")
Back to Archived Journals » Oncolytic Virotherapy » Volume 9
Treatment of an Alveolar Rhabdomyosarcoma Allograft with Recombinant Myxoma Virus and Oclacitinib
Authors Ashton LV , Graham B , Afzali MF, Gustafson D, MacNeill AL
Received 6 March 2020
Accepted for publication 5 May 2020
Published 26 May 2020 Volume 2020:9 Pages 17—29
DOI https://doi.org/10.2147/OV.S252727
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tommy Alain
Laura V Ashton,1 Barbara Graham,1 Maryam F Afzali,1 Daniel Gustafson,2 Amy L MacNeill1
1Department of Microbiology, Immunology, and Pathology, College of Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO, USA; 2Departiment of Clinical Sciences, College of Veterinary Medicine and Biomedical Sciences, Colorado State University, Fort Collins, CO, USA
Correspondence: Amy L MacNeill Email [email protected]
Purpose: Rhabdomyosarcomas (RMS) are difficult tumors to treat with conventional therapies. Publications indicate that oncolytic virotherapy (OV) could benefit cancer patients with tumors that are refractory to conventional treatments. It is believed that the efficacy of OV can be enhanced when used in combination with other treatments. This study evaluated the response of mice with aggressive alveolar RMS (ARMS) allografts to treatment with an OV [recombinant myxoma virus (MYXVΔserp2)] in combination with a Janus kinase (JAK) inhibitor (oclacitinib). Oclacitinib is known to inhibit JAK1 and JAK2 cell signaling pathways, which should limit the antiviral Type I interferon response. However, oclacitinib does not inhibit immune pathways that promote antigen presentation, which help stimulate an anti-cancer immune response.
Materials and Methods: To determine if MYXVΔserp2 and oclacitinib could improve outcomes in animals with ARMS, nude mice were inoculated subcutaneously with murine ARMS cells to establish tumors. Immune responses, tumor growth, and clinical signs in mice treated with combination therapy were compared to mice given placebo therapy and mice treated with OV alone.
Results: Combination therapy was safe; no viral DNA was detected in off-target organs, only within tumors. As predicted, viral DNA was detected in tumors of mice given oclacitinib and MYXVΔserp2 for a longer time period than mice treated with OV alone. Although tumor growth rates and median survival times were not significantly different between groups, clinical signs were less severe in mice treated with OV.
Conclusion: Our data indicate that MYXVΔserp2 treatment benefits mice with ARMS by reducing clinical signs of disease and improving quality of life.
Keywords: oncolytic virus, poxvirus, Janus kinase inhibitor, sarcoma
Introduction
Soft tissue sarcomas (STS) are a group of neoplasms that are locally invasive, difficult to completely excise surgically, and have variable response rates to adjunctive treatment with radiation and chemotherapy. Over half of the STS in people under the age of 20 are classified as rhabdomyosarcomas (RMS).1 RMS originate from pluripotent mesenchymal cells within skeletal muscle.2 The annual incidence of RMS is 4.5 cases per million people under 20 years of age, making RMS one of the most common tumors in children.3 There are two main subtypes of RMS, embryonic RMS and alveolar RMS (ARMS), which have histological and genetic differences.4 The 5-year survival rates for children with embryonic RMS and ARMS are approximately 73% and 48%, respectively.3 Given the high rate of disease recurrence, there is a demand for strategies to improve survival rates of patients with RMS without compromising quality of life. Recent advances in the field of oncolytic viral (OV) therapeutics have showcased the potential effectiveness and safety of this treatment modality. For example, an oncolytic herpes virus was recently approved for use in the United States to treat melanoma.5 Studies using attenuated herpes simplex virus and poxvirus therapies have demonstrated oncolytic effects of OVs in sarcoma cells lines and sarcoma xenograft models.6–8 Oncolytic virotherapy has fewer side effects than radiation or chemotherapy and may prove to be more effective at reducing recurrence of RMS.
Myxoma virus (MYXV) is an OV that replicates in cultured cancer cells from several animal species,9–12 but does not cause disease in humans or other vertebrates (with the exception of rabbits).13–18 MYXV is a leporipoxvirus and, like other poxviruses, it does not require a specific cellular receptor to infect cells. When poxviruses enter cells, gene transcription begins immediately and all replication steps occur in the cytoplasm. Mature poxviruses virions are released from infected cells before cell lysis occurs.19 These virions effectively enter neighboring cells, spreading the virus infection in susceptible cells throughout the tumor microenvironment.19 Importantly, poxviruses elicit strong cell-mediated and humoral immune responses that clear the virus from the body and prevent latent or recurrent infections from occurring.20,21
The efficacy of MYXV treatment has been demonstrated in several murine cancer xenograft and allograft models, but complete cures are rarely achieved with OV therapy alone.6,22-27 This may be due, in part, to limited viral replication within treated tumors. In mice, MYXV replicates within a tumor for approximately 4 days after virus injection.23 When MYXV is injected repeatedly, outcome is improved.6 The ability of MYXV to replicate in cancer cells has been shown to be due in part to a lack of interferon (IFN) response in those cells.11,28 In an attempt to improve the efficacy of MYXV oncolytic therapy, we used a recombinant MYXV with deletion of a viral anti-apoptotic protein, serp2 (MYXV∆serp2). Deletion of serp2 from MYXV markedly attenuates pathogenesis in rabbits29 and enhances the oncolytic effects cancer cells.9,10 Additionally, we evaluated if MYXV∆serp2 oncolytic effects could be improved, without the need of multiple injections, by strategically managing the antiviral IFN immune response in the tumor microenvironment by administering oclacitinib in combination with OV therapy.
Oclacitinib (Zoetis, Apoquel, Parsippany-Troy Hills, NJ, USA) is a Janus kinase (JAK) inhibitor that has shown safety and efficacy in the treatment of canine allergic disease without inhibiting response to vaccination.30 It effectively inhibits JAK1 and JAK2 activation, but has relatively weak activity against JAK3 and tyrosine kinase 2 (Tyk2).31 Interleukin-6 (IL-6) and IL-13 (which signal through JAK1/2 and Tyk2) are inhibited by oclacitinib therapy in dogs.31 Oclacitinib also is predicted to inhibit Type I IFN signaling which occurs through JAK1 and Tyk2 pathways. Cytokines of the innate immune response that do not signal through JAK1 [including IL-12, IL-23 and granulocyte-monocyte colony-stimulating factor (GM-CSF)] are not inhibited during oclacitinib administration.31 We theorized that short-term inhibition of Type I IFN signaling by oclacitinib could allow viral replication to occur in the tumor for a longer period of time, enhancing oncolysis, while cytokines involved in innate immune system continued to stimulate the antitumoral immune response.
Materials and Methods
Cells
Murine ARMS (U21089) cells were a generous gift from Dr. Charles Keller whose laboratory had established the primary cell culture from rhabdomyosarcomas that arose in conditional knockout mice with activation of an oncogene (Pax3:Fkhr) and mutation of a tumor suppressor gene.32 Rabbit-kidney epithelial (RK-13) cells were obtained from the American Type Culture Collection (ATCC CCL-37). Cell growth media was comprised of Minimal Essential Medium with Earle’s salts and 2 mM L-glutamine supplemented with 2 mM L-glutamine, 50 U/mL penicillin, and 50 µg/mL streptomycin (Hyclone, Logan, UT, USA); 0.1 mM nonessential amino acids and 1 mM sodium pyruvate (Corning, Corning, NY, USA); and 10% fetal bovine serum (FBS; VWR Life Science Seradigm, Radnor, PA, USA). Cells were maintained in a water-jacketed incubator at 5% CO2 and 37°C.
