")
Back to Journals » Clinical Ophthalmology » Volume 8
Treatment of corneal cystine crystal accumulation in patients with cystinosis
Authors Shams F, Livingstone I, Oladiwura D, Ramaesh K
Received 9 May 2014
Accepted for publication 4 July 2014
Published 10 October 2014 Volume 2014:8 Pages 2077—2084
DOI https://doi.org/10.2147/OPTH.S36626
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Fatemeh Shams, Iain Livingstone, Dilys Oladiwura, Kanna Ramaesh
Department of Ophthalmology, Gartnavel General Hospital, Glasgow, Scotland
Abstract: Cystinosis is a rare autosomal recessive disorder characterized by the accumulation of cystine within the cells of different organs. Infantile nephropathic cystinosis is the most common and severe phenotype. With the success of renal transplantation, these patients are now living longer and thus more long-term complications within different organs are becoming apparent. Ophthalmic manifestations range from corneal deposits of cystine crystals to pigmentary retinopathy. With increasing age, more severe ocular complications have been reported. Photophobia is a prominent symptom for patients. With prolonged survival and increasing age, this symptom, along with corneal erosions and blepharospasm, can become debilitating. This review revisits the basic pathogenesis of cystinosis, the ocular manifestations of the disease, and the treatment of corneal crystals.
Keywords: cystinosis, cysteamine, cornea, ocular, crystals, lysosome
Introduction
Cystinosis is a rare autosomal recessive disorder affecting the lysosomal storage system. It is caused by a disruption of the carrier-mediated system that transports cystine out of the lysosomes.1 Accumulation of cystine within the lysosomes results in the formation of characteristic crystals accumulating within numerous tissues, including the kidneys, eyes, bone marrow, liver, spleen, pancreas, thyroid, muscle, and brain.2 The estimated incidence is one in 100,000 live births.1
Abderhalden3 first described cases of cystinosis in 1903. Our clinical understanding of the disease did not mature, however, until the 1930s, when Fanconi’s renotubular syndrome was described.1 Since then, several phenotypes of cystinosis have been described, depending on the degree of severity of the disease and the age of onset.1 These have been classified into nephropathic and non-nephropathic phenotypes.
The nephropathic phenotype is further subdivided into the infantile form, the classic, most common type of cystinosis, and the intermediate (late-onset juvenile) form. The non-nephropathic form was previously called adult or benign type cystinosis, now termed ocular cystinosis.4
The classic infantile nephropathic cystinosis accounts for the majority of cases, and exhibits the most severe phenotype.4 Early symptoms include Fanconi’s renotubular syndrome, which develops in 95% of patients as early as 6–12 months of life.1 By the age of 10 years, 50% of patients develop hypothyroidism and photophobia, while other signs and symptoms such as impaired growth and rickets are also common.5 Although disease severity varies, end-stage renal failure invariably occurs within the first 10 years of life.6
Intermediate or late-onset cystinosis also involves renal disease; however, the onset of symptoms is typically during adolescence, with slower progression. Such patients may retain renal function into their fourth decade of life. In the ocular form of the disease, there is no systemic involvement. The main symptom is photophobia, and patients have biomicroscopic evidence of crystal accumulation affecting the cornea and conjunctiva.7,8
In the 1960s, with the advent of renal transplantation, the clinical course of cystinosis was transformed. Patients who had end-stage renal failure were given hope and had successful outcomes with renal transplantation, prolonging their lifespan significantly.2 However, the accumulation of cystine continued in nonrenal organs; hence, a predominately renal disease changed clinically into a more multisystemic disorder. As survival improved, patients developed more significant, nonrenal manifestations, such as myopathy, pulmonary involvement, swallowing difficulties, central nervous system complications, endocrinopathies, and more severe ophthalmic complications, including retinal damage.9
