")
Back to Journals » International Journal of General Medicine » Volume 9
Treatment of bleeding in acquired hemophilia A with the proper administration of recombinant activated factor VII: single-center study of 7 cases
Authors Saito M , Kanaya M, Izumiyama K, Mori A, Irie T, Tanaka M, Morioka M, Ieko M
Received 29 July 2016
Accepted for publication 27 September 2016
Published 3 November 2016 Volume 2016:9 Pages 393—399
DOI https://doi.org/10.2147/IJGM.S118422
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Makoto Saito,1 Minoru Kanaya,1 Koh Izumiyama,1 Akio Mori,1 Tatsuro Irie,1 Masanori Tanaka,1 Masanobu Morioka,1 Masahiro Ieko2
1Department of Internal Medicine and Hematology, Aiiku Hospital, Japan; 2Department of Internal Medicine, Health Sciences University of Hokkaido, Japan
Abstract: Recombinant activated factor VII (rFVIIa) is the bypassing agent used in the first-line hemostatic therapy for acquired hemophilia A (AHA); however, the occurrence of thrombotic complications in rFVIIa-treated AHA patients was recently reported to be 2.9–6.5%. Therefore, the investigation of the proper administration of rFVIIa for AHA is needed. In the present study, we retrospectively investigated the clinical features of AHA with regards to the use of rFVIIa (presence or absence of use and total amount) in 7 AHA patients encountered in our department for 7 years between January 2008 and December 2014. Ages were 63–89 years old (median: 79 years old), and there were 5 male and 2 female patients. The coexistence of cardiovascular risk factors and arteriosclerotic diseases, such as hypertension, diabetes mellitus, and cerebral infarction were present in 6 patients. Anemia progressed to less than 7 g/dL of hemoglobin and required red blood cell transfusion in 5 patients, showing “severe” hemorrhage. Factor VIII inhibitors were removed by immunological treatments in 6 patients. As a hemostatic therapy, rFVIIa was used in 4 patients. rFVIIa was not administered or was administered at a very low dose (20 mg) to 3 and 1 patient, respectively, and bleeding stopped as inhibitor titers decreased and disappeared in these patients. Inhibitors did not disappear in 1 patient and the control of hemostasis became poor and was accompanied by intestinal hemorrhage. Although a large amount of rFVIIa (265 mg in total) was administered, the patient bled to death. Therefore, bleeding may be stopped without the administration of rFVIIa in some AHA cases, while the dose of rFVIIa is not necessarily related to hemostatic effects in other cases. Since the main aim of AHA treatments is the removal of inhibitors, caution is needed to ensure that more than the necessary amount of rFVIIa is not administered.
Keywords: acquired hemophilia A, recombinant activated factor VII, thrombotic complications
Introduction
Acquired hemophilia A (AHA) is a hemorrhagic disease caused by reduced factor VIII activity due to the appearance of autoantibodies (inhibitors) against coagulation factor VIII.1,2 AHA is very rare, with an annual incidence of 1.5 in one million individuals; elderly individuals aged 60 years or older account for more than 80% of patients.2
The treatment of AHA is divided into hemostatic therapy for hemorrhage and immunological treatments aimed at eradicating inhibitors.3 Bypassing agents, i.e. recombinant activated factor VII (rFVIIa) and activated prothrombin complex concentrate (aPCC) are considered the first-line approach for the treatment of bleeding episodes.4–6 Especially, rFVIIa is the most frequently used,7,8 suggesting its high efficacy (more than 80%).9,10 It is administered according to the administration dose and method for congenital hemophilia A patients possessing inhibitors (bolus administrations of 90 μg/kg every 2–3 hours until sufficient hemostasis is achieved);5,6,10–12 it has not yet been established whether this administration method is applicable without modifications for hemostasis in AHA. Severe thrombotic complications have been recently reported in 2.9–6.5% of rFVIIa-treated AHA patients.7,9 Moreover, rFVIIa is very expensive and places pressure on health economics.