Viruses
Wild-type MYXV (Lausanne strain) was originally acquired from Dr. Grant McFadden. Construction and characterization of recombinant MYXV expressing tandem dimer tomato red,33 and MYXVΔserp234 have been previously described. MYXVΔserp2 was amplified in RK-13 cells, sucrose pad-purified, and re-suspended in sterile phosphate buffered saline (PBS) prior to injection into mice bearing ARMS. Virus titers [plaque/foci-forming units (pfu)/mL] of MYXV and MYXVΔserp2 were determined by incubating RK-13 cells with virus inoculum for 30 minutes 5% CO2 and 37°C then adding a solid overlay of 1-part 2× growth media and incubating for 4 days at 5% CO2 and 37°C. Virus titers, the number of viral foci that formed in RK-13 cells per mL of virus added, were calculated to determine pfu/mL of virus.
Growth Rate of MYXV in U21089 Cells
The growth rate of MYXV and MYXVΔserp2 in U21089 cells was evaluated as follows. Growth media (detailed above) was removed from wells of U21089 cells when they were 80% confluent (1.8 ×106 cells/well). Cells were inoculated with MYXV or MYXVΔserp2 at a multiplicity of infection (moi) of 0.01 pfu/cell in media lacking FBS. Cells were incubated with virus for 1 h at 5% CO2 and 37°C. Viral inoculum was removed, cells were rinsed with PBS, and growth media was added to wells. Cells were scraped into growth media at designated time-points post-inoculation, centrifuged at 400 ×g for 15 min, washed in PBS, re-suspended in media lacking FBS, frozen and thawed 3 times, and sonicated. Viral lysates were serially diluted in media lacking FBS and incubated on RK-13 cells for 30 min. A solid overlay of 1-part 2× growth media and 1 part 1% agarose was placed on the infected cells. Viral foci were counted 4 days later. Pfu/per U21089 cell was calculated and plotted versus time.
Cytolysis by MYXV in U21089 Cells
Confluent U21089 cells were incubated with MYXV or MYXVΔserp2 (moi = 1) or mock-infected with media without FBS as described above. Cytopathic effects of virus inoculation, including clustering of cells, disruption of nuclear shape and size, and release of cells from the monolayer into the media, were observed microscopically at 24, 48, and 72 h post-inoculation (hpi). Cell-titer blue reagent (Promega, Madison, WI, USA) was added to the cell culture media to detect viable cells at 24, 48, and 72 hpi. A Synergy H1 microplate reader (BioTek, Winooski, VT, USA) was used to detect differences in the amount of cell-titer blue reagent that was metabolized by the cells (fluorescence excitation = 560 nm; emission = 590 nm).
Mouse Studies
All mouse studies were approved by the Institutional Animal Care and Use Committee at Colorado State University (protocol #17-7709A). American Association for Laboratory Animal Science guidelines were used for the treatment and housing of the mice. Veterinary Medical Association guidelines for humane euthanasia were followed. Murine ARMS allografts were established in six- to eleven-week-old, B6.Cg-Foxn1nu/J mice by injecting 3 ×106 U21089 ARMS cells subcutaneously (SQ) in the lateral aspect of the thigh. Seven days after tumor cells were injected, either placebo (30 µL 0.5% methylcellulose/0.25% Tween 20 solution) or 45 mg/kg oclacitinib diluted in 30 µL 0.5% methylcellulose/0.25% Tween 20 solution was administered orally (PO) twice daily for 14 days. The oclacitinib dose was chosen based upon its published clinical effectiveness in mice.35,36 Intratumoral (IT) injection of PBS or 106 pfu of sucrose-purified MYXV∆serp2 in 100 µL PBS was administered 16 days after tumor cells were injected. A second dose of PBS or MYXV∆serp2 was injected IT two weeks after the first dose, if the mouse survived to that time point. A timeline for injections and oral treatments is diagramed in Figure 1. In total, 36 mice were given placebo PO and PBS IT (PBS), 36 mice were given placebo PO and 106 pfu MYXV∆serp2 IT (MYXVΔserp2), and 37 mice were given oclacitinib PO and 106 pfu MYXV∆serp2 IT (O+MYXVΔserp2). Twelve mice in each treatment group were humanely euthanized and necropsied on Days 4 and 7. Twelve mice in the PBS and MYXVΔserp2 treatment groups and 13 mice in the O+MYXVΔserp2 treatment group were euthanized when a criterion for euthanasia was reached. Criteria for euthanasia included tumor volume ≥ 3500 mm3, ulceration of tumors, lethargy or signs of pain, or loss of > 25% of the maximum body weight of the mouse.
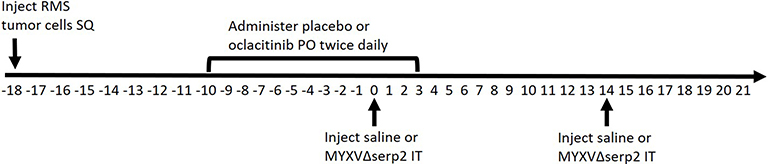 |
Figure 1 Diagram of the timeline (in days) of oral (PO) and intratumoral (IT) treatments given to mice bearing subcutaneous (SQ) rhabdomyosarcomas (RMS). Day 0 was designated as the first day that IT treatment was administered. Subsets of mice were euthanized on Days 4 and 7 for sample collection. The shortest and longest survival times were Day 11 and Day 31, respectively. |
Assessment of Clinical Condition
Individual mice were monitored daily for weight, tumor diameter, and clinical appearance. The number of mice in each treatment group with limb swelling, hematoma formation, erythema, ecchymosis, ulceration, and/or impaired mobility was tallied on Days 0, 4, 7, and 14. Contingency data of affected and unaffected animals in each treatment group at each time-point were compiled and analyzed.
Analysis of Viral Spread and Organ Pathology
All major internal organs and the tumor site were examined and collected during necropsy for assessment of gross appearance, isolation of viral DNA using droplet digital PCR (ddPCR; BioRad, Hercules, CA, USA), and histopathology. For PCR, frozen tissues were weighed then placed into 1.5 mL sterilized tubes containing 2.0 mm zirconia beads. 250 µL PBS was added to each sample. Samples were homogenized for 5 min using a Mini-Beadbeater (BioSpec, Atkinson, NH, USA). DNA was extracted from homogenized tissue sections using the QIAGEN DNeasy blood and tissue kit (Qiagen NV, Venlo, Netherlands) according to the manufacturer’s instructions. Purity and concentration were measured using 260nm/280nm ratios on a BioTek Take3 Micro-Volume Plate and the BioTek Synergy H1 Multi-Mode Reader. Approximately 100 ng of DNA was combined with QX200™ ddPCR™ EvaGreen Supermix (BioRad, Hercules, CA, USA) to perform ddPCR. Primer sets used for detection of MYXVΔserp2 by ddPCR are indicated in Table 1. Confirmation of MYXVΔserp2 in ddPCR positive tissues was performed using conventional PCR using 100ng of DNA in OneTaq® 2X Master Mix (New England Biolabs, Ipswich, MA, USA). Amplicons were annealed at 52°C, elongated at 68°C, and the product was visualized on a 2% agarose gel. For histopathology, tissues were placed into cassettes and stored in 10% buffered formalin for at least 1 week. Tissues were then paraffin-embedded, sectioned, and stained with hematoxylin and eosin (H&E) by the Histopathology Section of the Veterinary Diagnostic Laboratory at Colorado State University. Tissue sections were evaluated by a veterinary pathologist (ALM) using light microscopy.