In the 1970s the focus of treatment of cystinosis moved from supportive treatment to therapy, focusing on the lysosomal accumulation of cystine via the oral drug cysteamine. This was first used in the treatment of cystinosis in 1976.10 A cystine-depleting free thiol, cysteamine has been shown to reduce cystine levels both in vitro and in vivo.10 Early treatment with cysteamine has been shown to delay deterioration in renal function, improve growth, and delay nonrenal complications.11 Its mechanism of action is via its entry into the lysosome through a specific transporter, and interaction with cystine, where it forms a cysteamine–cysteine mixed disulfide compound. This compound is then able to exit the lysosome via an intact cationic amino acid transporter.12,13 This transporter, PQLC2, has been shown to be a member of the PQ loop family of proteins, which also includes the cystine carrier protein, cystinosin (Figure 1).14
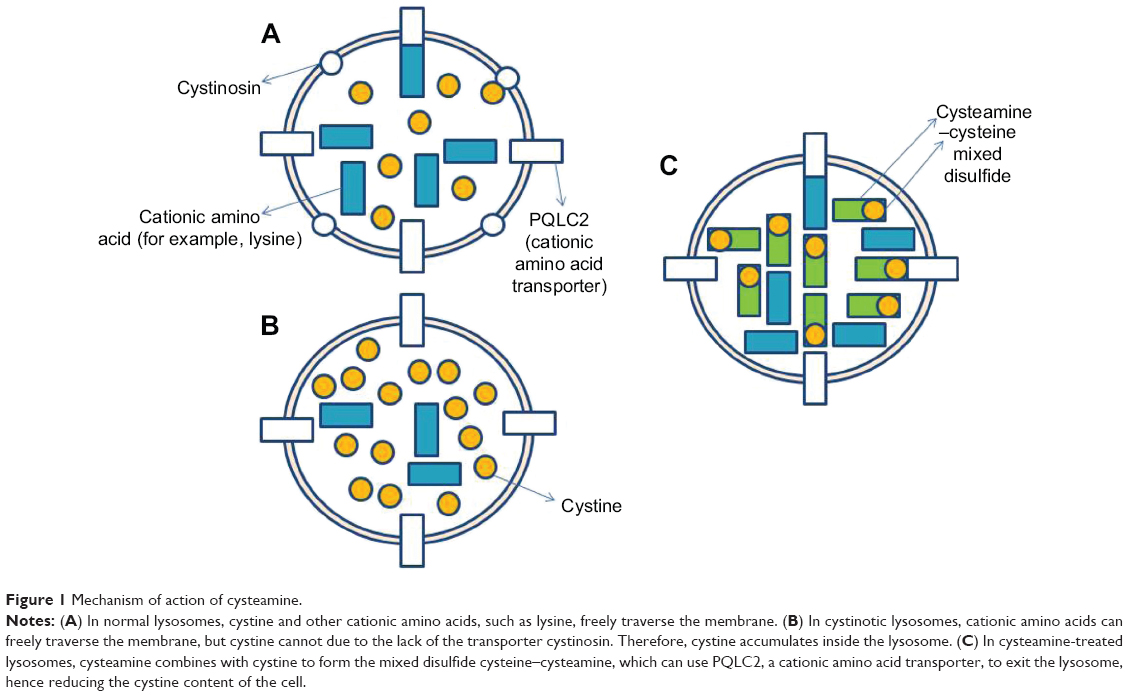 | Figure 1 Mechanism of action of cysteamine. |
Genetic features
The gene implicated in cystinosis, CTNS, has been localized to chromosome 17p13.15 It has 12 exons and encodes the protein cystinosin, an integral lysosomal membrane protein. Cystinosin is a cystine carrier within the membrane of lysosmes, containing 367 amino acids and seven transmembrane domains.16 All patients with cystinosis appear to have a mutation in CTNS.1
The most common mutation of CTNS is a 57 kb deletion.2 This is thought to have first arisen around 1,500 years ago, and it affects nearly half of the American and European population with cystinosis.17 To date, more than 50 different mutations have been described in the CTNS gene.18,19 Patients with classic nephropathic cystinosis have homozygous mutations that lead to functional loss of the protein, such as a deletion mutation. Less severe phenotypes tend to have heterozygous mutations, with one severe mutation (such as a nonsense mutation) and one less severe mutation (such as a splice-site mutation).8
Patients that never manifest clinical symptoms, yet who are heterozygotes for cystinosis, typically have around half the normal cystine-transporting capacity.20 This allows cystine levels in lysosomes to be maintained to near-normal levels.21 In general, it is thought that the severity of the disease correlates directly with cystine storage, and inversely with the amount of residual transport capacity.22 Other environmental factors and possible modifying genes may also play a role in influencing phenotype.
Pathogenesis
Cystine crystals formed within lysosomes are poorly water soluble. While crystallization is a clear sign of impaired cystine transport, the extent of crystal formation does not correlate with the severity of tissue damage. The liver, for example, shows high levels of crystal formation, yet only rarely causes clinical symptoms.2 Some organs have a more extensive functional reserve or higher cell turnover rate, which could explain this difference.2
It is still unclear how cystine accumulation leads to the pathology seen in nephropathic cystinosis.2 Fanconi syndrome occurs without significant cystine crystal accumulation in the renal tubules, and it does not seem to respond as well to cystine-depleting medication.23 One hypothesis revolves around cell apoptosis based on the fact that the lysosomes play a part in programmed cell death.24
By using a mouse model of cystinosis (CTNS−/− knockout mice) Cherqui et al (in 2002) found that the cystinotic mice did not show signs of the renotubular defect seen early on in life in humans, even though the kidneys showed high levels of cystine accumulation.25 The authors suggest that the renal defect seen in humans is secondary to metabolic consequences, rather than a direct effect of cystine storage.25 To add to this hypothesis, a previous study in rabbit models has shown that the lysosomes of the cystine-loaded proximal tubules have significantly lower intracellular adenosine triphosphate (ATP) concentration, suggesting that the mitochondrial oxidative phosphorylation process responsible for ATP production is impaired in cystinotic proximal tubular cells.26
A more recent study looking at cultured human cystinotic fibroblasts also showed a significant decrease in intracellular ATP content.27 However, in this study, the mitochondrial energy-generating capacity and Na/K-ATPase activity were intact in these cells, thus bringing into question the hypothesis of altered mitochondrial ATP synthesis as a keystone for the pathogenesis of cystinosis.
Mitochondrial swelling has been described in renal biopsies from cystinotic patients.28,29 Further research in animal models may give us a better understanding of the disease etiopathogenesis.
