")
Back to Journals » Vascular Health and Risk Management » Volume 13
Treatment of pulmonary hypertension with left heart disease: a concise review
Authors Desai A, Desouza SA
Received 28 April 2016
Accepted for publication 2 December 2016
Published 6 November 2017 Volume 2017:13 Pages 415—420
DOI https://doi.org/10.2147/VHRM.S111597
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Daniel Duprez
Anish Desai, Shilpa A Desouza
Division of Pulmonary and Critical Care Medicine, Winthrop-University Hospital, Mineola, NY, USA
Abstract: Pulmonary hypertension (PH) is defined by a mean pulmonary artery pressure ≥ 25 mmHg, as determined by right heart catheterization. Pulmonary arterial hypertension (PAH) can no longer be considered an orphan disease given the increase in awareness and availability of new drugs. PH carries with it a dismal prognosis and leads to significant morbidity and mortality. Symptoms can range from dyspnea, fatigue and chest pain to right ventricular failure and death. PH is divided into five groups by the World Health Organization (WHO), based on etiology. The most common cause of PH in developed countries is left heart disease (group 2), owing to the epidemic of heart failure (HF). The data regarding prevalence, diagnosis and treatment of patients with group 2 PH is unclear as large, prospective, randomized controlled trials and standardized protocols do not exist. Current guidelines do not support the use of PAH-specific therapy in patients with group 2 PH. Prostacyclins, endothelin receptor antagonists, phosphodiesterase-5 inhibitors and guanylate cyclase stimulators have been tried in treatment of patients with HF and/or group 2 PH with mixed results. This review summarizes and critically appraises the evidence for diagnosis and treatment of patients with group 2 PH/HF and suggests directions for future research.
Keywords: pulmonary hypertension, left heart disease, diagnosis, treatment
Introduction
Pulmonary hypertension (PH) is defined by a mean pulmonary artery pressure (mPAP) ≥ 25 mmHg, as determined by right heart catheterization (RHC).1,2 PH is classified into five groups, based on etiology, according to the 5th World Symposium held in Nice, France, in 2013.3 Group 1 PH is differentiated from group 2 PH by presence of pulmonary arterial wedge pressure (PAWP) ≤ 15 mmHg, at end-expiration or when averaged over several respiratory cycles.2,4 Symptoms range from fatigue, dyspnea and chest pain to right ventricular (RV) failure and death. Pulmonary arterial hypertension (PAH) can no longer be considered as an orphan disease owing to a tremendous increase in awareness and availability of new drugs, fueled in part by the increasing number of chest computed tomography scans and echocardiograms being performed. At present, there are five approved classes of drugs for the treatment of PH: endothelin receptor antagonists, phosphodiesterase-5 inhibitors, prostacyclin analogs, calcium channel blockers and soluble guanylate cyclase stimulators. Almost all the clinical trials performed to test the efficacy of these drugs included patients with group 1 PH. Curative treatment exists only for group 4 PH (chronic thromboembolic disease), which is pulmonary endarterectomy. Standardized protocols and data are sparse for treatment of group 2, 3 and 5 PH. This review focuses on the challenges in determination of prevalence, diagnosis and treatment of patients with group 2 PH.
Definitions and prevalence
Heart failure (HF) is a national epidemic with a prevalence of more than five million cases, and more than half a million new cases are diagnosed each year.5 PH secondary to left heart disease (PH-LHD; group 2 PH) is defined as an mPAP ≥ 25 mmHg and a PAWP > 15 mmHg.6 PH-LHD leads to retrograde transmission of elevated filling pressures, mainly driven by left ventricular diastolic or systolic dysfunction.7,8 This sustained elevation of pressure leads to pulmonary capillary stress failure, arterial remodeling, impaired vascular reactivity and endothelial dysfunction, which are similar to the changes seen in group 1 PH.9 The presence of these pathological changes led to terms such as “out-of-proportion” or “reactive” PH, in order to explain the disproportionate increase in mPAP than expected from the underlying LHD. The European Society of Cardiology (ESC) and the European Respiratory Society (ERS) divided PH-LHD into isolated post-capillary PH (Ipc-PH) and combined post-capillary and pre-capillary PH (Cpc-PH) based on diastolic pressure gradient (DPG) and pulmonary vascular resistance (PVR).1 Ipc-PH was defined as DPG < 7 mmHg and/or PVR ≤ 3 Wood units (WU), and Cpc-PH was defined as DPG ≥ 7 mmHg and PVR > 3 WU.
