")
Back to Journals » Clinical Ophthalmology » Volume 8
Treatment of allergic conjunctivitis with bepotastine besilate ophthalmic solution 1.5%
Authors Bergmann M, Williams J, Gomes P, Mclaughlin J
Received 22 April 2014
Accepted for publication 10 June 2014
Published 13 August 2014 Volume 2014:8 Pages 1495—1505
DOI https://doi.org/10.2147/OPTH.S66637
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Mark T Bergmann,1 Jon I Williams,2 Paul J Gomes3
On behalf of the Bepotastine Besilate Ophthalmic Solutions Clinical Study Group
1Eye Care Associates of Greater Cincinnati, Inc., Cincinnati, OH, USA; 2Bausch + Lomb, Irvine, CA, USA; 3Ora, Inc., Andover, MA, USA
Purpose: To examine the pooled per-protocol ocular end points from two conjunctival allergen challenge (CAC) clinical trials of the dual-action antihistamine bepotastine besilate ophthalmic solution (BBOS) 1.5%.
Methods: Two Phase III, placebo-controlled, double-masked, randomized clinical trials were conducted at a total of six separate centers using the CAC model of allergic conjunctivitis. The same study design was employed for both clinical trials, with subjects randomly assigned to either BBOS 1.5% (n=78) or placebo (n=79) treatment. Each subject received one eye drop of the test agent bilaterally at different study visits 15 minutes, 8 hours, or 16 hours prior to a CAC. Primary ocular end points included changes in ocular itching reported at 3, 5, and 7 minutes and conjunctival hyperemia assessed at 7, 15, and 20 minutes following each CAC. Secondary ocular end points included chemosis as well as episcleral and ciliary hyperemia judged by investigators, and tearing (scored as either absent or present) and eyelid swelling judged by subjects.
Results: A statistically significant reduction in ocular itching was observed for BBOS 1.5% treatment compared to placebo at all time points (P<0.0001), while measures for onset and 8-hour persistence of action also reached clinical significance (ie, ≥1.0 unit difference) at a majority of time points. In addition, a significant reduction in conjunctival hyperemia was achieved at a majority of time points during the onset of action CAC test. Secondary end points were also significantly improved compared to placebo, most prominently for reduced tearing at all study visits and reduced eyelid swelling at the onset of action and 8-hour study visits. Adverse events were generally mild and transient.
Conclusion: BBOS 1.5% rapidly reduced CAC-induced ocular itching with duration of effectiveness of at least 8 hours after dosing. Certain secondary signs of inflammation were also significantly reduced.
Keywords: allergen challenge, antihistamine, itching, Bepreve
Introduction
Allergic conjunctivitis (AC) is the most common allergic ocular disorder, affecting up to 60 million Americans annually.1,2 The hallmarks of AC include a combination of ocular itching and conjunctival hyperemia (redness),2,3 which are often accompanied by chemosis, lid swelling, and tearing, as well as rhinorrhea and other nasal symptoms.1–3 The most common instigators of this allergic condition are either seasonal pollens or perennial allergens, including, but not limited to, dust mites, molds, or animal dander.3,4 Allergic responses generally occur in two phases. The early or immediate (acute) phase occurs within minutes of allergen exposure, and results from antigen-evoked mast cell degranulation and subsequent histamine release. This is followed by a late or delayed (chronic) phase, hours or days after exposure, that is manifested by proinflammatory mediators and recruitment of immune cells including eosinophils and neutrophils.3,4
AC is classified as a type 1, IgE-mediated hypersensitivity immune reaction and occurs in individuals previously exposed to the offending allergen.4,5 The signs and symptoms of AC are primarily caused by the action of mast cell-derived histamine, which acts on H1 and H2 histamine receptors in the surrounding ocular tissues.6,7 For this reason, medications that stabilize mast cells and reduce degranulation and those that directly antagonize the action of histamine at its receptors have become the first-line therapy for AC.8,9 Medications of this class, dual-action antihistamines, have both an improved safety profile and increased therapeutic effect compared to older topical agents (eg, pheniramine maleate) used in the treatment of AC.3,5,8,9