Ocular manifestations
Accumulation of cystine crystals within the cornea and conjunctiva are pathognomonic of cytinosis. Burki30 first described the presence of these crystals in 1941 as a myriad of small, white, shining sequin-like crystals, which are deposited on the superficial layers of the parenchyma, while respecting the limits of the epithelium and endothelium.4,31
The crystals are typically obvious on slit-lamp biomicroscopy, appearing as needle-shaped, reflective corneal opacities. They tend to diffusely span the entire cornea. While it was initially thought that the endothelium was spared, case reports have since demonstrated crystals within the epithelium, stroma, and endothelium (Figures 2 and 3).32–35
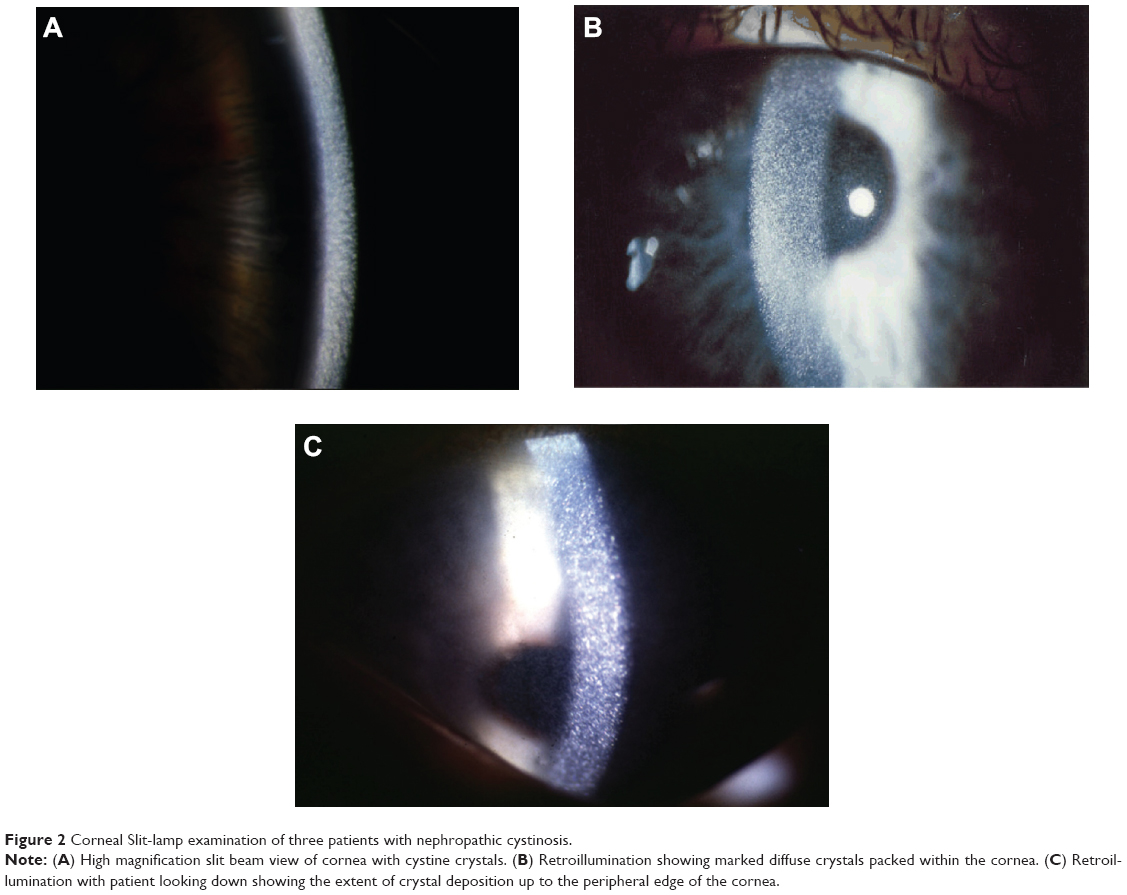 | Figure 2 Corneal Slit-lamp examination of three patients with nephropathic cystinosis. |
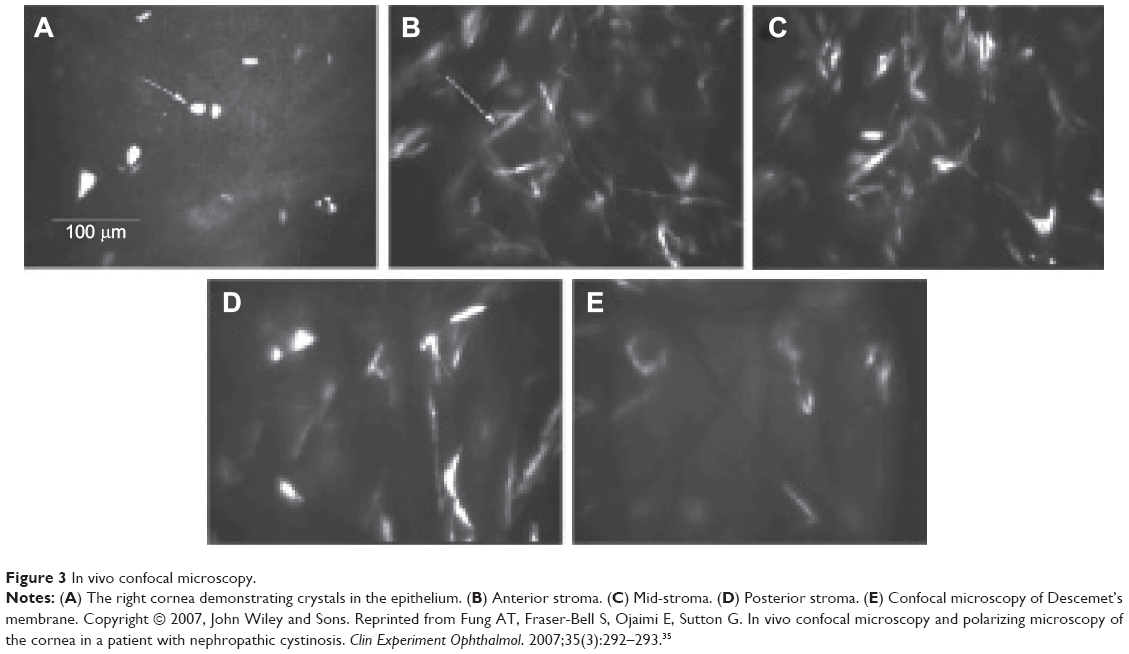 | Figure 3 In vivo confocal microscopy. |
The buildup of corneal crystals begins in infancy, and is evident on slit-lamp examination in all nephropathic cystinosis patients by 16 months of age.36 There is progressive accumulation of corneal crystals with age, albeit at a variable rate. By the age of 12 years, most affected corneas evince marked crystal formation.36
Deposition of the crystals begins in the peripheral cornea, progressing centripetally with age.4 The increase in crystal density culminates with a hazy cornea, which can occasionally be seen by the naked eye in older untreated patients. The crystals themselves, however, have minimal effect on visual acuity,37 though rarely contribute to band keratopathy, with consequent impact on visual acuity when the central cornea becomes involved.2
The aggregation of corneal crystals is typically asymptomatic initially, though most patients develop photophobia within the first decade of life,38 with severity varying with ambient light levels, and some experiencing associated blepharospasm.4 Superficial punctate keratopathy, foreign body sensation, and pain have also been reported, predominantly in older patients.39,40 This is thought to be secondary to crystals interrupting Bowman’s membrane, inciting inflammation within the epithelium and basement membrane.1,32 There has also been documentation of loss of contrast sensitivity, decreased corneal sensation, increased glare, and corneal thickening in patients with nephropathic cystinosis.41,42