 |
Table 1 Primer Sequences and Specific PCR Parameters Used to Detect MYXVΔserp2 DNA in Tissues |
Analysis of the Immune Response
The immune response to treatment was assessed using histopathology, multiplex serum cytokine analysis, and flow cytometry of cells from the spleen. Tumor histopathology sections were processed as previously described.6 Briefly, percent necrosis and hemorrhage were estimated. The numbers of inflammatory cells and mitotic figures were counted in ten 400× microscopic fields and averaged. The grading scheme indicated in Table 2 was followed. Serum cytokine analysis and flow cytometric assays to determine systemic inflammatory responses and activation of lymphoid populations also followed protocols previously described.37 Briefly, a multiplex bead array was used to detect the concentrations of 25 cytokines in mouse serum as directed by the manufacturer (BioRad, 23-plex Bio-Plex Pro™, Hercules, CA, USA; Invitrogen, ProcartaPlex mouse IFNalpha/IFNbeta Assay, Vienna, Austria). For flow cytometry, nucleated cells were isolated from spleens and lymph nodes, incubated with fluorescent dye-conjugated antibodies (CD3, CD11c, CD14, CD20, CD25, MHCII, and/or NK1.1) that targeted lymphocyte surface markers (BD Biosciences, San Jose, CA, USA), and detected using a CyAn ADP flow cytometer (Beckman Coulter, Brea, CA, USA).
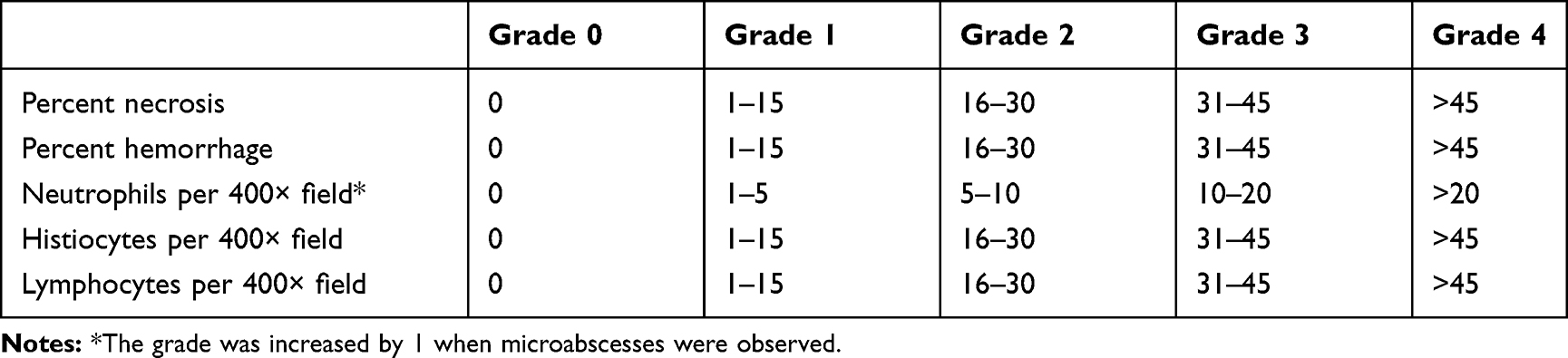 |
Table 2 Tumor Histology Grading Schemes |
Treatment Efficacy
The efficacy of treatment was evaluated by observation of clinical signs of disease, calculation of tumor volume and tumor growth rate, determination of median survival time, detection of viral load, and comparison of histopathology. Survival data were recorded from the time of the initial IT injection (Day 0) until euthanasia and plotted using a Kaplan-Meier curve.
Determination of Serum Oclacitinib Concentration
Sera from mice in each treatment group that were euthanized at Day 4 or Day 7 were pooled so that 6 samples were analyzed: Day 4 PBS, Day 7 PBS, Day 4 MYXVΔserp2, Day 7 MYXVΔserp2, Day 4 O+MYXVΔserp2, and Day 7 O+MYXVΔserp2. The serum concentration of oclacitinib maleate salt was measured using high performance liquid chromatography.
Statistics
Cell viability percentages, viral titers, serum oclacitinib concentration, histological grades, flow cytometry results, tumor volumes, and tumor growth rates were compared by t-tests using GraphPad Prism software version 8.1.0. The occurrence of clinical signs in mice at Day 0, 4, and 7 were compared with a Chi-square and Fisher’s exact test using GraphPad Prism software version 8.1.0. Median survival times were compared with a Cox proportional hazards model using GraphPad Prism software version 8.1.0. Serum cytokine data were analyzed by a statistician (BG) using Dunn’s test for comparisons on rank between groups. Associations of sex to cytokine levels were tested separately for each treatment using Welsh’s t-tests. Outlier tests were conducted using the car package version 3.0.2. Analyses were calculated using R open software version 3.6.0. This program with the fisheries stock analysis package version 0.8.23 was used for Dunn’s test analyses.
Results
MYXV Replicated in and Induced Lysis of U21089 Cells
Microscopic cytopathic effects and recombinant protein expression in murine U21089 rhabdomyosarcoma cell cultures suggested that the cells are semi-permissive for MYXV infection (Figure 2A). Logarithmic growth of virus was detected in U21089 cells, confirming that MYXV replication occurs in U21089 cells (Figure 2B). The rates of MYXV and MYXVΔserp2 growth in U21089 cells were not significantly different. No significant difference in cell viability was observed when MYXV and MYXVΔserp2 infection were compared at any time-point (P-values ≥ 0.181, Figure 2C). Importantly, MYXV and MYXVΔserp2 infection significantly decreased cell viability when compared to mock-infected cells (Figure 2C) at 24 (P-values ≤ 0.023), 48 (P-values ≤ 0.005), and 72 (P-values ≤ 0.002) hpi.
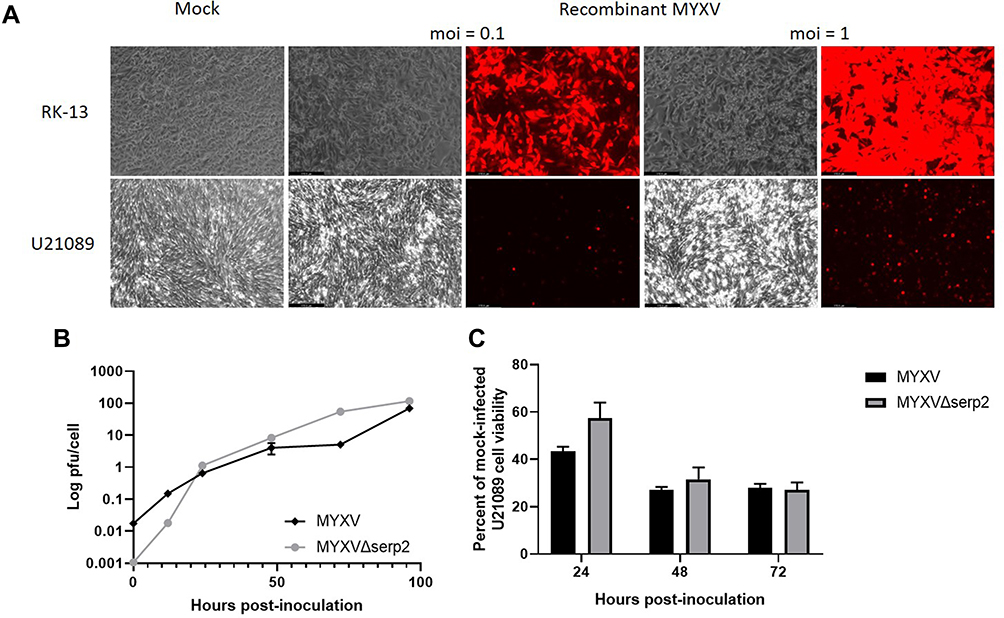 |
Figure 2 (A) Photomicrographs showing cytopathic effects (phase) and recombinant protein expression (fluorescence) 48 h post-inoculation (hpi) of RK-13 cells and U21089 cells with recombinant MYXV expressing tomato red fluorescent protein [multiplicity of infection (moi) = 0.1 and 1 virus particle/cell]. (B) Multi-step viral growth curves. U21089 cells were inoculated with MYXV or MYXVΔserp2 (moi = 0.01). Samples were collected at 0, 12, 24, 48, 72, and 96 hpi then plaqued onto RK-13 cells. The number of viral plaque-forming units (pfu) per U21089 cell was calculated from the number of foci that formed in RK-13 cell monolayers. (C) Viability of U21089 cells after inoculation with myxoma virus (MYXV) or MYXVΔserp2 (moi = 1) expressed as a percentage of viable mock infected U21089 cells. Error bars indicate standard error of the mean. |
U21089 Cells Formed Rapidly Growing ARMS Tumors in Nude Mice
Murine ARMS allografts were established in six- to eleven-week-old, B6.Cg-Foxn1nu/J (nude) mice by injecting 3x106 U21089 ARMS cells SQ in the lateral aspect of the thigh. Tumors formed in 100% (109/109) of the nude mice. The timeline for engraftment and treatments are shown in Figure 1. Some individual variation in tumor size at time of the first IT injection PBS or MYXVΔserp2 (designated Day 0) was observed, however the average tumor volumes of the three treatment groups (PBS, MYXVΔserp2, and O+MYXVΔserp2) were not significantly different (Figure 3A; P-values > 0.22). The rate of tumor growth from the time of U21089 cell injection until euthanasia was similar for mice in all treatment groups (Figure 3B; P-values > 0.17).