Bepotastine besilate is an anti-allergic agent with multiple mechanisms of action that has been extensively investigated in both preclinical and clinical studies.10 Bepotastine besilate is a dual-action agent, a highly selective histamine H1-receptor antagonist10,11 with potent mast cell-stabilizing effects.12–17 The anti-inflammatory actions of bepotastine besilate include inhibition of leukotriene B418 and attenuating eosinophil chemotaxis and activation.13–17 Bepotastine has also been shown to inhibit the biosynthesis of proinflammatory IL-5 in vitro.19 Oral formulations of bepotastine besilate have been well-characterized in Japan in both preclinical13,15–17 and clinical reports,20–23 as well as in several clinical safety studies.24–26 Positive results from these studies led to approval by the Japanese Ministry of Health and Welfare in July 2000 of an oral bepotastine besilate formulation (Talion® tablet; Mitsubishi Tanabe Pharma Corporation, Osaka, Japan)27 for the treatment of allergic rhinitis and subsequently (in 2002) for urticaria and several types of dermal prurituses.23,24,28–30 Based on the history of clinical effectiveness accumulated since the approval in Japan,31,32 bepotastine besilate was reformulated for investigation of its topical use in the treatment of AC.
Two clinical trials examined the efficacy, safety, and comfort of a topical formulation of bepotastine besilate for treatment of AC.33,34 Both studies used the conjunctival allergen challenge (CAC) model, a rigorously validated approach to assess the efficacy of ophthalmic anti-allergics.35,36 The CAC-based clinical trials (one single site, n=10733 and one multisite, n=13034) established that bepotastine besilate opthalmic solution (BBOS) (either 1.0% or 1.5%) provided a statistically and clinically significant reduction in ocular itching for up to 8 hours post-instillation in both trials as well as statistically significant reductions in conjunctival hyperemia associated with AC.33,34 Additionally, several of the secondary end points for both trials showed a statistically significant improvement with BBOS compared to placebo that approached the level of clinical significance in at least one CAC trial. Thus, while results from these trials established the clinical effectiveness of BBOS 1.5%, leading to its specific approval for treatment of ocular itching associated with AC, it is possible that the true breadth of BBOS’s efficacy in treating the signs and symptoms of AC was underestimated by the relatively small sample size in each CAC study.
To address this hypothesis, subject data from the two clinical CAC trials were pooled to create a larger population and then reanalyzed according to the statistical analysis plan established in both CAC study protocols. In addition to the primary end points of ocular itching and conjunctival hyperemia, the goal of this post hoc analysis was to reexamine secondary end points that approached clinical or statistical significance in the earlier analyses.
Methods
Study design and approval
Data for this pooled analysis were collected over the course of two Phase III clinical CAC trials; a single-center study (NCT00424398) and a multicenter study (NCT00586664). Data resulting from individual analyses of these studies have been provided elsewhere.33,34 Both trials were double-masked, placebo-controlled, randomized trials that employed essentially identical study protocols with enrolled subjects 10 years of age or older. Both trials were approved by independent institutional review boards prior to initiation of the trial: IntegReview (Austin, TX, USA) for the single-site trial and Coast IRB, LLC (Colorado Springs, CO, USA) for the multisite trial. Both trials were conducted in accordance with the International Conference on Harmonization (ICH) Good Clinical Practice guidelines37 and the Declaration of Helsinki.38 Written informed consent (or assent and parental/guardian permission in subjects <18 years of age) was obtained from all subjects before any clinical study procedures were conducted. All ophthalmic examinations and study-related procedures were conducted by clinically trained researchers who had prior experience with the CAC methodology. Test agents included in these post hoc analyses were the vehicle (placebo) and BBOS 1.5%.
