry to the study by week 20. For this, a standardized corticosteroid-tapering protocol was permitted from week 9 to week 28 patients who achieved at least an adapted PedACR50 response. Corticosteroids were used by 128 of 177 patients (72.3%) patients at study entry. Forty-two of 128 patients (32.8%) were able to stop corticosteroids on treatment with canakinumab and 57 of 128 patients could reduce their steroid doses. The mean daily dose of corticosteroids decreased from 0.34 mg/kg to 0.05 mg/kg. Thus, the primary goal of this part of the study was reached. The secondary endpoint of the study was the proportion of patients achieving an adapted PedACR30/50/70/90/100 response or inactive disease. At day 15, 81.3%, 74.1%, 58.3%, 34.5%, 18.0%, and 16.4% already fulfilled these response criteria (Figure 3).
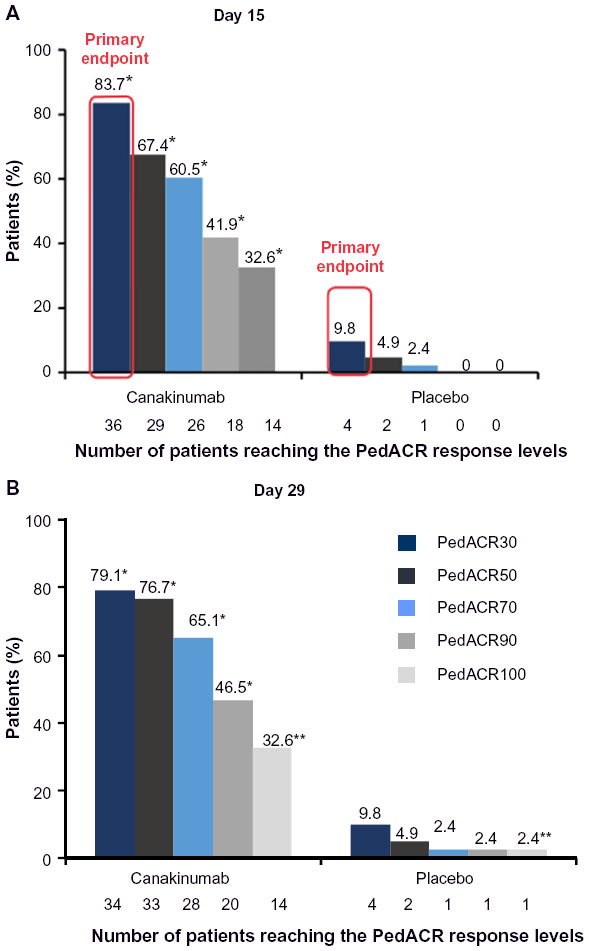 | Figure 3 Response according to PedACR30, PedACR50, and PedACR100 criteria in a double-blind study in patients with systemic-onset juvenile idiopathic arthritis. |
If patients improved by reaching the goal of decreasing pre-existing steroid doses or discontinued steroids, they were then transferred into the second part of this study, ie, the double-blind, placebo-controlled withdrawal phase. One hundred patients qualified and were admitted to this randomized part of the study in which occurrence of a predefined flare of the disease was the primary outcome criterion. In this part, the median time to onset of a disease relapse was 236 days (95% confidence interval 141–449) in the placebo cohort and was not determinable for canakinumab, since less than 50% of the patients ever had a relapse. This corresponds to a relative risk reduction of 63% (Figure 4). Patients who flared were immediately transferred to the open-label extension part of the study. The withdrawal phase was terminated after 37 patients had flared. Subsequently, all patients were transferred from an observational study to an ongoing open-label extension.