Therefore, we have carefully administered rFVIIa in hemostatic therapy for AHA patients with concerted efforts to avoid its excess administration. In the present study, we retrospectively analyzed the clinical features of 7 AHA cases treated at our institution and investigated the proper administration of rFVIIa.
Patients and methods
Written informed consent was obtained from all patients or their next of kin to publish this paper and any accompanying images in accordance with the Declaration of Helsinki. The study was approved by the Aiiku Hospital Ethics Committee. Subjects comprised 7 AHA patients treated at our department between January 2008 and December 2014 (7 years). Their clinical features were retrospectively analyzed. All 7 patients consulted with our department for hemorrhage of an unknown cause with accompanying anemia and the prolongation of activated partial thromboplastin time (APTT).
Age, sex, underlying diseases, hemorrhagic sites, the APTT value (seconds), hemoglobin (Hb) (g/dL) at the time of the most advanced anemia after the onset and its degree, and the presence or absence and amount of red blood cell (RBC) transfusions on admission were reviewed. We defined hemorrhage as the progression of anemia to less than 7.0 g/dL of Hb requiring RBC transfusion, or life-threatening organ hemorrhage as “severe”, and other hemorrhaging as “non-severe”. Factor VIII activity levels (%), inhibitor titers (Bethesda units; BU/mL), drugs for immunological treatments, and changes in inhibitors as well as the relationships between the factor VIII activity levels/inhibitor titers and severity of hemorrhage were investigated. The use of rFVIIa for hemostatic therapy (presence or absence of its use and total amount) was also investigated with regards to its association with the severity of hemorrhage and outcomes of patients.
(We have never used aPCC as a bypassing agent nor replacement therapy, such as factor VIII and 1-desamino-8-D-arginine-vasopressin [DDAVP].)
Results
Ages at the diagnosis of AHA were 63–89 years old (median: 79 years old); all patients were elderly (Table 1). There were 5 male and 2 female patients. As underlying diseases, 6 patients excluding 1 patient (Case 4), had concomitant cardiovascular risk factors and arteriosclerotic diseases, such as hypertension, diabetes mellitus, and cerebral infarction, and 4 patients had dementia. Regarding the hemorrhagic site, subcutaneous hemorrhage occurred in all 7 patients, and intramuscular, intestinal, and post-dental treatment gingival hemorrhaging (4, 2, and 2 patients, respectively) were also observed (Figure 1). APTT was 81.4–100.1 seconds (median: 93.0 seconds), which was markedly prolonged.
 | Table 1 Clinical features of our 7 AHA patients Notes: Cases 1–5: severe hemorrhage, Cases 6 and 7: non-severe hemorrhage. #Later, the inhibitor appeared again, but it was removed finally. Abbreviations: AAA, abdominal aortic aneurysm; AHA, acquired hemophilia A; DM, diabetes mellitus; HT, hypertension; CKD, chronic kidney disease; HCC, hepatocellular carcinoma; RBC, red blood cell; PSL, prednisolone; CPA, cyclophosphamide; RTX, rituximab; VCR, vincristine; rFVIIa, recombinant activated factor VII; Hb, hemoglobin. |
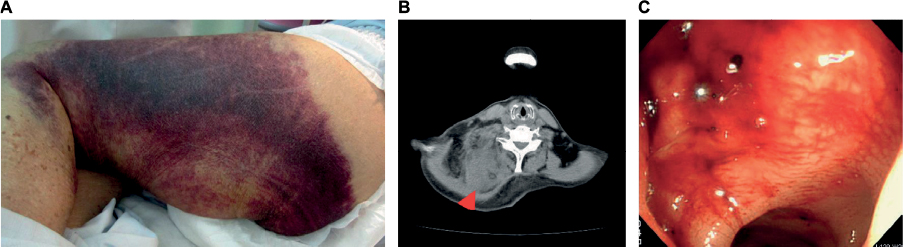 | Figure 1 Hemorrhagic sites. Notes: (A) Subcutaneous, (B) intramuscular (red arrow head, trapezius muscle), (C) intestinal (sigmoid colon) hemorrhage. |
Anemia progressed to 4.3–6.9 g/dL of Hb (4.0–7.0g/dL decrease for 5 days-2 weeks) and necessitated RBC transfusions (2–12 units) in 5 patients (Cases 1–5), showing “severe” hemorrhage. In 2 patients (Cases 6 and 7), Hb was maintained at at least 8.0 g/dL or higher (1.8–2.6 g/dL decrease for 9 days-2 weeks) and no RBC transfusion was necessary, being “non-severe” cases.