Aside from corneal involvement, crystal accumulation within the conjunctiva, iris, ciliary body, choroid, fundus, and optic nerve have been reported.40,43 Pigmentary retinopathy has been well described, preceding corneal involvement, and it has been reported as early as 5 weeks of age in the context of nephropathic cystinosis.44–46
The most common posterior segment abnormality described is a depigmentation and pigmentary mottling of the fundus, with a wide variation in terms of extent between subjects.46–48 Damaged retinal pigment epithelium with pigment migration and the destruction of the photoreceptor’s outer layers has been reported in cystinotic patients.49
With the development of cystine-depleting agents and the success of renal transplantation in cystinosis, patient survival has significantly improved, and more complex late-onset complications have emerged.4 Examples include superficial punctate keratopathy, filamentory keratopathy,50 band keratopathy, and peripheral corneal neovascularization.51 Anecdotal reports of other ocular morbidities presumed secondary to cystinosis include pupil-block glaucoma secondary to angle abnormalities, posterior synechiae associated with iris thickening,52 iris transillumination, crystals on the anterior lens capsule,43 and a vascular membrane in the papillary space.40
Furthermore, in a large cross-sectional study by Tsilou et al50 75% of patients aged 20–29 years, and 87% of patients above 30 years with nephropathic cystinosis had some form of anterior segment complication distinct from the typical corneal and conjunctival crystal accumulation.
Reported posterior segment complications include worsening retinopathy with acuity reduction, constricted visual fields, and disturbed rod/cone function on electroretinography.53
Treatment
The oral cystine-depleting agent, cysteamine, has become the treatment of choice in cystinosis. It has been proven to delay disease progression, improve growth, and slow down the damage to nonrenal organs, such as the thyroid.54 It does not, however, seem to have an effect on the Fanconi renotubular syndrome secondary to cystinosis and, therefore, patients still require electrolyte replacement and nutritional support.2 The response to cystine-depleting therapy is variable, and it has thus been proposed that genetic heterogeneity and influences of unknown genetic/environmental factors may have an impact on the progression of the disease.2,55 Regarding ophthalmic manifestations in cystinosis, Tsilou et al53 showed that the frequency of cystinotic retinopathy correlated positively with time off cysteamine therapy, and negatively with time on cysteamine therapy.