Multiple studies have established that development of PH in patients with HF with preserved ejection fraction (HF-pEF) is indicative of worse outcomes.10,11 It is difficult to calculate the exact prevalence of PH-LHD as the largest studies performed in this population relied only on echocardiographic criteria. Data from these studies put the prevalence of PH-LHD between 25% and 79% in patients with HF-pEF and HF with reduced ejection fraction (HF-rEF).12,13 Given the extremely high prevalence of HF in the general population, perhaps the most common cause of PH is LHD (group 2 PH).
Diagnosis
The first challenge in treatment of PH-LHD is establishing the correct diagnosis. As previously mentioned, most studies performed to determine the prevalence of PH-LHD did not necessitate RHC as an inclusion criterion. We recommend patients suspected of group 2 PH, especially prior to consideration of PAH-specific therapy, undergo RHC for accurate diagnosis and risk stratification. Pulmonary vasoreactivity testing to identify patients who will respond to calcium channel blockers is indicated only in cases of idiopathic PH, heritable PH and drug-induced PH.1 In other cases, results might be misleading, and vasoreactivity testing may actually be detrimental in group 2 PH patients.
If treatment with PAH-specific drugs is being considered, the goal should be to isolate patients with Cpc-PH. Different hemodynamic values obtained from RHC including PVR, transpulmonary pressure gradient (TPG; the difference between mPAP and PAWP) and diastolic pressure gradient (DPG; the difference between diastolic PAP and PAWP) have been proposed to identify this subset of patients.
Guidelines issued by the ESC and the ERS in 2009 determined a TPG value of > 12 mmHg to diagnose patients with PH out of proportion to their LHD (Cpc-PH).6 Gerges et al analyzed RHC data retrospectively from more than 3,000 patients.14 Around 30% of patients had group 2 PH, and out of these, 490 cases were diagnosed as Cpc-PH. Cpc-PH was defined as mPAP ≥ 25 mmHg, PAWP > 15 mmHg and TPG > 12 mmHg or PVR > 3 WU. Out of the 490 cases of Cpc-PH, patients with DPG ≥ 7 mmHg (n = 179) had a worse survival when compared to patients with DPG < 7 mmHg (n = 311). The authors suggested that DPG might be a better indicator of Cpc-PH than TPG based on observations that patients with LHD and a TPG > 12 mmHg may normalize their pulmonary hemodynamics after cardiac transplant or with an infusion of nitroprusside or diuretics. Furthermore, Naeije et al proved that a DPG of > 5 mmHg is more accurate than TPG for predicting Cpc-PH, as DPG is not influenced by changes in stroke volume and pulmonary blood flow.15 DPG appears to be superior to TPG in diagnosing Cpc-PH based on available evidence, but there are no trials comparing DPG and TPG. TPG and DPG values should not be taken in isolation, and the diagnosis of Cpc-PH should be supported by the patient’s history and physical examination and echocardiographic findings, in addition to hemodynamic values obtained during RHC. A diagnosis of Cpc-PH in group 2 PH patients is established when the DPG is > 7 mmHg and/or the PVR is > 3 WU, in the appropriate clinical scenario.
Often, it is difficult to differentiate between group 1 and group 2 PH, especially in cases of HF-pEF. In such cases, the clinician will have to resort to dynamic testing, in the form of exercise or fluid loading, during RHC. Low-intensity exercise (20−25 W) has been shown in small trials to distinguish between HF-pEF and noncardiac causes of dyspnea.16–18 Borlaug et al at the Mayo Clinic studied the effects of exercise during RHC in 55 consecutive patients and found that PAWP and mPAP increased significantly (PAWP > 25 mmHg) in patients with HF-pEF, when compared with patients with dyspnea due to other causes.16 Fluid loading by administration of normal saline (10 ml/kg or 500 ml) has also been shown to differentiate between group 1 and group 2 PH.17,19,20 HF-pEF patients will demonstrate an increase in PAWP and/or left ventricular end diastolic pressure (LVEDP; > 15 mmHg) after fluid administration, while group 1 PH patients may exhibit an increase in mPAP and the PAWP may remain ≤ 15 mmHg.