The factor VIII activity level was <1% in 3 patients, and the factor VIII inhibitor titer was >50 BU/mL in 4 patients. Regarding immunological treatments, prednisolone (PSL), cyclophosphamide (CPA), and rituximab (RTX) were administered to 6, 3, and 2 patients, respectively, and inhibitors disappeared in 6 patients. No relationship was noted between factor VIII activity levels/inhibitor titers and the severity of hemorrhage (Figure 2).
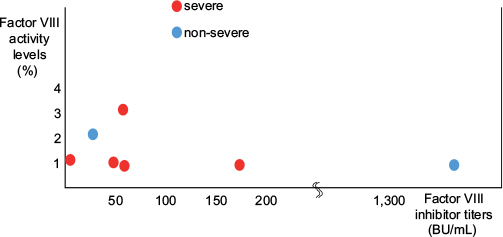 | Figure 2 Relationship between factor VIII activity levels/inhibitor titers and the severity of hemorrhage. |
Though rFVIIa was not administered to the 2 “non-severe” cases, hemostasis was possible and the patients have been alive for more than 2 years after their treatment. In Case 7, the factor VIII inhibitor titer was markedly high (1,364.9 BU/mL), for which PSL, CPA, and RTX were administered, and the treatment for the removal of inhibitors was prolonged (1 year and 5 months). Subcutaneous, intramuscular (Figure 1B), and nasal hemorrhage not requiring RBC transfusion were noted, but hemostasis was achieved without the administration of rFVIIa. In 2 out of the 5 “severe” cases, inhibitors rapidly decreased and hemostasis was achieved without the administration of rFVIIa (Case 5) (Figure 3A) or by the administration of a very small amount of rFVIIa (a total of 20.0 mg was administered for 24 hours divided into 5 times by bolus injection of 100 μg/kg at 0, 3, 6, 12, and 24 h) (Case 4) (Figure 3B). In Case 5, RTX was used as a single agent for immunological treatment. However, these patients died within a short time due to the aggravation of an underlying disease (hepatocellular carcinoma) and concomitant infection (pneumonia), respectively. Of the 3 patients (Cases 1–3) treated with a sufficient amount of rFVIIa, inhibitors did not disappear in 1 (Case 3), in whom hemostasis was almost achieved including intestinal hemorrhage during the administration of rFVIIa, but hemorrhagic signs recurred during drug withdrawal and hemostasis control became poor (Figure 1A and C), and the patient bled to death despite a large amount of rFVIIa (265 mg in total) (Figure 3C). In Case 1, the patient suddenly died due to the rupture of an abdominal aortic aneurysm nearly 1 year after the completion of the rFVIIa treatment, and rFVIIa was not considered to be directly involved in the death of this patient because the rupture occurred a long time after the treatment. rFVIIa was administered to 4 out of 7 AHA patients, and no thrombotic complications occurred within a short time in any of these patients.