The oral formulation of cysteamine does not appear to have an effect on the established corneal cystine crystals. The avascular properties of the cornea are thought to prevent cystine clearance through the oral administration of cysteamine.1 With improved disease management and patient survival, photophobia has become the prominent symptom reported in the majority of predominantly older patient groups.40 The photophobia is often severe and occasionally debilitating, frequently associated with blepharospasm, as well as recurrent corneal erosions. In the late 1980s, topical preparations of cysteamine were found to be nontoxic in animal studies, and found to reduce the number of corneal crystals present.40 Indeed, numerous single-center trials have shown that topical cysteamine can effectively dissolve corneal crystals and, therefore, significantly improve symptoms of photophobia, blepharospasm, and pain, irrespective of the age at which the drops were commenced.36,56
The problem with the topical formulation of cysteamine is twofold. Firstly, while it has been shown to be effective in reducing corneal crystals, administration needs to be particularly frequent, either every hour while awake,57 or at least six times a day,58 but not four times a day.59 Secondly, cysteamine oxidizes to its disulfide form, cystamine, at room temperature. A randomized controlled trial60 comparing cysteamine drops to its oxidized disulfide form, cystamine, showed that cystamine was not as effective in reducing corneal cystine crystals. This study looked at eleven patients with nephropathic cystinosis. Each patient underwent an initial baseline ophthalmic examination and slit-lamp images were taken at each visit. The corneal crystal density was graded based on these images by two independent masked graders. This grading system has previously been described in the literature.56 One eye of each patient was then randomized to either cysteamine 0.5% drops or cystamine 0.5% drops. Patients were followed up at 6–8-month intervals and information on the symptoms of photophobia and foreign body sensation, possible side effects; differences between the two eyes, and treatment compliance were attained. Six patients were noted to have significantly different corneal crystal density scores as early as their first follow up. In each case, the better eye was the eye treated with cysteamine 0.5% drops. Five of these six patients with significant reduction in corneal crystal density also reported subjective improvement in their symptoms of photophobia.
Therefore, to maintain efficacy, the cysteamine drops have to be frozen, or kept away from oxygen, to prevent oxidization. Current formulations of drops have to be kept cool and used within 1 week of opening. The instability of the formula at room temperature makes packaging and distribution of the drops difficult. This is one of the reasons why cysteamine drops have yet to be licensed in the United Kingdom (currently used off-license), and why they have only recently been approved by the Food and Drug Administration in the United States (Cystaran®; Sigma-Tau Pharmaceuticals, Inc., Gaithersburg, MD, USA).
Such problems of storage and frequency make adherence an issue for many patients. Recent studies have focused on developing a formulation of cyteamine drops that are not only stable at room temperature, but also efficacious at lower frequencies.
Tsilou et al61 reported a randomized clinical trial evaluating the safety and efficacy of a new topical formulation where cysteamine is maintained as a free thiol, and which is stable at room temperature for up to 7 months. The new formulation was obtained from Sigma-Tau Pharmaceuticals, Inc., and consisted of a 0.55% cysteamine hydrochloride solution with monosodium phosphate 1.85%, disodium EDTA 0.10%, and benzalkonium chloride 0.01%. Despite a promising safety profile, it was deemed less effective than the standard topical treatment (0.55% cysteamine hydrochloride solution with benzalkonium chloride 0.01%).61
Labbé et al62 have also recently studied a new gel formulation of cysteamine, Cystadrops® (Orphan Europe, Paris, France). This formulation is reported to be stable for 7 days after opening, when refrigerated at night, and when kept at room temperature during the day. The study was carried out as an open-label, dose-response study, though the number of patients was small (number =8); follow up continued for 4 years. After an initial reduction in corneal crystal density comparable to the standard formulation of cysteamine drops, crystal density remained stable within the 4-year follow-up period on a reduced regime of three instillations of the gel form.62 Further controlled trials are required to establish the place of this treatment in clinical practice.
Other modalities with the potential for extended delivery of ocular drugs include fornix inserts, punctual plugs, subconjunctival inserts, and contact lenses. Hsu et al63 combined in vitro experimental results with mathematical models to evaluate the feasibility of daily-use contact lenses loaded with cyteamine to circumvent the frequent installation, and the authors observed that loading daily contact lenses with vitamin E in addition to cyteamine increased the release duration of the drug, optimizing the drug stability by reducing the oxidation rate. The authors asserted that single-use, daily contact lenses worn for 2 hours a day have the potential to achieve the same therapeutic effect as an hourly instillation of cysteamine drops.63 The clinical place of this potential innovation remains a subject of future study.
Penetrating keratoplasty has previously been used in rare cases to treat intractable symptoms of photophobia/blepharospasm and pain.40,41 However, with recent improvement in the control of these symptoms using drops, this treatment is rarely necessary in the absence of such complications such as band keratopathy involving the visual axis. Furthermore, recurrent corneal crystal deposition within the graft following penetrating keratoplasty has been reported.41 At present, the mainstay of treatment of corneal cystine crystals is through the use of cysteamine hydrochloride drops at 0.55% or 0.44%, instilled hourly or six times a day.
Curative therapy for hereditary genetic diseases requires the addition of the gene to many cells, usually within different tissue compartments, or the replacement of injured and dying cells by cells expressing the functional version of the gene.64
Cystinosin is a lysosomal transmembrane protein that cannot be secreted.65 The therapeutic efficacy of stem cell transplantation for cystinosis requires the local integration of donor cells with the functional protein, which would then reverse the accumulation of cystine in the tissue.