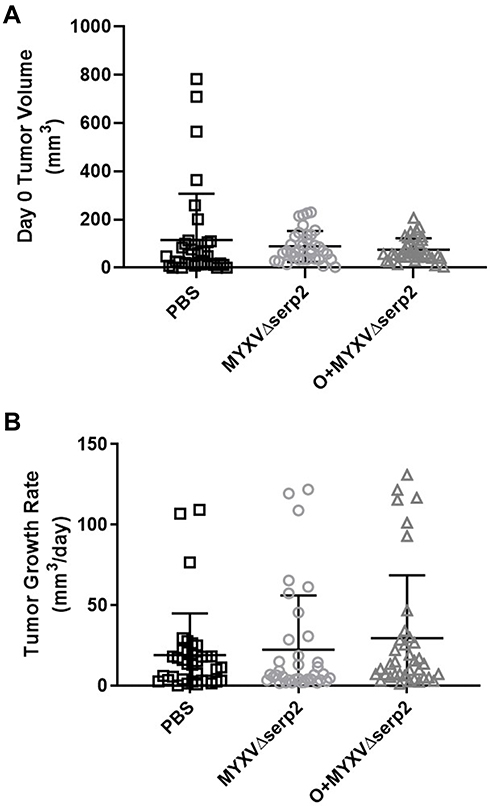 |
Figure 3 Tumor growth. (A) Tumor volume on the day of the first intratumoral injection (Day 0) and (B) average tumor growth rate calculated from U21089 cell injection until euthanasia in mice treated with phosphate buffered saline (PBS), MYXVΔserp2, and oclacitinib with MYXVΔserp2 (O+MYXVΔserp2). Error bars indicate mean and standard deviation. |
Clinical Findings in Mice with ARMS Were Less Severe in Mice Treated with OV
The gross appearance of tumors on Day 2 varied slightly between individual mice (Figure 4A–C). Ecchymoses at the base of the tumors (Figure 4D), on thighs (Figure 4E), or on abdomens occurred in all mice that met a criterion for euthanasia. Hematomas formed within tumors in 59% (64/109) of the mice. Hematomas ruptured forming ulcers (Figure 4F) that necessitated euthanasia of 32% (12/37) of the mice that were euthanized after Day 7. The ratio of mice showing clinical signs at Day 0 was significantly higher in the O+MYXVΔserp2 group (Figure 5A; P-value = 0.02), but no significance was noted between treatment groups by Day 4 (Figure 5B; P-value = 0.72). Table 3 summarizes the reasons that mice with ARMS had to be euthanized during this study. In mice evaluated beyond Day 7, 41% (15/37) mice were euthanized when their calculated tumor volume was ≥ 3500 mm3. Importantly, greater than 50% of mice in the MYXVΔserp2 treated and O+MYXVΔserp2 treated groups were surrendered past Day 7 due to the size of the tumor compared to 17% (2/12) of mice in the PBS group. This reflects the finding that clinical signs of disease that necessitated euthanasia (eg lethargy, hunched posture, difficulty moving, weight loss) were less common in mice treated with MYXVΔserp2.
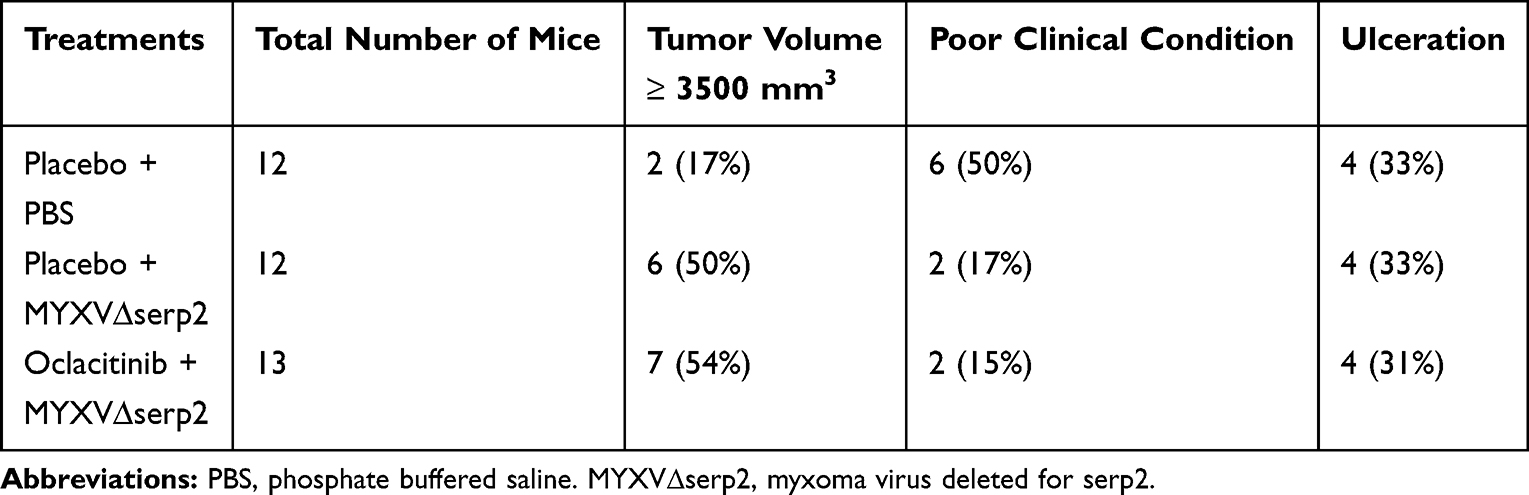 |
Table 3 Reason for Euthanasia of Mice with Rhabdomyosarcoma Allografts |
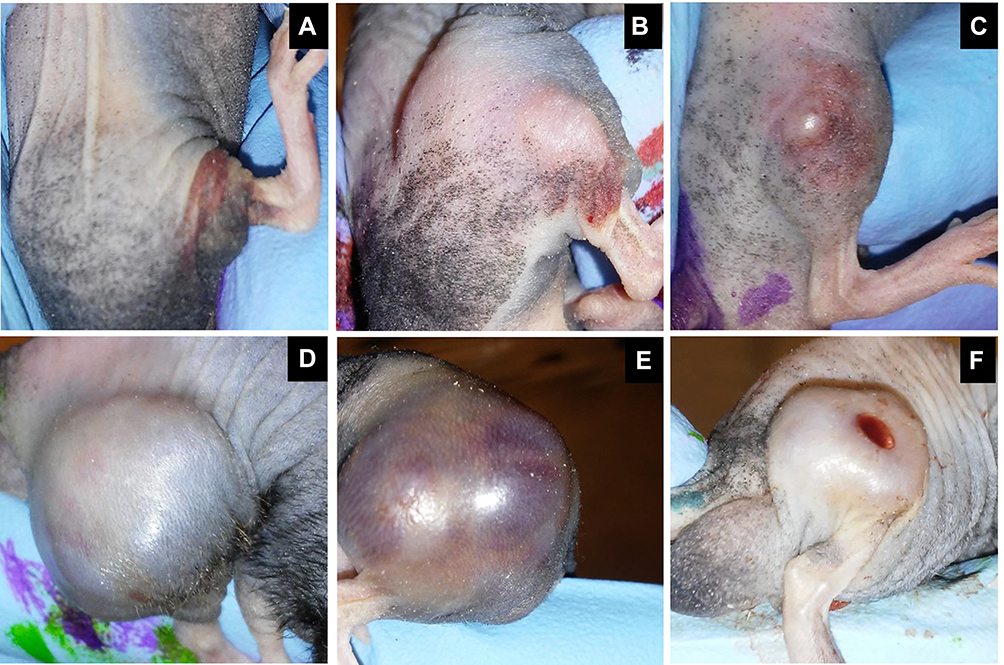 |
Figure 4 Representative images of subcutaneous rhabdomyosarcoma (RMS) tumors on Day 2 (A-C) and Day 19 (D-F) after the first intratumoral MYXVΔserp2 injection. Clinical signs associated with the tumors included erythema (A, B, D), swelling (B, E), ecchymosis (C, D, E), significant tumor development (D, E), and ulceration (F). |
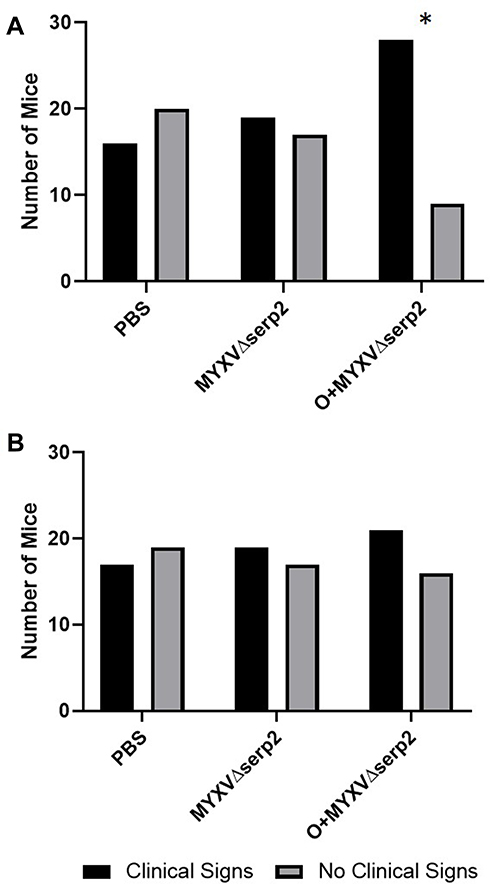 |