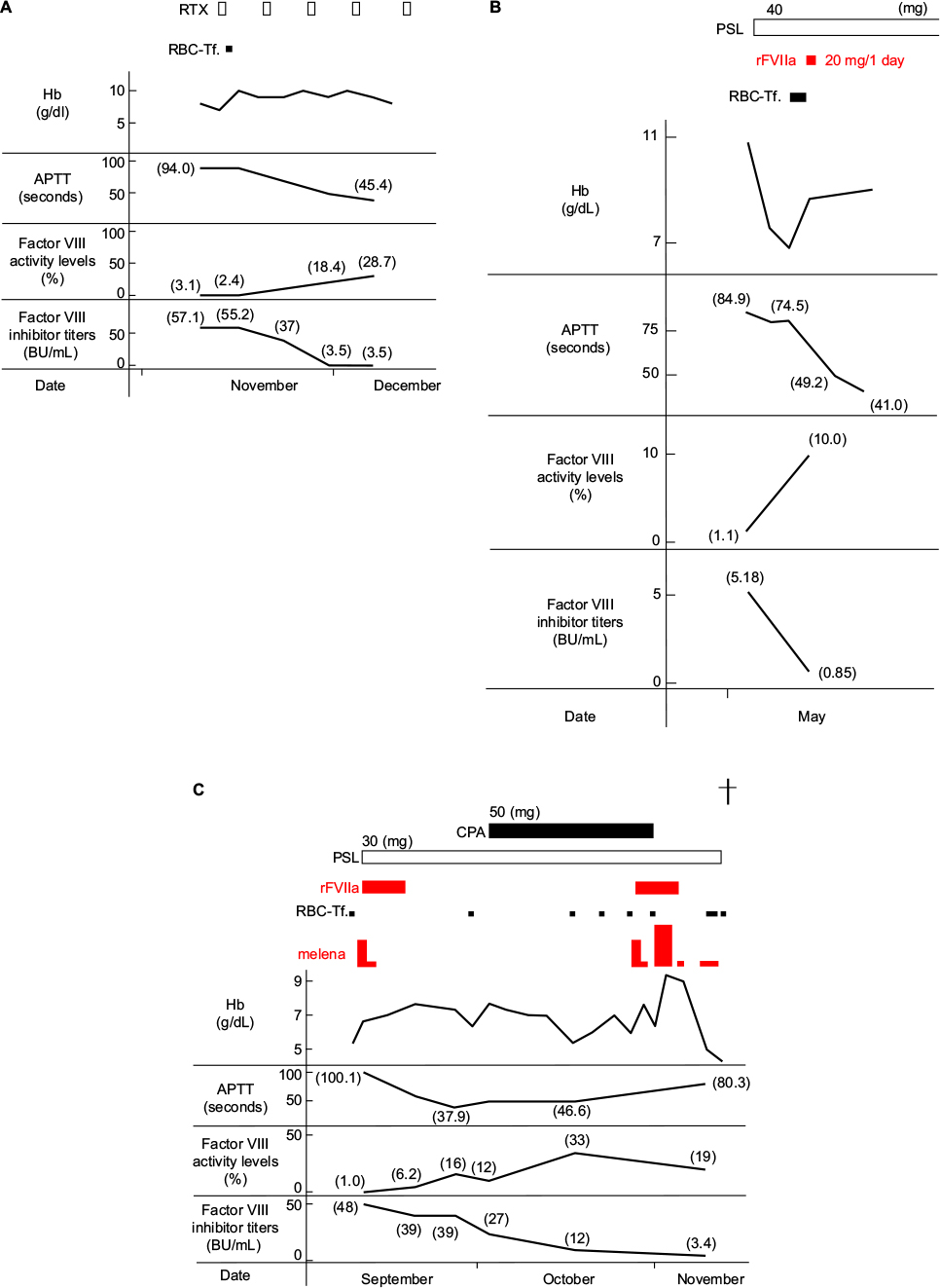 | Figure 3 Clinical course. Notes: (A) Case 5, five courses of RTX once weekly as immunological treatments reduced the factor VIII inhibitor to 3.5 BU/ml, and hemostasis was achieved. (B) Case 4, inhibitors rapidly decreased with the administration of PSL, and hemostasis was achieved by the use of a very small amount of rFVIIa (a total of 20.0 mg was administered for 24 hours). (C) Case 3, inhibitors did not disappear with the administration of PSL, the patient bled to death despite a large amount of rFVIIa (265 mg in total), because hemostasis control including intestinal hemorrhage became poor during rFVIIa withdrawal (the black cross symbol indicates death). Abbreviations: RBC-Tf, red blood cell transfusion; RTX, rituximab; APTT, activated partial thromboplastin time; PSL, prednisolone; rFVIIa, recombinant activated factor VII; CPA, cyclophosphamide; Hb, hemoglobin. |
Discussion
As shown in the present study, the development of AHA is most frequent in a population of elderly patients at high risk of cardiovascular and arteriosclerotic diseases.8,13 Accordingly, thrombotic complications have been reported to develop in AHA patients (2.9–6.5% of rFVIIa-treated cases)7,9 after the high dose and long-term administration of rFVIIa14 or when concomitantly administered with aPCC for a short interval.15 The suitable dose and method of administration of FVIIa for AHA have not been sufficiently investigated and it currently remains unclear whether it may be administered similarly to that for younger, congenital hemophilia patients with inhibitors. In the largest available collection of treatment records, the European Acquired Haemophilia (EACH2) registry,7 hemostatic therapy was not required in 144 of 482 AHA patients (30%), particularly for “non-severe” cases (severity criteria were slightly less strict than our criteria: an Hb level less than 8 g/dL or a decrease in Hb by more than 2 g/dL was regarded as “severe”, and Case 6 in our study is classified as “severe”), and the median total amount of rFVIIa used per patient was 84 mg (24–216 mg). In our Case 1, the first AHA patient treated by our institution, rFVIIa was administered at a high dose (177.8 mg) approximately 1 year before the rupture of an abdominal aneurysm. After this case, we reconsidered the dose and method of use of rFVIIa for AHA, and have made efforts to not excessively administer rFVIIa. Therefore, this research was not just a simple retrospective study, but also has the value of being a prospective pilot study to evaluate the outcomes of suppressed administration of rFVIIa within a possible range for the treatment of AHA.