Dynamic testing during RHC does have some limitations. Most trials evaluating dynamic testing were of single center and retrospective nature. The changes in PAWP, mPAP and right atrial pressure are variable, and no absolute cut-offs have been proposed. Treatment decisions based on exercise testing or volume loading during RHC should be made only after taking into account various other factors including echocardiographic findings, patient demographics and presence of other comorbid conditions. In cases of discrepancy, RHC should be followed by measurement of LVEDP.
Treatment
This section reviews the different therapeutic options for group 2 PH patients. Treatment for HF including pharmacotherapy, minimally invasive catheterization techniques and surgeries (e.g. valve replacements, assist devices, arrhythmia ablations and bypass grafting) should be performed foremost. General treatment measures such as oxygen supply when indicated, supervised exercise program, immunizations, and optimizing comorbid conditions are applicable to all patients with PH. Anticoagulation has not been studied in group 2 PH patients.
After the successful implementation of epoprostenol in treatment of group 1 PH patients, the FIRST study evaluated its role in patients with class IIIB/IV HF and decreased left ventricular ejection fraction (EF).21 Although patients in the epoprostenol group demonstrated a significant increase in cardiac index (CI), decrease in PAWP and decrease in systemic vascular resistance, the trial was terminated early as there was a trend toward increased mortality in the treatment arm.
Five prospective, randomized, double-blind clinical trials evaluated the role of endothelin receptor antagonists (bosentan = 3 and darusentan = 2) in treatment of HF and/or PH.22–26 Only one trial included patients with suspected PH, as determined by echocardiography.26 This study, performed by Kaluski et al demonstrated no change in the systolic pulmonary artery pressure at 20 weeks, after treatment with bosentan. The only trial to show a positive statistically significant end point was the one performed by Lüscher et al, where the authors demonstrated an increase in CI at three weeks after treatment with bosentan.22 None of the above-mentioned studies demonstrated long-term improvement in a clinically significant outcome. Two trials were terminated early secondary to increased risk of HF, most likely secondary to increased fluid retention observed with bosentan.23,25
Phosphodiesterase-5 inhibitors have also been extensively studied for treatment of HF and group 2 PH. Table 1 summarizes the findings of the different randomized controlled trials (RCTs).27–32 RHC was performed, and hemodynamic outcomes were reported in four of the six studies.27,29,30,32 Three studies included patients with HF-pEF.29,31,32 Four studies demonstrated a statistically significant improvement in the primary outcome including peak exercise capacity, mPAP, pulmonary artery systolic pressure, PAWP and exercise oscillatory breathing.27–30 Of note, of the four studies demonstrating benefits of sildenafil, three included patients with HF-rEF.27,28,30 These observations give more credence to the theory proposed by Forfia and Borlaug which indicated that RV dysfunction might have to be necessary to ensure a response to phosphodiesterase inhibitors.33 Furthermore, Wu et al demonstrated in a meta-analysis that additional treatment with sildenafil in patients with HF-rEF resulted in an improvement in exercise capacity, quality of life and pulmonary hemodynamics.34