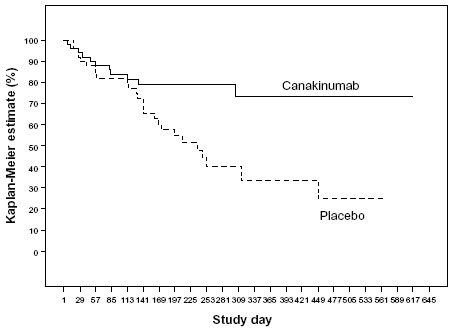 | Figure 4 Occurrence of relapses during second double blind study phase. |
Thirty-nine (78%) patients remained flare-free in the canakinumab arm versus 24 (48%) in the placebo arm, with a statistically significant 63% relative risk reduction for experiencing a soJIA flare (hazard ratio 0.37; 95% confidence interval 0.17–0.78; P=0.0043). Corticosteroids were used by 128/177 (72.3%) of patients at study entry.
At the end of the second part of the second trial, 41/50 (82%) patients in the canakinumab group had a minimum adapted PedACR70 response and 31/50 (62%) had inactive disease status compared with 31/50 (62%) and 16/50 (32%), respectively, in the placebo group (Figure 5).
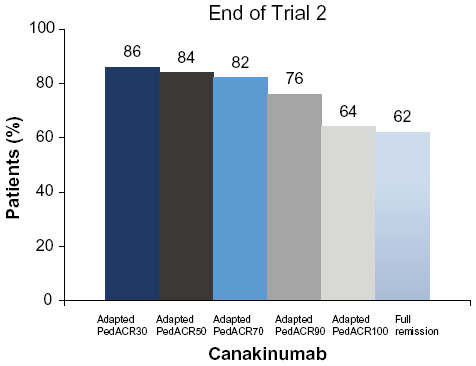 | Figure 5 Responses to canakinumab therapy in trial 2. |
Canakinumab in other immune-mediated diseases
In a three-part, 48-week, double-blind, placebo-controlled, randomized withdrawal study of canakinumab in patients with cryopyrin-associated autoinflammatory syndrome, treatment with subcutaneous canakinumab once every 8 weeks was associated with a rapid remission of symptoms in most patients.23 Following this study, canakinumab became approved as the first IL-1 blocker in 2011.
In a two-part, open-label, single-arm Phase II study, the long-term therapeutic effect of canakinumab was demonstrated in subjects aged 4–16 years with colchicine-resistant familial Mediterranean fever and a history of at least three documented attacks in the 3 months prior to enrolment.24
Treatment with canakinumab in an 8-year-old girl with a clinical picture of hyperimmunoglobulinemia D syndrome, that had started before the end of the first year of life, resulted in disappearance of febrile attacks and a considerable improvement in the patient’s quality of life during a 12-month follow-up period. In this case report, the drug was well tolerated, and no side effects were observed.25 The efficacy and safety of canakinumab has been assessed in a pilot study of 20 patients with active tumor necrosis factor receptor-associated periodic fever syndrome. In this study, canakinumab produced a rapid and highly effective clinical and serological benefit which was maintained for at least 4 months.26 A randomized, double-blind, placebo-controlled study of canakinumab in patients with these hereditary autoinflammatory disorders (tumor necrosis factor receptor-associated periodic fever syndrome, hyperimmunoglobulinemia D syndrome, familial Mediterranean fever) is currently recruiting.
In a 9-month study, monthly injection of canakinumab was an effective and well tolerated treatment for Schnitzler’s syndrome, a chronic disabling autoinflammatory disorder characterized by chronic urticaria, paraproteinemia, and severe systemic inflammation,27 as shown in other case reports.28
In a report of three adult patients with Behçet’s disease refractory to treatment with corticosteroids and different combinations of immunosuppressant agents who received canakinumab, prompt and sustained clinical efficacy was demonstrated, and canakinumab was seen as a therapeutic option for resistant or refractory Behçet’s disease.29 In this single-center, open-label pilot study, the effects of canakinumab on clinical signs and symptoms, quality of life, inflammation markers, and cytokine levels were investigated in ten patients with active urticarial vasculitis, and the efficacy and safety of canakinumab was documented.31
Khanna et al systematically reviewed the published data on the pharmacologic and nonpharmacologic agents used for the treatment of acute gouty arthritis, and found evidence to suggest the efficacy of treatment of acute gout with canakinumab, on a comedication with nonsteroidal anti-inflammatory drugs, selective cyclooxygenase type 2 inhibitors, corticosteroids, and colchicine.32
Safety of canakinumab in soJIA
As far as we can see at the moment, adverse events with canakinumab are manageable. In most cases, the adverse events are infections. Nevertheless, in one study, macrophage activation syndrome occurred in two patients, one of whom died.35 Macrophage activation syndrome is a well-known potential lethal complication in autoinflammatory diseases, and especially in soJIA. Despite this report, canakinumab can be considered as well tolerated. Even in patients with comorbidities like type 2 diabetes mellitus, the adverse events reported were manageable,38 although long-term observation and further studies are needed.