Figure 5 Contingency graphs indicating the number of mice with and without clinical signs of limb swelling, hematoma formation, erythema, and/or ecchymosis in each treatment group on (A) the day of the first intratumoral injection (Day 0) and (B) Day 4. On Day 0, the ratio of mice treated with oclacitinib with MYXVΔserp2 (O+MYXVΔserp2) that had clinical signs was higher than other treatment groups (*P-value = 0.02). |
Histologic Parameters Revealed Minor Differences Between Treatment Groups
No histologic abnormalities were observed in organs other than lymph nodes. This supported ddPCR data that indicated O+MYXVΔserp2 treatment does not induce unwanted spread of MYXVΔserp2 in off-target organs (Table 4). Lymphoid tissue was hyperplastic in low numbers of mice from all treatment groups. Tumors were unencapsulated and were composed of spindle cells arranged in streams and bundles. Cells were separated by a small amount of collagenous matrix. Cells had a small to moderate amount of wispy eosinophilic cytoplasm and a large round to oval nucleus with clumped chromatin and one to four prominent round nucleoli. Marked anisocytosis and anisokaryosis were noted. There were several binucleated cells and 0–3 mitotic figures per 400× microscopic field. Several tumor sections had areas of necrosis and some were hemorrhagic. Neutrophils were commonly associated with necrotic areas of the tumors and ulceration of the overlying dermis. Small pockets of neutrophils, consistent with microabscesses, were present within several tumor sections. Histiocytic and lymphocytic inflammation were not observed. Average grades for necrosis (Figure 6A), hemorrhage (Figure 6B), and neutrophilic inflammation (Figure 6C) within tumors were not significantly different between treatment groups in samples collected on Day 4. On Day 7, significantly decreased average hemorrhage and neutrophil grades were observed in O+MYXVΔserp2-treated mice as compared to MYXVΔserp2-treated mice (P-values = 0.05 and 0.03, respectively). In mice that met a criterion for euthanasia, animals in the O+MYXVΔserp2 treatment group had a significantly higher average necrosis grade as compared to mice treated with PBS (P-value = 0.02). These differences did not appear to alter tumor volume, tumor growth rate, or median survival time.