By using CTNS knockout (CTNS−/−) mice66 as a model for cystinosis, Syres et al67 showed that bone marrow cell (BMC) transplantation leads to a major decrease in the cystine content of all tissues tested. This was reflected by a significant decrease in the development and progression of kidney injury and the reduction in the number of mice with corneal cystine crystals. BMC-derived cells were detected in the eye, brain, spleen, heart, muscle, and liver, with an ensuing decrease in the cystine content of all these organs. This study demonstrates the potential use of BMC transplantation for the multisystemic complications of cystinosis.
The exact mechanism by which cystinosin-expressing cells replace or fuse with cells that accumulate cystine crystals in each tissue remains to be defined. A study by Iglesias et al64 has more recently implicated the shedding and fusing of microvesicles containing wild-type cystinosin protein and messenger RNA by stem cells as a plausible mechanism.
The rare nature of cystinosis limits the potential for large clinical trials; although cell therapy shows great promise, progress has not yet extended beyond the laboratory, with no human trials in progress at the time of writing. The mainstay of treatment still remains the reduction of cystine accumulation with oral cysteamine, as well as symptom control.
Conclusion
Over the recent decades, our understanding of cystinosis has significantly improved. With the success of renal transplantation and the development of cysteamine therapy, both mortality and morbidity have reduced. While timely treatments can limit complications of the disease, a definitive cure remains elusive. With prolonged survival, more complex late manifestations of the disease are emerging.
Corneal crystals are evident in all patients with cystinosis, and they are typically symptomatic in most by the age of 12 years. Photophobia and blepharospasm are significant, debilitating symptoms in many patients. Oral cysteamine has no effect on the accumulation of crystals within the cornea and, hence, topical cyteamine drops have been developed. Although topical treatment appears to reduce the corneal deposit accumulation, with evidence of improved symptoms the frequency of use and storage regime presents significant barriers to adherence.
Disclosure
The authors report no conflicts of interest in this work.
References
Gahl WA, Thoene JG, Schneider JA. Cystinosis. N Engl J Med. 2002; 347(2):111–121. | ||
Nesterova G, Gahl W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol. 2008;23(6):863–878. | ||
Abderhalden E. Familiare cystindiathese. Hoppe Seylers Z Physiol Chem. 1903;38:557–561. | ||
Tsilou E, Zhou M, Gahl W, Sieving PC, Chan CC. Ophthalmic manifestations and histopathology of infantile nephropathic cystinosis: report of a case and review of the literature. Surv Ophthalmol. 2007;52(1):97–105. | ||
Gahl WA, Schneider JA, Thoene JG, Chesney R. Course of nephropathic cystinosis after age 10 years. J Pediatr. 1986;109(4):605–608. | ||
Manz F, Gretz N. Progression of chronic renal failure in a historical group of patients with nephropathic cystinosis. European Collaborative Study on Cystinosis. Pediatr Nephrol. 1994;8(4):466–471. | ||
Cogan DG, Kuwabara T, Kinoshita J, Sheehan L, Merola L. Cystinosis in an adult. J Am Med Assoc. 1957;164(4):394–396. | ||
Anikster Y, Lucero C, Guo J, et al. Ocular nonnephropathic cystinosis: clinical, biochemical, and molecular correlations. Pediatr Res. 2000; 47(1):17–23. | ||
Theodoropoulos DS, Krasnewich D, Kaiser-Kupfer MI, Gahl WA. Classic nephropathic cystinosis as an adult disease. JAMA. 1993; 270(18): 2200–2204. | ||
Thoene JG, Oshima RG, Crawhall JC, Olson DL, Schneider JA. Cystinosis. Intracellular cystine depletion by aminothiols in vitro and in vivo. J Clin Invest. 1976;58(1):180–189. | ||
Brodin-Sartorius A, Tête MJ, Niaudet P, et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012;81(2):179–189. | ||
Pisoni RL, Thoene JG, Christensen HN. Detection and characterization of carrier-mediated cationic amino acid transport in lysosomes of normal and cystinotic human fibroblasts. Role in therapeutic cystine removal? J Biol Chem. 1985;260(8):4791–4798. | ||
Gahl WA, Thoene JG, Schneider JA. Cystinosis: a disorder of lysosomal membrane transport. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. Vol 3. New York, NY: McGraw-Hill; 2001:5085–5108. | ||
Jézégou A, Llinares E, Anne C, et al. Heptahelical protein PQLC2 is a lysosomal cationic amino acid exporter underlying the action of cysteamine in cystinosis therapy. Proc Natl Acad Sci U S A. 2012; 109(50):E3434–E3443. | ||