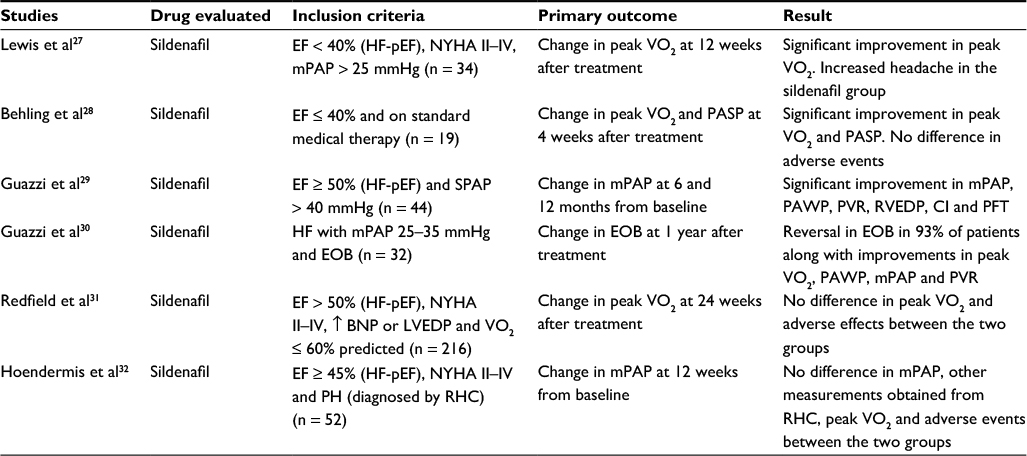 | Table 1 Summary of trials involving phosphodiesterase-5 inhibitors in HF and group 2 PH Abbreviations: HF, heart failure; PH, pulmonary hypertension; EF, ejection fraction; HF-pEF, heart failure with preserved ejection fraction; NYHA, New York Heart Association; mPAP, mean pulmonary artery pressure; VO2, exercise capacity; PASP, pulmonary artery systolic pressure; SPAP, systolic pulmonary artery pressure; PAWP, pulmonary arterial wedge pressure; PVR, pulmonary vascular resistance; RVEDP, right ventricular end diastolic pressure; CI, cardiac index; PFT, pulmonary function test; EOB, exercise oscillatory breathing; BNP, brain natriuretic peptide; LVEDP, left ventricular end diastolic pressure; RHC, right heart catheterization. |
Riociguat, the only drug available in the newest class (soluble guanylate cyclase stimulator), has also been evaluated for treatment of group 2 PH. LEPHT was a phase IIb, double-blind, RCT which studied the effects of riociguat in patients with HF-pEF (EF ≥ 40%) and mPAP ≥ 25 mmHg, as determined by RHC.35 The primary outcome was change in mPAP 16 weeks after treatment. At 16 weeks, mPAP was similar in both groups, but significant increases in CI and quality of life and decrease in PVR were seen, when compared to placebo. There were no significant adverse effects observed with riociguat. Another placebo-controlled, double-blind, RCT evaluated the role of single-dose riociguat in altering pulmonary hemodynamics.36 Thirty-six patients with HF-pEF (EF > 50%) and group 2 PH diagnosed by RHC were evaluated six hours after administration of a single dose of riociguat or placebo. There was no change in mPAP (primary end point), although riociguat significantly increased stroke volume, and decreased systolic blood pressure and RV end diastolic area.
Atrial septostomy (AS) involves creating a communication between both the atria to facilitate unloading of the right-sided cardiac chambers. This procedure has been shown to improve pulmonary hemodynamics, although the 30-day mortality can be as high as 25%.37,38 AS has almost exclusively been studied in group 1 PH patients, and currently, it is considered as a palliative treatment or a bridge to lung transplant. AS has the potential to paradoxically worsen group 2 PH in patients with HF-pEF by increasing preload to the left ventricle.
In conclusion, we could find 14 RCTs (one with prostacyclin analogs, five with endothelin receptor antagonists, six with sildenafil and two with riociguat) that evaluated the role of PH-specific therapy in HF and/or group 2 PH. Out of these, one trial involving epoprostenol and two with bosentan had to be terminated early secondary to an increased trend toward mortality or complications. Most trials consisted of small sample sizes, were of single-center nature and did not elaborate on long-term clinical outcomes as well as side effects. Additionally, all trials had different inclusion criteria, and a few did not mandate a diagnosis of PH by RHC prior to inclusion, thus making it very difficult to derive standard conclusions about group 2 PH treatment. Data regarding riociguat in treatment of group 2 PH is not robust as one trial was a phase IIb RCT, while the other evaluated pulmonary hemodynamics six hours after administration of one dose of riociguat. Till date, no RCTs have been performed evaluating the role of PH-specific drugs in patients with Cpc-PH. Various RCTs involving macitentan, sildenafil and other drugs are ongoing to address some of these problems, and results should be out soon.1,39 Secondary to the limitations discussed, current guidelines do not support the use of PAH-specific therapy in patients with group 2 PH.
Future directions
RCTs requiring PH diagnosis by RHC as an inclusion criterion prior to patient enrollment are needed. Additionally, the various pharmacotherapies should be studied in patients with group 2 PH with DPG ≥ 7 mmHg and/or PVR > 3 WU. Physiological improvement may not always correlate with clinical improvement, and more trials focusing on clinical outcomes including mortality, frequency of hospital admissions and quality of life, are needed.