Conclusion
The more we know about the pathophysiology of inflammation and its pathways, the more specific targets we can find to control it. IL-1 is a global player in inflammation, and by blocking it we have a way to block inflammation very specifically with few side effects. With improving genetics and panels of ILs, we might in future even know in advance which patients to treat with which drug in the form of personalized medicine. Canakinumab seems to be one of the drugs for this approach.
In summary, canakinumab seems to be very effective in the treatment of soJIA. Its subcutaneous application is of benefit for patients who need to treated for a prolonged period of time.
Disclosure
JP reports no conflict of interest in this work. GH declares research grants from AbbVie Inc./Abbott Laboratories, Pfizer, Inc./Wyeth, and Hoffman-La Roche AG/Chugai Pharmaceutical Co., Ltd.; and consulting fees and speakers honoraries from AbbVie Inc., Chugai Pharmaceutical Co., Ltd., Genzyme Corporation, Pfizer, Inc., Novartis International AG, Hoffman-La Roche AG, Sandoz, and Swedish Orphan Biovitrum AB. GH reports no further conflicts of interest in this work.
References
Petty RE, Southwood TR, Baum J, et al. Revision of the proposed classification criteria for juvenile idiopathic arthritis: Durban, 1997. J Rheumatol. 1998;25:1991–1994. | |
Manners PJ, Bower C. Worldwide prevalence of juvenile arthritis why does it vary so much? J Rheumatol. 2002;29:1520–1530. | |
Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377:2138–2149. | |
Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369: 767–778. | |
Martini A, Ravelli A, Di Fuccia G, Rosti V, Cazzola M, Barosi G. Intravenous iron therapy for severe anaemia in systemic-onset juvenile chronic arthritis. Lancet. 1994;344:1052–1054. | |
Packham JC, Hall MA. Long-term follow-up of 246 adults with juvenile idiopathic arthritis: functional outcome. Rheumatology (Oxford). 2002;41:1428–1435. | |
De Benedetti F, Alonzi T, Moretta A, et al. Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin-like growth factor-I: a model for stunted growth in children with chronic inflammation. J Clin Invest. 1997;99:643–650. | |
Ravelli A, Magni-Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005;146:598–604. | |
Lovell DJ. Update on treatment of arthritis in children: new treatments, new goals. Bull NYU Hosp Jt Dis. 2006;64:72–76. | |
Horneff G, Schmeling H, Biedermann T, et al; for the Paediatric Rheumatology Collaborative Group. The German etanercept registry for treatment of juvenile idiopathic arthritis. Ann Rheum Dis. 2004;63: 1638–1644. | |
Russo RA, Katsicas MM, Zelazko M. Etanercept in systemic juvenile idiopathic arthritis. Clin Exp Rheumatol. 2002;20:723–726. | |
Quartier P, Taupin P, Bourdeaut F, et al. Efficacy of etanercept for the treatment of juvenile idiopathic arthritis according to the onset type. Arthritis Rheum. 2003;48:1093–1101. | |
Gartlehner G, Hansen RA, Jonas BL, Thieda P, Lohr KN. Biologics for the treatment of juvenile idiopathic arthritis: a systematic review and critical analysis of the evidence. Clin Rheumatol. 2008;27:67–76. | |
Yokota S, Imagawa T, Mori M, et al. Efficacy and safety of tocilizumab in patients with systemiconset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;371:998–1006. | |
De Benedetti F, Brunner HI, Ruperto N, et al. Efficacy and safety of tocilizumab in patients with systemic juvenile idiopathic arthritis (sJIA):12-week data from the phase 3 TENDER trial. Ann Rheum Dis. 2010;69:146. | |
Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479–1486. | |
Gattorno M, Pelagatti MA, Meini A, et al. Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2008;58:1516–1520. | |