McDowell GA, Town MM, van’t Hoff W, Gahl WA. Clinical and molecular aspects of nephropathic cystinosis. J Mol Med (Berl). 1998; 76(5):295–302. | ||
Kalatzis V, Cherqui S, Antignac C, Gasnier B. Cystinosin, the protein defective in cystinosis, is a H(+)-driven lysosomal cystine transporter. EMBO J. 2001;20(21):5940–5949. | ||
Shotelersuk V, Larson D, Anikster Y, et al. CTNS mutations in an American-based population of cystinosis patients. Am J Hum Genet. 1998;63(5):1352–1362. | ||
Kleta R, Anikster Y, Lucero C, et al. CTNS mutations in African American patients with cystinosis. Mol Genet Metab. 2001;74(3):332–337. | ||
Kalatzis V, Cohen-Solal L, Cordier B, et al. Identification of 14 novel CTNS mutations and characterization of seven splice site mutations associated with cystinosis. Hum Mutat. 2002;20(6):439–446. | ||
Gahl WA, Bashan N, Tietze F, Schulman JD. Lysosomal cystine counter-transport in heterozygotes for cystinosis. Am J Hum Genet. 1984;36(2):277–282. | ||
Scriver CR. Human genetics: lessons from Quebec populations. Annu Rev Genomics Hum Genet. 2001;2:69–101. | ||
Attard M, Jean G, Forestier L, et al. Severity of phenotype in cystinosis varies with mutations in the CTNS gene: predicted effect on the model of cystinosin. Hum Mol Genet. 1999;8(13):2507–2514. | ||
Racusen LC, Wilson PD, Hartz PA, Fivush BA, Burrow CR. Renal proximal tubular epithelium from patients with nephropathic cystinosis: immortalized cell lines as in vitro model systems. Kidney Int. 1995; 48(2):536–543. | ||
Park M, Helip-Wooley A, Thoene J. Lysosomal cystine storage augments apoptosis in cultured human fibroblasts and renal tubular epithelial cells. J Am Soc Nephrol. 2002;13(12):2878–2887. | ||
Cherqui S, Sevin C, Hamard G, et al. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol Cell Biol. 2002;22(21):7622–7632. | ||
Coor C, Salmon RF, Quigley R, Marver D, Baum M. Role of adenosine triphosphate (ATP) and NaK ATPase in the inhibition of proximal tubule transport with intracellular cystine loading. J Clin Invest. 1991; 87(3):955–961. | ||
Levtchenko EN, Wilmer MJ, Janssen AJ, et al. Decreased intracellular ATP content and intact mitochondrial energy generating capacity in human cystinotic fibroblasts. Pediatr Res. 2006;59(2):287–292. | ||
Jackson JD, Smith FG, Litman NN, Yuile CL, Latta H. The Fanconi syndrome with cystinossis. Electron microscopy of renal biopsy specimens from five patients. Am J Med. 1962;33:893–910. | ||
Spear GS, Slusser RJ, Tousimis AJ, Taylor CG, Schulman JD. Cystinosis. An ultrastructural and electron-probe study of the kidney with unusual findings. Arch Pathol. 1971;91(3):206–221. | ||
Burki E. Ueber die Cystinkrankheit im Kleinkindesalter unter besonderer Berücksichtigung des Augenbefundes [About the Cystinkrankheit in infancy with special reference to eye findings]. Ophthalmologica. 1941;101:331–342 | ||
Scouras J, Faggioni R. Ocular manifestations of cystinosis. Ophthalmologica. 1969;159(1):24–30. | ||
Alsuhaibani AH, Khan AO, Wagoner MD. Confocal microscopy of the cornea in nephropathic cystinosis. Br J Ophthalmol. 2005;89(11): 1530–1531. | ||
Grupcheva CN, Ormonde SE, McGhee C. In vivo confocal microscopy of the cornea in nephropathic cystinosis. Arch Ophthalmol. 2002; 120(12):1742–1745. | ||
Okami T, Nakajima M, Higashino H, Aoki T. [Ocular manifestations in a case of infantile cystinosis]. Nihon Ganka Gakkai Zasshi. 1992;96(10): 1341–1346. Japanese. | ||
Fung AT, Fraser-Bell S, Ojaimi E, Sutton G. In vivo confocal microscopy and polarizing microscopy of the cornea in a patient with nephropathic cystinosis. Clin Experiment Ophthalmol. 2007;35(3):292–293. | ||
Gahl WA, Kuehl EM, Iwata F, Lindblad A, Kaiser-Kupfer MI. Corneal crystals in nephropathic cystinosis: natural history and treatment with cysteamine eyedrops. Mol Genet Metab. 2000;71(1–2):100–120. | ||
Gahl WA, Thoene JG, Schneider JA, O’Regan S, Kaiser-Kupfer MI, Kuwabara T. NIH conference. Cystinosis: progress in a prototypic disease. Ann Intern Med. 1988;109(7):557–569. | ||
Schneider JA, Katz B, Melles RB. Update on nephropathic cystinosis. Pediatr Nephrol. 1990;4(6):645–653. | ||
Elder MJ, Astin CL. Recurrent corneal erosion in cystinosis. J Pediatr Ophthalmol Strabismus. 1994;31(4):270–271. | ||
Kaiser-Kupfer MI, Caruso RC, Minkler DS, Gahl WA. Long-term ocular manifestations in nephropathic cystinosis. Arch Ophthalmol. 1986;104(5):706–711. | ||