The key point to successfully treating AHA is to remove inhibitors as rapidly as possible using immunological therapeutic drugs because there is a risk of severe, fatal hemorrhage during the period in which inhibitors are present.5,6,16 When serious hemorrhagic symptoms develop before the completion of inhibitor removal, hemostatic therapy is necessary.7 In our case series, 2 patients (Cases 6 and 7) in the “non-severe” group did not develop serious life-threatening hemorrhagic symptoms and the administration of rFVIIa was unnecessary. Even with “severe” hemorrhage, when inhibitors may be rapidly decreased and removed, hemostasis may be achieved without the administration of rFVIIa (Case 5) or by its administration at a very low dose (20 mg) (Case 4). However, the risk of bleeding to death is high (9.1–12.5%) in AHA.2,17 In 1 patient (Case 3) with hemorrhage from the colon, hemorrhagic tendency accompanied by melena was protracted and the patient bled to death despite a large amount of rFVIIa (265 mg in total) being administered. We should have changed the administration of rFVIIa for aPCC during the clinical course. This patient had repeated serious hemorrhaging even though the factor VIII inhibitor titer was reduced to 3.4 BU/mL. In contrast, in Case 7, the factor VIII inhibitor
titer was abnormally high (1,364.9 BU/mL), whereas the severity of hemorrhage was not recognized. Therefore, neither the factor VIII activity level, nor the titer of the factor VIII inhibitor was associated with severity of hemorrhage.4 A biomarker with which the degree of hemorrhagic risk may be objectively evaluated in each AHA patient needs to be developed.
When and for how long the AHA patients should be administered rFVIIa as hemostatic therapy have not been fully established. Since no optimum biomarker to objectively represent the hemorrhagic risk or monitoring test for bypassing agent is available, judgments are subjectively made by each attending physician in many cases. rFVIIa has to be rapidly administered for direct life-threatening serious hemorrhage, such as brain, intestinal, urogenital bleeding or hematoma causing airway obstruction. In contrast, for “non-severe” AHA cases with no organic-bleeding or with chronic hemorrhagic symptoms without the progression of anemia, we propose that active rFVIIa administration may be avoided because the occurrence of subsequent fatal hemorrhage is less likely. Even for “severe” hemorrhage not directly life-threatening in AHA cases, the necessity of the administration of rFVIIa is investigated by reasoning regarding the time of the disappearance of inhibitors from the velocity of reductions in inhibitors, and comparing it with the current severity of hemorrhage, because hemostasis is expected when inhibitors are removed. After the initiation of hemostatic therapy with rFVIIa, when symptoms accompanying hemorrhage, such as pain and swelling, remit or the progression of anemia stops, the administration of rFVIIa more than necessary can be discontinued.