Conclusion
PH carries a poor prognosis with significant morbidity and mortality. Group 2 PH is perhaps the most common as well as an underdiagnosed cause of PH. Current guidelines do not recommend treatment with PAH-specific drugs in this patient population. Patients with group 2 PH, especially Cpc-PH, should be referred to specialized centers for individualized treatment decisions.
Acknowledgment
The authors are grateful to Dr. Jean-Luc Vachiery for his valuable insights.
Disclosure
The authors report no conflicts of interest in this work.
References
Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37(1):67–119. | ||
Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl): D42–D50. | ||
Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013; 62(25 Suppl):D34–D41. | ||
Kovacs G, Avian A, Pienn M, Naeije R, Olschewski H. Reading pulmonary vascular pressure tracings. How to handle the problems of zero leveling and respiratory swings. Am J Respir Crit Care Med. 2014; 190(3):252–257. | ||
Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology Foundation; American Heart Association. 2009 focused update incorporated into the ACC/AHA 2005 guidelines for the diagnosis and management of heart failure in adults. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines developed in collaboration with the International Society for Heart and Lung Transplantation. J Am Coll Cardiol. 2009;53(15):e1–e90. | ||
Task Force for Diagnosis and Treatment of Pulmonary Hypertension of European Society of Cardiology (ESC); European Respiratory Society (ERS); International Society of Heart and Lung Transplantation (ISHLT), Galiè N, Hoeper MM, Humbert M, et al. Guidelines for diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2009;34(6):1219–1263. | ||
Vachiéry JL, Adir Y, Barberà JA, et al. Pulmonary hypertension due to left heart diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D100–D108. | ||
Rosenkranz S, Gibbs JS, Wachter R, De Marco T, Vonk-Noordegraaf A, Vachiéry JL. Left ventricular heart failure and pulmonary hypertension. Eur Heart J. 2016;37(12):942–954. | ||
Guazzi M, Borlaug BA. Pulmonary hypertension due to left heart disease. Circulation. 2012;126(8):975–990. | ||
Melenovsky V, Hwang SJ, Lin G, Redfield MM, Borlaug BA. Right heart dysfunction in heart failure with preserved ejection fraction. Eur Heart J. 2014;35(48):3452–3462. | ||
Mohammed SF, Hussain I, AbouEzzeddine OF, et al. Right ventricular function in heart failure with preserved ejection fraction: a community-based study. Circulation. 2014;130(25):2310–2320. | ||
Bursi F, McNallan SM, Redfield MM, et al. Pulmonary pressures and death in heart failure: a community study. J Am Coll Cardiol. 2012;59(3):222–231. | ||
Damy T, Goode KM, Kallvikbacka-Bennett A, et al. Determinants and prognostic value of pulmonary arterial pressure in patients with chronic heart failure. Eur Heart J. 2010;31(18):2280–2290. | ||
Gerges C, Gerges M, Lang MB, et al. Diastolic pulmonary vascular pressure gradient: a predictor of prognosis in “out-of-proportion” pulmonary hypertension. Chest. 2013;143(3):758–766. | ||
Naeije R, Vachiery JL, Yerly P, Vanderpool R. The transpulmonary pressure gradient for the diagnosis of pulmonary vascular disease. Eur Respir J. 2013;41(1):217–223. | ||
Borlaug BA, Nishimura RA, Sorajja P, Lam CS, Redfield MM. Exercise hemodynamics enhance diagnosis of early heart failure with preserved ejection fraction. Circ Heart Fail. 2010;3(5):588–595. | ||
McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol. 2015;65(18):1976–1997. | ||
Hager WD, Collins I, Tate JP, et al. Exercise during cardiac catheterization distinguishes between pulmonary and left ventricular causes of dyspnea in systemic sclerosis patients. Clin Respir J. 2013;7(3):227–236. | ||
Robbins IM, Hemnes AR, Pugh ME, et al. High prevalence of occult pulmonary venous hypertension revealed by fluid challenge in pulmonary hypertension. Circ Heart Fail. 2014;7(1):116–122. | ||
Fox BD, Shimony A, Langleben D, et al. High prevalence of occult left heart disease in scleroderma-pulmonary hypertension. Eur Respir J. 2013;42(4):1083–1091. | ||