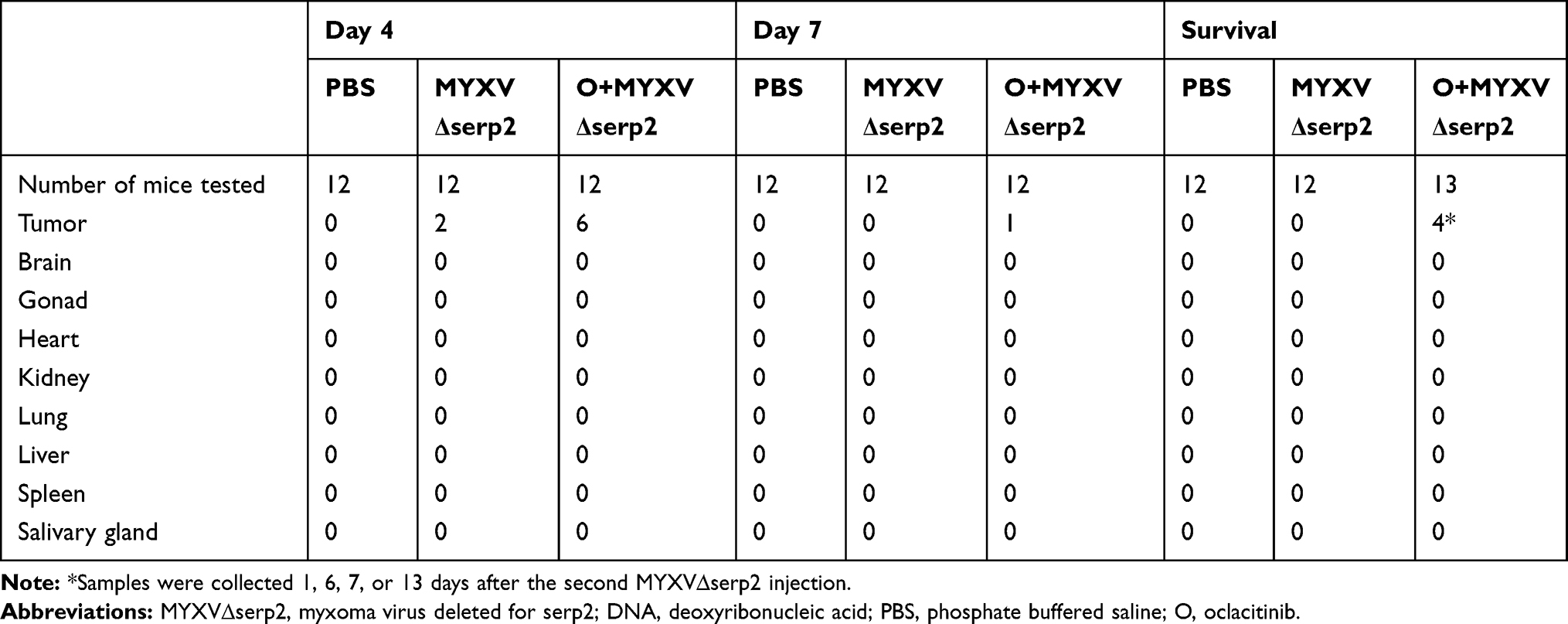 |
Table 4 Number of Samples in Which MYXV∆serp2 DNA Was Detected in Mouse Tissues by Droplet Digital PCR |
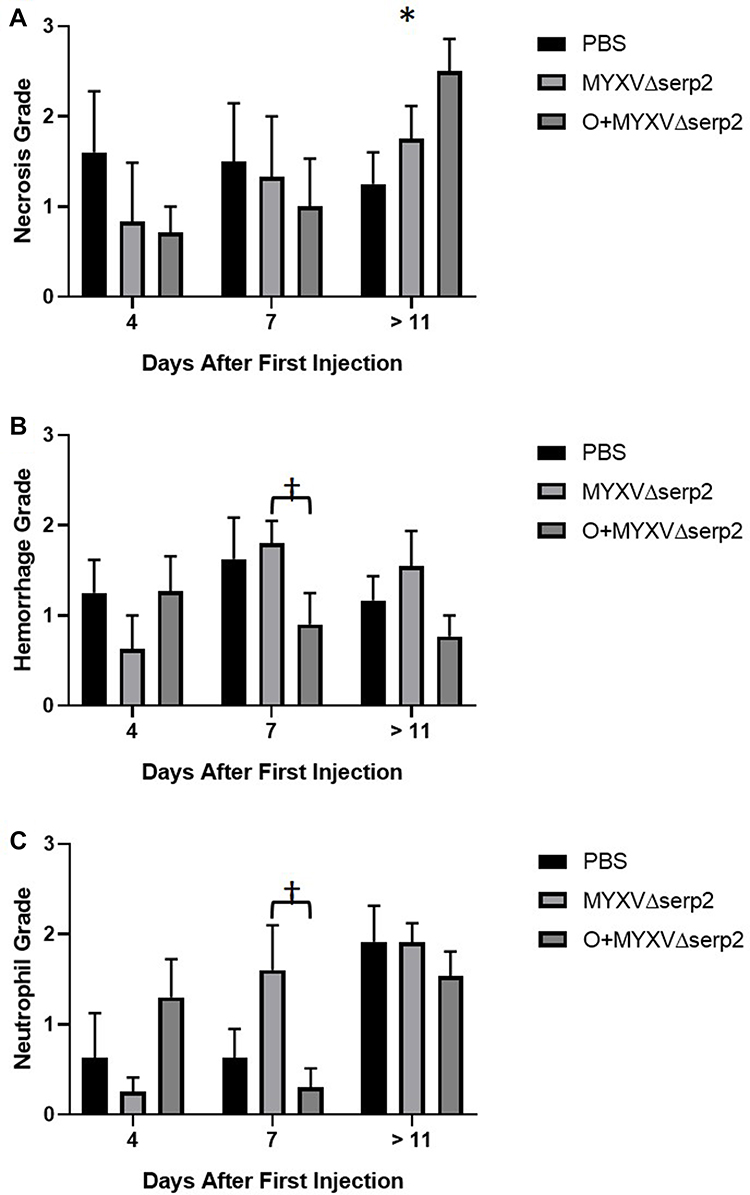 |
Figure 6 Average histologic grades. (A) Necrosis, (B) hemorrhage, and (C) neutrophil grades were determined in histologic sections of tumors collected at the time of euthanasia. Error bars indicate standard error of the mean. *P-value of the t-test comparing phosphate buffered saline (PBS) to oclacitinib with MYXVΔserp2 (O+MYXVΔserp2) treatment = 0.02. †P-values of the t-tests comparing MYXVΔserp2 to O+MYXVΔserp2 treatment ≤ 0.05. |
Serum Cytokine Concentrations Were Not Affected by Treatment
Serum cytokine concentrations were determined in all treatments groups of mice on Days 4 and 7, and in mice that met a criterion for euthanasia. No significant differences were observed in any of the following cytokine concentrations: IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-17, eotaxin, GM-CSF, G-CSF, interferon-α (IFN-α), IFN-β, IFN-γ, keratinocyte chemoattractant (CXCL1), monocyte chemoattractant protein-1, macrophage inflammatory protein-1α (MIP-1α), MIP-1β, tumor necrosis factor-α, or regulated on activation normal T cell expressed and secreted (CCL5). Graphs of cytokine concentrations can be viewed behind the Colorado State University firewall with this link: https://bgraham-csu.shinyapps.io/bjg_macneill_cytokines/.
Splenocyte Subsets Were Similar in All Treatment Groups
In mice euthanized on Day 7, cells were isolated from spleens, erythrocytes were lysed, nucleated splenocytes were incubated with antibodies against leukocyte antigens, and cells were analyzed using flow cytometry. Results were gated on intact, singlet cells. When treatment groups were compared, no significant differences were observed in cells consistent with B cells (< 42% of splenocytes), natural killer (NK) cells (< 0.74% of splenocytes), or macrophages (< 1.7% of splenocytes). As expected, NKT cell and T cell numbers were extremely low (< 0.030% of splenocytes).
Median Survival Time in Mice with ARMS Tumors Was Not Improved by Treatment
Mice treated with O+MYXV∆serp2 were significantly younger than mice in the other treatment groups (Figure 7A), but this did not cause a significant difference in median survival time (Figure 7B). Likewise, no significant differences in median survival time were observed between male and female mice in any treatment group (Figure 7C).