Katz B, Melles RB, Schneider JA. Recurrent crystal deposition after keratoplasty in nephropathic cystinosis. Am J Ophthalmol. 1987; 104(2):190–191. | ||
Katz B, Melles RB, Schneider JA, Rao NA. Corneal thickness in nephropathic cystinosis. Br J Ophthalmol. 1989;73(8):665–668. | ||
Mungan N, Nischal KK, Héon E, MacKeen L, Balfe JW, Levin AV. Ultrasound biomicroscopy of the eye in cystinosis. Arch Ophthalmol. 2000;118(10):1329–1333. | ||
Broyer M, Guillot M, Gubler MC, Habib R. Infantile cystinosis: a reappraisal of early and late symptoms. Adv Nephrol Necker Hosp. 1981; 10:137–166. | ||
François J. Metabolic tapetoretinal degenerations. Surv Ophthalmol. 1982;26(6):293–333. | ||
Wong VG, Lietman PS, Seegmiller JE. Alterations of pigment epithelium in cystinosis. Arch Ophthalmol. 1967;77(3):361–369. | ||
Richard G, Kroll P. [Retinal changes in cystinosis]. Ophthalmologica. 1983;186(4):211–218. German. | ||
Wong VG. Ocular manifestations in cystinosis. Birth Defects Orig Artic Ser. 1976;12(3):181–186. | ||
Dufier JL, Dhermy P, Gubler MC, Gagnadoux MF, Broyer M. Ocular changes in long-term evolution of infantile cystinosis. Ophthalmic Paediatr Genet. 1987;8(2):131–137. | ||
Tsilou ET, Rubin BI, Reed GF, Iwata F, Gahl W, Kaiser-Kupfer MI. Age-related prevalence of anterior segment complications in patients with infantile nephropathic cystinosis. Cornea. 2002;21(2):173–176. | ||
Yamamoto GK, Schulman JD, Schneider JA, Wong VG. Long-term ocular changes in cystinosis: observations in renal transplant recipients. J Pediatr Ophthalmol Strabismus. 1979;16(1):21–25. | ||
Wan WL, Minckler DS, Rao NA. Pupillary-block glaucoma associated with childhood cystinosis. Am J Ophthalmol. 1986;101(6):700–705. | ||
Tsilou ET, Rubin BI, Reed G, et al. Nephropathic cystinosis: posterior segment manifestations and effects of cysteamine therapy. Ophthalmology. 2006;113(6):1002–1009. | ||
Kimonis VE, Troendle J, Rose SR, Yang ML, Markello TC, Gahl WA. Effects of early cysteamine therapy on thyroid function and growth in nephropathic cystinosis. J Clin Endocrinol Metab. 1995;80(11): 3257–3261. | ||
Cherqui S. Cysteamine therapy: a treatment for cystinosis, not a cure. Kidney Int. 2012;81(2):127–129. | ||
Kaiser-Kupfer MI, Gazzo MA, Datiles MB, Caruso RC, Kuehl EM, Gahl WA. A randomized placebo-controlled trial of cysteamine eye drops in nephropathic cystinosis. Arch Ophthalmol. 1990;108(5):689–693. | ||
Jones NP, Postlethwaite RJ, Noble JL. Clearance of corneal crystals in nephropathic cystinosis by topical cysteamine 0.5%. Br J Ophthalmol. 1991;75(5):311–312. | ||
Bradbury JA, Danjoux JP, Voller J, Spencer M, Brocklebank T. A randomised placebo-controlled trial of topical cysteamine therapy in patients with nephropathic cystinosis. Eye (Lond). 1991;5(Pt 6): 755–760. | ||
MacDonald IM, Noel LP, Mintsioulis G, Clarke WN. The effect of topical cysteamine drops on reducing crystal formation within the cornea of patients affected by nephropathic cystinosis. J Pediatr Ophthalmol Strabismus. 1990;27(5):272–274. | ||
Iwata F, Kuehl EM, Reed GF, McCain LM, Gahl WA, Kaiser-Kupfer MI. A randomized clinical trial of topical cysteamine disulfide (cystamine) versus free thiol (cysteamine) in the treatment of corneal cystine crystals in cystinosis. Mol Genet Metab. 1998;64(4):237–242. | ||
Tsilou ET, Thompson D, Lindblad AS, et al. A multicentre randomised double masked clinical trial of a new formulation of topical cysteamine for the treatment of corneal cystine crystals in cystinosis. Br J Ophthalmol. 2003;87(1):28–31. | ||
Labbé A, Baudouin C, Deschênes G, et al. A new gel formulation of topical cysteamine for the treatment of corneal cystine crystals in cystinosis: the Cystadrops OCT-1 study. Mol Genet Metab. 2014;111(3): 314–320. | ||
Hsu KH, Fentzke RC, Chauhan A. Feasibility of corneal drug delivery of cysteamine using vitamin E modified silicone hydrogel contact lenses. Eur J Pharm Biopharm. 2013;85(3 Pt A):531–540. | ||
Iglesias DM, El-Kares R, Taranta A, et al. Stem cell microvesicles transfer cystinosin to human cystinotic cells and reduce cystine accumulation in vitro. PLoS One. 2012;7(8):e42840. | ||
Town M, Jean G, Cherqui S, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18(4):319–324. | ||
Nevo N, Chol M, Bailleux A, et al. Renal phenotype of the cystinosis mouse model is dependent upon genetic background. Nephrol Dial Transplant. 2010;25(4):1059–1066. | ||
Syres K, Harrison F, Tadlock M, et al. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood. 2009;114(12):2542–2552. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.