Califf RM, Adams KF, McKenna WJ, et al. A randomized controlled trial of epoprostenol therapy for severe congestive heart failure: the Flolan International Randomized Survival Trial (FIRST). Am Heart J. 1997;134(1):44–54. | ||
Lüscher TF, Enseleit F, Pacher R, et al; Heart Failure ET(A) Receptor Blockade Trial. Hemodynamic and neurohumoral effects of selective endothelin A (ET(A)) receptor blockade in chronic heart failure: the Heart Failure ET(A) Receptor Blockade Trial (HEAT). Circulation. 2002;106(21):2666–2672. | ||
Kalra PR, Moon JC, Coats AJ. Do results of the ENABLE (Endothelin Antagonist Bosentan for Lowering Cardiac Events in Heart Failure) study spell the end for non-selective endothelin antagonism in heart failure? Int J Cardiol. 2002;85(2–3):195–197. | ||
Anand I, McMurray J, Cohn JN, et al; EARTH investigators. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the EndothelinA Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364(9431):347–354. | ||
Packer M, McMurray J, Massie BM, et al. Clinical effects of endothelin receptor antagonism with bosentan in patients with severe chronic heart failure: results of a pilot study. J Card Fail. 2005;11(1):12–20. | ||
Kaluski E, Cotter G, Leitman M, et al. Clinical and hemodynamic effects of bosentan dose optimization in symptomatic heart failure patients with severe systolic dysfunction, associated with secondary pulmonary hypertension—a multi-center randomized study. Cardiology. 2008;109(4):273–280. | ||
Lewis GD, Shah R, Shahzad K, et al. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation. 2007;116(14):1555–1562. | ||
Behling A, Rohde LE, Colombo FC, Goldraich LA, Stein R, Clausell N. Effects of 5’-phosphodiesterase four-week long inhibition with sildenafil in patients with chronic heart failure: a double-blind, placebo-controlled clinical trial. J Card Fail. 2008;14(3):189–197. | ||
Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation. 2011;124(2):164–174. | ||
Guazzi M, Vicenzi M, Arena R. Phosphodiesterase 5 inhibition with sildenafil reverses exercise oscillatory breathing in chronic heart failure: a long-term cardiopulmonary exercise testing placebo-controlled study. Eur J Heart Fail. 2012;14(1):82–90. | ||
Redfield MM, Chen HH, Borlaug BA, et al; RELAX Trial. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309(12):1268–1277. | ||
Hoendermis ES, Liu LC, Hummel YM, et al. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: a randomized controlled trial. Eur Heart J. 2015;36(38):2565–2573. | ||
Forfia PR, Borlaug BA. Letter by Forfia and Borlaug regarding article, “Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study”. Circulation. 2012;125(8):e408; author reply e409–e410. | ||
Wu X, Yang T, Zhou Q, Li S, Huang L. Additional use of a phosphodiesterase 5 inhibitor in patients with pulmonary hypertension secondary to chronic systolic heart failure: a meta-analysis. Eur J Heart Fail. 2014;16(4):444–453. | ||
Bonderman D, Ghio S, Felix SB, et al; Left Ventricular Systolic Dysfunction Associated with Pulmonary Hypertension Riociguat Trial (LEPHT) Study Group. Riociguat for patients with pulmonary hypertension caused by systolic left ventricular dysfunction: a phase IIb double-blind, randomized, placebo-controlled, dose-ranging hemodynamic study. Circulation. 2013;128(5):502–511. | ||
Bonderman D, Pretsch I, Steringer-Mascherbauer R, et al. Acute hemodynamic effects of riociguat in patients with pulmonary hypertension associated with diastolic heart failure (DILATE-1): a randomized, double-blind, placebo-controlled, single dose study. Chest. 2014;146(5):1274–1285. | ||
Reichenberger F, Pepke-Zaba J, McNeil K, Parameshwar J, Shapiro LM. Atrial septostomy in the treatment of severe pulmonary arterial hypertension. Thorax. 2003;58(9):797–800. | ||
Kurzyna M, Dabrowski M, Bielecki D, et al. Atrial septostomy in treatment of end-stage right heart failure in patients with pulmonary hypertension. Chest. 2007;131(4):977–983. | ||
Hoeper MM, McLaughlin VV, Dalaan AM, Satoh T, Galiè N. Treatment of pulmonary hypertension. Lancet Respir Med. 2016;4(4):323–336. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.