Inclusion criteria
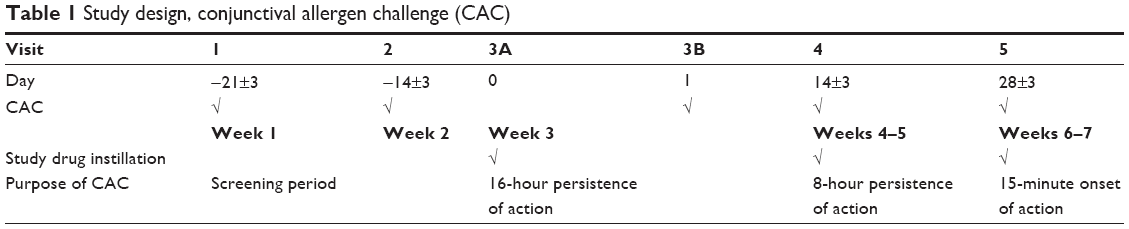
Male and female subjects ≥10 years of age were initially recruited if they elicited a positive skin-test reaction to common seasonal or perennial allergens (ie, cat dander, grasses, trees, or ragweed). Subjects also had to have a positive bilateral CAC reaction (≥2 units itching and ≥2 units redness in two of three vessel beds) within 10 minutes of instillation of the last allergen titration at visit 1 and a similarly positive bilateral CAC reaction at two or more time points at visit 2 (Table 1). Subjects were further required to have a best corrected visual acuity (BCVA) of 0.7 logMAR (ie, 20/100 vision) or better in each eye and an intraocular pressure (IOP) in the range of 5 to 22 mmHg in both eyes. A negative urinary pregnancy test result was required at the first and last study visit as was the use of a medically acceptable contraceptive method for female subjects of childbearing potential. Additional stipulations included a willingness to avoid the use of disallowed medications and contact lenses for the duration of each clinical trial.
 | Table 1 Study design, conjunctival allergen challenge (CAC) |
Exclusion criteria
Potential subjects were excluded based upon criteria established prior to the study. They were excluded if they were not willing to attend all study visits, had a known contraindication or sensitivity to the use of any study drugs or their components, or had any active ocular disease or other significant illness (eg, narrow-angle glaucoma, clinically significant blepharitis, or severe cardiovascular disease) that could affect their safety or study parameters during the trial. They were also excluded if they had undergone refractive surgery within the 6 months before screening, were concurrently enrolled in another study of an investigational drug or device, or were enrolled in another study within 30 days of the study. In addition, subjects could be excluded from enrollment following results of visits 1 and 2, as described in the study protocol.
Study medications
Both the single-site and multisite CAC studies used identically formulated ophthalmic solutions. Both BBOS 1.5% and placebo were manufactured by Alliance Medical Products, Inc. (Irvine, CA, USA) and were packaged such that they were indistinguishable to investigators and subjects in order to maintain masking.
Study protocol
There were five study visits for enrolled subjects in each CAC trial. At each study visit, the medical and medication histories of the subjects were updated, the inclusion and exclusion criteria were reviewed, and (when applicable) tolerability to an assigned investigational product was assessed. Additionally, visual acuity was measured, slit-lamp biomicroscopy was performed, and baseline ocular allergic sign and symptom assessments were recorded before any investigational product instillation or CAC test. The study design is depicted in Table 1. Following each CAC session, subject responses were scored according to an established set of grading scales.35,36 Subjects were asked to grade the degree of ocular itching on a 0–4 scale with half-unit increments allowed (0= no itch, 4= severe itch) at 3, 5, and 7 minutes after the CAC assessment. Conjunctival hyperemia and secondary end points including episcleral or ciliary hyperemia, chemosis, and eyelid swelling were each evaluated on a 0–4-unit scale, with half-unit increments allowed, at 7, 15, and 20 minutes after a CAC. Ocular hyperemia and chemosis were graded by the investigator, while eyelid swelling was graded by the subject. Tearing was separately scored by subjects as either absent or present at 7, 15, and 20 minutes after a CAC.