Gattorno M, Piccini A, Lasiglie D, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58: 1505–1515. | |
De Benedetti F, Martini A. Is systemic juvenile rheumatoid arthritis an interleukin 6 mediated disease? J Rheumatol. 1998;25:203–207. | |
Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70:747–754. | |
Nigrovic PA, Mannion M, Prince FH, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63:545–555. | |
Verbsky JW, White AJ. Effective use of the recombinant interleukin 1 receptor antagonist anakinra in therapy resistant systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2004;31:2071–2075. | |
Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al; Canakinumab in CAPS Study Group. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360:2416–2425. | |
Hashkes P, Butbul Aviel Y, Lubin S, Ben-Dayan E, Tseng L, Brik R. A76: long-term efficacy of canakinumab in childhood colchicine resistant familial Mediterranean fever. Arthritis Rheumatol. 2014;66 Suppl 11:S108. | |
Tsitsami E, Papadopoulou C, Speletas M. A case of hyperimmunoglobulinemia D syndrome successfully treated with canakinumab. Case Rep Rheumatol. 2013;2013:79502. | |
Gattorn M, Obici L, Meini A, et al. Efficacy and safety of canakinumab in patients with TNF receptor associated periodic syndrome (TRAPS). Ann Rheum Dis. 2012;71 Suppl 3:289. | |
de Koning HD, Schalkwijk J, van der Ven-Jongekrijg J, Stoffels M, van der Meer JW, Simon A. Sustained efficacy of the monoclonal anti-interleukin-1 beta antibody canakinumab in a 9-month trial in Schnitzler’s syndrome. Ann Rheum Dis. 2013;72:1634–1638. | |
Vanderschueren S, Knockaert D. Canakinumab in Schnitzler syndrome. Semin Childhood Arthritis Rheum. 2013;42:413–416. | |
Vitale A, Rigante D, Caso F, et al. Inhibition of interleukin-1 by canakinumab as a successful mono-drug strategy for the treatment of refractory Behçet’s disease: a case series. Dermatology. 2014;228:211–214. | |
Jaeger T, Andres C, Grosber M, et al. Pyoderma gangrenosum and concomitant hidradenitis suppurativa – rapid response to canakinumab (anti-IL-1β). Eur J Dermatol. 2013;23:408–410. | |
Krause K, Mahamed A, Weller K, Metz M, Zuberbier T, Maurer M. Efficacy and safety of canakinumab in urticarial vasculitis: an open-label study. J Allergy Clin Immunol. 2013;132:751–754. | |
Khanna PP, Gladue HS, Singh MK, et al. Treatment of acute gout: a systematic review. Semin Arthritis Rheum. 2014;44:31–38. | |
Geusau A, Mothes-Luksch N, Nahavandi H, et al. Identification of a homozygous PSTPIP1 mutation in a patient with a PAPA-like syndrome responding to canakinumab treatment. JAMA Dermatol. 2013;149:209–215. | |
Kimura Y, Morgan Dewitt E, Beukelman T, et al; members of the Juvenile Idiopathic Arthritis Disease Specific Research Committee of the Childhood Arthritis and Rheumatology Research Alliance. Adding canakinumab to the childhood arthritis and rheumatology research alliance consensus treatment plans for systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken). 2014;66:1430–1431. | |
Barsotti S, Neri R, Iacopetti V, et al. Successful treatment of refractory adult-onset still disease with canakinumab: a case report. J Clin Rheumatol. 2014;20:121. | |
Ruperto N, Quartier P, Wulffraat N, et al; Paediatric Rheumatology International Clinical Trials Organisation. A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 2012;64:557–567. | |
Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367:2396–2406. | |
Howard C, Noe A, Skerjanec A, et al. Safety and tolerability of canakinumab, an IL-1beta inhibitor, in type 2 diabetes mellitus patients: a pooled analysis of three randomised double-blind studies. Cardiovasc Diabetol. 2014;13:94. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.