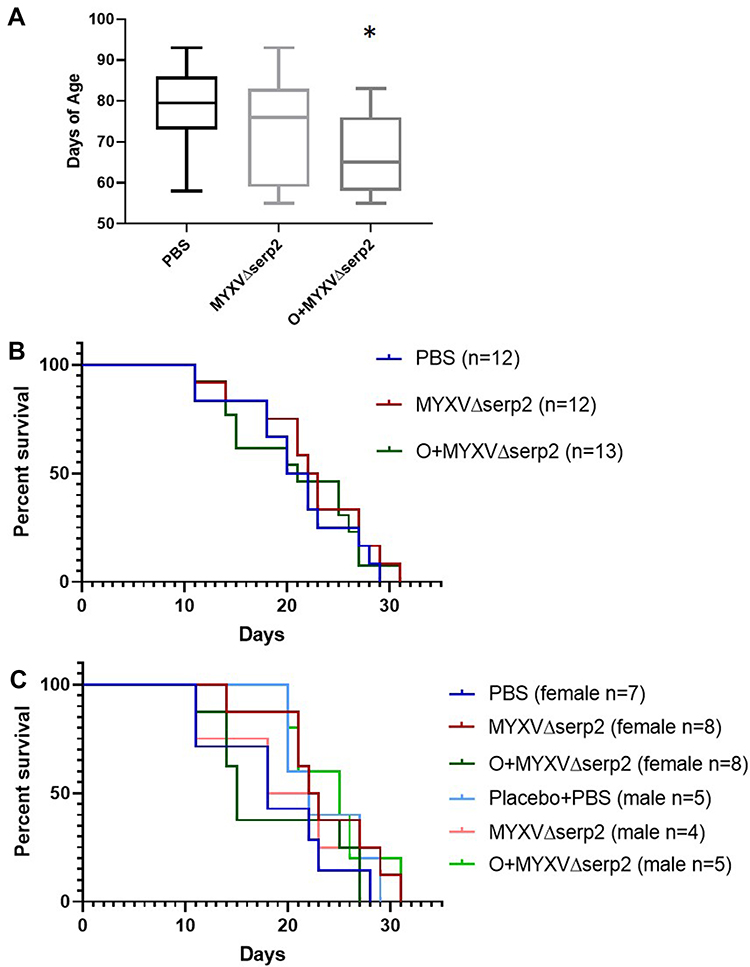 |
Figure 7 Graphs of (A) age on the day of U21089 cell injection and (B and C) survival outcomes. (A) Mice treated with oclacitinib with MYXVΔserp2 (O+MYXVΔserp2) were significantly younger than mice in other treatment groups (*P-value < 0.02). However, there were no significant differences between (B) median survival times in treatment groups. (C) Likewise, no differences were calculated when median survival times of male and female mice in each treatment group were analyzed independently. |
MYXVΔserp2 Replication Time in ARMS Tumors Was Extended by Oclacitinib Treatment
Oclacitinib maleate salt was detected in the serum of mice treated with oclacitinib on Day 4 (the first day after oral oclacitinib dosing was completed), but not in mice given placebo. By Day 7, serum oclacitinib concentrations were not detectable in any mice. MYXVΔserp2 was detected by ddPCR within the tumors of oclacitinib-treated mice up to 13 days after virus inoculation but was not detected beyond Day 4 in mice treated with MYXVΔserp2 alone (Table 4).
Discussion
MYXV∆serp2 replication was prolonged in ARMS tumors when combined with oclacitinib treatment as compared to MYXV∆serp2 treatment alone. Increasing the length of time that a recombinant MYXV replicated within a tumor microenvironment was a primary goal of this study. Increased MYXVΔserp2 replication time in tumors of O+MYXVΔserp2-treated mice may be due to inhibition of IFN signaling in the tumor microenvironment by oclacitinib. Oclacitinib inhibits JAK1 and JAK2 activation.31 Inhibition of JAK1 activation is expected to prevent IFN signaling in response to virus replication.38 Although many tumor cells do not mount and effective type I IFN response to MYXV infection in cell culture,11,28 healthy cells in the tumor microenvironment may release IFNs that signal tumor cells to upregulate production of antiviral IFN stimulated genes. Serum concentrations of IFNs and other cytokines were not significantly altered in mice treated with oclacitinib. However, most of the mice in this study had very low concentrations of serum cytokines which may have prevented detection of small changes in O+MYXVΔserp2-treated mice. It is likely that systemic effects of oclacitinib are less important that effects localized to the tumor microenvironment. Unfortunately, due to the lack of treatment efficacy, detection of IFN concentrations within the tumor microenvironment was not pursued.
We hypothesized that the longer MYXV∆serp2 was able to replicate in tumors, the more effective treatment would be. This was based upon previously published data that showed MYXV replicated for < 7 days in subcutaneous murine B16F10 melanoma allografts23 and that frequent injection of MYXV greatly improved outcomes in mice with subcutaneous CCL-136 embryonic RMS xenografts.6 This study indicates that prolonged virus replication is not sufficient to improve outcomes in a U21089 ARMS allograft model. Although MYXVΔserp2 DNA was isolated from tumors for up to 13 days after virus injection in mice treated with O+MYXVΔserp2, combination treatment did not decrease the tumor growth rate or median survival time of the mice.
The ARMS allografts that formed following subcutaneous injection of U21089 cells were quickly growing tumors that often became necrotic and hemorrhagic early in the course of disease. Tumors such as these are very challenging to treat with IT injections due to the inability to target viable tumor tissue around the areas of necrosis and hemorrhage. It is possible that other models of cancer would be more amendable to combination oclacitinib and OV therapy. We are currently treating dogs that have spontaneously arising soft tissue sarcomas with combination oclacitinib and MYXVΔserp2 therapy. Dogs that have residual disease following tumor excision are being enrolled in this study. We hypothesize that dogs who mount an adaptive immune response against MYXVΔserp2, will have a prolonged disease-free interval as compared to dogs that do not receive OV therapy.
The most significant finding in this study was the decreased severity of clinical signs in mice treated with MYXVΔserp2. Only 2 of 12 mice treated with MYXVΔserp2 and 2 of 13 mice treated with combination MYXVΔserp2 and oclacitinib had to be euthanized due to poor clinical condition (eg lethargy, hunched posture, difficulty moving, significant loss of weight), whereas 6 of 12 mice treated with placebo reached these criteria for euthanasia. This suggests that OV therapy may help patients with ARMS be more comfortable and experience fewer medical complications during the course of their disease. Additional studies are warranted to evaluate other combination therapies with MYXVΔserp2 to improve outcomes for RMS patients.
Conclusion
Our data indicate that MYXVΔserp2 treatment benefits mice with ARMS by reducing clinical signs of disease and improving quality of life.
Acknowledgments
The Elsa U Pardee Foundation generously funded this study. We thank the undergraduate (Mikaela Maldonado, Garin Wilson) and veterinary (Kayla Gravelle) students who helped with daily evaluation of the mice.
Disclosure
Ms Barbara Graham reports grants from The Elsa U Pardee Foundation, during the conduct of the study. The authors report no conflicts of interest in this work.
References
1. Casanova M, Meazza C, Favini F, Fiore M, Morosi C, Ferrari A. Rhabdomyosarcoma of the extremities: a focus on tumors arising in the hand and foot. Pediatr Hematol Oncol. 2009;26(5):321–331. doi:10.1080/08880010902964367
2. Gurria JP, Dasgupta R. Rhabdomyosarcoma and extraosseous ewing sarcoma. Children. 2018;5(12):165. doi:10.3390/children5120165
3. Ognjanovic S, Linabery A, Charbonneau G, Ross J. Trends in childhood RMS incidence and survival in the US. Cancer. 2009;115(18):4218–4226. doi:10.1002/cncr.24465.Trends
4. Hoang NT, Acevedo LA, Mann MJ, Tolani B. A review of soft-tissue sarcomas: translation of biological advances into treatment measures. Cancer Manag Res. 2018;10:1089–1114. doi:10.2147/CMAR.S159641
5. Pol J, Bloy N, Obrist F, et al. Trial watch:: oncolytic viruses for cancer therapy. Oncoimmunology. 2014;3:e28694. doi:10.4161/onci.28694
6. Kinn VG, Hilgenberg VA, MacNeill AL. Myxoma virus therapy for human embryonal rhabdomyosarcoma in a nude mouse model. Oncolytic Virother. 2016;5:59–71. doi:10.2147/OV.S108831
7. Gentschev I, Adelfinger M, Josupeit R, et al. Preclinical evaluation of oncolytic vaccinia virus for therapy of canine soft tissue sarcoma. PLoS One. 2012;7(5):e37239. doi:10.1371/journal.pone.0037239
8. Cinatl J
9. MacNeill AL, Moldenhauer T, Doty R, Mann T. Myxoma virus induces apoptosis in cultured feline carcinoma cells. Res Vet Sci. 2012;93(2):1036–1038. doi:10.1016/j.rvsc.2011.10.016
10. Urbasic AS, Hynes S, Somrak A, et al. Oncolysis of canine tumor cells by myxoma virus lacking the serp2 gene. Am J Vet Res. 2012;73(8):1252–1261. doi:10.2460/ajvr.73.8.1252
11. Wang F, Ma Y, Barrett JW, et al. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol. 2004;5(1):1266–1274. doi:10.1038/ni1132
12. Woo Y, Kelly KJ, Stanford MM, et al. Myxoma virus is oncolytic for human pancreatic adenocarcinoma cells. Ann Surg Oncol. 2008;15(8):2329–2335. doi:10.1245/s10434-008-9924-z
13. Fenner F, Woodroffe GM. The pathogenesis of infectious myxomatosis: the mechanism of infection and the immunological response in the European rabbit (Oryctolagus cuniculus). Br J Exp Pathol. 1953;34(4):400–411.