In conclusion, the main aim of the treatments of AHA is the removal of inhibitors; hemostasis may be achieved without the administration of rFVIIa in some cases, and the dose of rFVIIa is not necessarily related to hemostatic effects in other cases. When rFVIIa has to be administered, its effective use at a lower dose needs to be considered within a possible range because its excess administration may cause thrombotic complications. We investigated the proper administration of rFVIIa based on a retrospective single-center study involving 7 patients. Since the number of patients was limited, a prospective study involving a large number of AHA patients is required.
Disclosure
The authors report no conflicts of interest in this work.
References
Delgado J, Jimenez-Yuste V, Hernandez-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol. 2003;121(1):21–35. | ||
Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood. 2007;109(5):1870–1877. | ||
Kessler CM, Nemes L. Acquired inhibitors to factor VIII. In: Rodriguez EC, Lee CA, editors. Inhibitors in patients with haemophilia. Blackwell Publishing; 2002:98–112. | ||
Collins PW, Chalmers E, Hart D, et al. Diagnosis and management of acquired coagulation inhibitors: a guideline from UKHCDO. Br J Haematol. 2013;162(6):758–773. | ||
Huth-Kühne A, Baudo F, Collins P, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica. 2009;94(4):566–575. | ||
Collins P, Baudo F, Huth-Kühne A, et al. Consensus recommendations for the diagnosis and treatment of acquired hemophilia A. BMC Res Notes. 2010;3:161. | ||
Baudo F, Collins P, Huth-Kühne A, et al. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) Registry. Blood. 2012;120(1):39–46. | ||
Di Capua M, Coppola A, Nardo A, et al. Management of bleeding in acquired haemophilia A with recombinant activated factor VII: does one size fit all? A report of four cases. Blood Transfus. 2015;13(2): | ||
Sumner MJ, Geldziler BD, Pedersen M, Seremetis S. Treatment of acquired haemophilia with recombinant activated FVII: a critical appraisal. Haemophilia. 2007;13(5):451–461. | ||
Hay CR, Negrier C, Ludlam CA. The treatment of bleeding in acquired haemophilia with recombinant factor VIIa: a multicentre study. Thromb Haemost. 1997;78(6):1463–1467. | ||
Bech RM. Recombinant factor VIIa in joint and muscle bleeding episodes. Haemostasis. 1996;26 Suppl 1:135–138. | ||
Scharrer I. Recombinant factor VIIa for patients with inhibitors to factor VIII or IX or factor VII deficiency. Haemophilia. 1999;5(4): | ||
Zanon E, Milan M, Brandolin B, et al. High dose of human plasma-derived FVIII-VWF as first-line therapy in patients affected by acquired haemophilia A and concomitant cardiovascular disease: four case reports and a literature review. Haemophilia. 2013;19(1):e50–53. | ||
Abshire T, Kenet G. Safety update on the use of recombinant factor VIIa and the treatment of congenital and acquired deficiency of factor VIII or IX with inhibitors. Haemophilia. 2008;14(5):898–902. | ||
Gringeri A, Mannucci PM; Italian Association of Haemophilia Centres. Italian guidelines for the diagnosis and treatment of patients with haemophilia and inhibitors. Haemophilia. 2005;11(6):611–619. | ||
Collins P, Baudo F, Knoebl P, et al. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood. 2012;120(1):47–55. | ||
Tanaka I, Amano K, Taki M, et al. [A 3-year consecutive survey on current status of acquired inhibitors against coagulation factors in Japan: analysis of prognostic factors]. Nihon kessen shiketsu gakkai shi. 2008;19(1):140–152 Japanese [with English abstract]. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.