14. Fenner F. Adventures with poxviruses of vertebrates. FEMS Microbiol Rev. 2000;24:123–133. doi:10.1111/j.1574-6976.2000.tb00536.x
15. Gorski J, Mizak B, Chrobocinska M. Control of rabbit myxomatosis in Poland. Rev Sci Tech. 1994;13(3):869–879. doi:10.20506/rst.13.3.803
16. McCabe VJ, Tarpey I, Spibey N. Vaccination of cats with an attenuated recombinant myxoma virus expressing feline calicivirus capsid protein. Vaccine. 2002;20(19):2454–2462. doi:10.1016/S0264-410X(02)00186-X
17. McCabe VJ, Spibey N. Potential for broad-spectrum protection against feline calicivirus using an attenuated myxoma virus expressing a chimeric FCV capsid protein. Vaccine. 2005;23(46–47):5380–5388. doi:10.1016/j.vaccine.2005.05.038
18. Pignolet B, Boullier S, Gelfi J, et al. Safety and immunogenicity of myxoma virus as a new viral vector for small ruminants. J Gen Virol. 2008;89(Pt 6):1371–1379. doi:10.1099/vir.0.83595-0
19. Thorne SH, Bartlett DL, Kirn DH. The use of oncolytic vaccinia viruses in the treatment of cancer: a new role for an old ally? CurrGene Ther. 2005;5:429–443.
20. Chaudhri G, Panchanathan V, Buller RML, et al. Polarized type 1 cytokine response and cell-mediated immunity determine genetic resistance to mousepox. Proc Natl Acad Sci U S A. 2004;101(24):9057–9062. doi:10.1073/pnas.0402949101
21. Xu R, Johnson AJ, Liggitt D, Bevan MJ. Cellular and humoral immunity against vaccinia virus infection of mice. J Immunol. 2004;172(1):6265–6271. doi:10.4049/jimmunol.172.10.6265
22. Tosic V, Thomas DL, Kranz DM, et al. Myxoma virus expressing a fusion protein of interleukin-15 (IL15) and IL15 receptor alpha has enhanced antitumor activity. PLoS One. 2014;9(10):e109801. doi:10.1371/journal.pone.0109801
23. Doty RA, McFadden G, Roy EJ, MacNeill AL. Histological evaluation of intratumoral myxoma virus treatment in an immunocompetent mouse model of melanoma. Oncolytic Virother. 2013;2:1–17. doi:10.2147/OV.S37971
24. Lun X, Alain T, Zemp FJ, et al. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Res. 2010;70(2):598–608. doi:10.1158/0008-5472.CAN-09-1510
25. Stanford MM, Shaban M, Barrett JW, et al. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol Ther. 2008;16(1):52–59. doi:10.1038/sj.mt.6300348
26. Thomas DL, Doty R, Tosic V, et al. Myxoma virus combined with rapamycin treatment enhances adoptive T cell therapy for murine melanoma brain tumors. Cancer Immunol Immunother. 2011;60(10):1461–1472. doi:10.1007/s00262-011-1045-z
27. Lun X, Yang W, Alain T, et al. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005;65(2):9982–9990. doi:10.1158/0008-5472.CAN-05-1201
28. Bartee E, McFadden G. Human cancer cells have specifically lost the ability to induce the synergistic state caused by tumor necrosis factor plus interferon-beta. Cytokine. 2009;47(3):199–205. doi:10.1016/j.cyto.2009.06.006
29. MacNeill AL, Turner PC, Moyer RW. Mutation of the Myxoma virus SERP2 P1-site to prevent proteinase inhibition causes apoptosis in cultured RK-13 cells and attenuates disease in rabbits, but mutation to alter specificity causes apoptosis without reducing virulence. Virology. 2006;356(1–2):12–22. doi:10.1016/j.virol.2006.07.049
30. Zoetis. APQ1213211 © 2014 Zoetis Inc. All rights reserved. Available from: https://www.zoetisus.com/products/dogs/apoquel/pdf/apoqueltechnicalmonograph.pdf.
31. Gonzales AJ, Bowman JW, Fici GJ, Zhang M, Mann DW, Mitton-Fry M. Oclacitinib (APOQUEL®) is a novel Janus kinase inhibitor with activity against cytokines involved in allergy. J Vet Pharmacol Ther. 2014;37(4):317–324. doi:10.1111/jvp.12101
32. Taniguchi E, Nichijo K, McCleish A, et al. PDGFR-A is a therapeutic target in alveolar rhabdomyosarcoma. Oncogene. 2008;27(51):51. doi:10.1038/onc.2008.255
33. Liu J, Wennier S, Reinhard M, Roy E, MacNeill A, McFadden G. Myxoma virus expressing interleukin-15 fails to cause lethal myxomatosis in European rabbits. J Virol. 2009;83(11):5933–5938. doi:10.1128/JVI.00204-09
34. Nathaniel R, MacNeill AL, Wang YX, Turner PC, Moyer RW. Cowpox virus CrmA, Myxoma virus SERP2 and baculovirus P35 are not functionally interchangeable caspase inhibitors in poxvirus infections. J Gen Virol. 2004;85:1267–1278. doi:10.1099/vir.0.79905-0
35. Fukuyama T, Ganchingco JR, Bäumer W. Demonstration of rebound phenomenon following abrupt withdrawal of the JAK1 inhibitor oclacitinib. Eur J Pharmacol. 2017;794(November2016):20–26. doi:10.1016/j.ejphar.2016.11.020
36. Fukuyama T, Ehling S, Cook E, Bäumer W, Baumer W. Topically administered janus-kinase inhibitors tofacitinib and oclacitinib display impressive antipruritic and anti-inflammatory responses in a model of allergic dermatitis. J Pharmacol Exp Ther. 2015;354(3):394–405. doi:10.1124/jpet.115.223784
37. MacNeill AL, Moldawer LL, Moyer RW. The role of the cowpox virus crmA gene during intratracheal and intradermal infection of C57BL/6 mice. Virology. 2009;384(1):151–160. doi:10.1016/j.virol.2008.10.041
38. O’Shea JJ. Targeting the Jak/STAT pathway for immunosuppression. Ann Rheum Dis. 2004;63(SUPPL. 2):67–71. doi:10.1136/ard.2004.028290
39. Liu G, Friggeri A, Yang Y, Park YJ, Tsuruta Y, Abraham E. miR-147, a microRNA that is induced upon toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc Natl Acad Sci U S A. 2009;106(37):15819–15824. doi:10.1073/pnas.0901216106
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.