Subjects who met initial entry criteria participated in the first two study visits as part of the final eligibility criteria. All subjects underwent an allergen titration at visit 1, at which time a drop of allergen solution was instilled bilaterally, and subjects were observed and scored after 10 minutes. This process was repeated with increasing allergen concentration until the subjects elicited a positive response (grade ≥2 units) for both ocular itching and conjunctival hyperemia. Subjects who did not reach this predefined value were excluded from the remainder of the study. One week later (visit 2), subjects were challenged with the same concentration of allergen, and those subjects with reproducible responses of a ≥2 unit score for both ocular itching and conjunctival hyperemia were selected for randomization.
At visit 3A, enrolled subjects were randomly assigned to one of three treatment groups, BBOS 1.0%, BBOS 1.5%, or vehicle (BBOS 1.0% results are not discussed in this article). One drop of the assigned formulation was instilled in the conjunctival cul-de-sac of each eye. The study drug vials were masked to the subjects and investigators; labeling of all investigational products was identical and included the subject number. At visit 3B, 16 hours (±30 minutes) after study drug instillation at visit 3A, a CAC test was conducted for each subject. Scores recorded for this visit provided an evaluation of the 16-hour persistence of drug action.
In two additional trial visits at approximately 2-week intervals, test agent instillation was followed by a CAC at either 8 hours (±30 minutes; visit 4) or at 15 minutes (visit 5) post-dosing. These visits provided an assessment of an 8-hour persistence of action and the onset of action, respectively.
Outcome measures
The primary efficacy end points for the pooled data set were subject-assessed ocular itching and investigator-assessed conjunctival hyperemia in both eyes at each visit. Scores for each end point were calculated by subtracting scores measured for BBOS 1.5% treatments from scores measured for placebo treatments at visits 3B, 4 and 5. Investigator-scored secondary outcome measures included ciliary hyperemia, episcleral hyperemia, and chemosis. Tearing and eyelid swelling were subject-graded; tearing was graded as either present or absent.
Safety and adverse events (AEs)
At visit 1, subjects signed an informed consent (informed assents for minors) prior to any study procedures. Medical and medication histories were then obtained, inclusion and exclusion criteria were reviewed, and subjects were informed about the procedures for AE reporting. Visit 1 also included a visual acuity measurement using an Early Treatment Diabetic Retinopathy Study (ETDRS) chart, slit-lamp biomicroscopy examination, dilated fundoscopy, and an IOP measurement. The fundoscopy and IOP assessments were repeated at visit 5; the other procedures were repeated at every visit before the instillation of any investigational product or CAC test. Records of concurrent medications were updated at each return visit, and AEs were registered as they were reported. AEs were recorded according to the categories in the Medical Dictionary for Regulatory Activities (MedDRA).39
Data analysis and statistical methods
Demographics
Demographic data were pooled as part of the dataset analyses. The mean, median, minimum, and maximum data values for the per-protocol (PP) population were used to summarize the quantitative demographic variables, which were then analyzed between treatment groups with either a two-sided t-test or the Fisher’s exact test.
Primary efficacy variables
The primary efficacy outcome analyses presented here are for the PP population. For the purposes of these analyses of pooled data, the PP population included subjects who attended visit 3A/3B or visit 4 and also attended visit 5. All ocular datasets other than tearing used the average scores of both eyes of a subject from each post-CAC assessment to calculate individual subject scores.
Statistical significance of between-group differences in mean ocular itching and conjunctival hyperemia scores (placebo versus BBOS 1.5%) was assessed using a nonparametric Wilcoxon rank-sum test for each post-CAC measurement during each visit. Treatment differences for either primary variable were considered statistically significant for the PP population at a study visit, at a two-sided α of P≤0.0125 for a majority of time points at visit 5, or at a two-sided α of P≤0.00625 for a majority of time points at visit 3B or visit 4. The values used for these nonparametric assessments were adjusted for multiple comparisons using conservative Bonferroni corrections.
An overall assessment of treatment group differences for each primary efficacy variable was also carried out across all time points at each visit using an analysis of covariance (ANCOVA) with repeated measures, with eyes nested within a subject as the cluster unit, and using a linear contrast model. Clinical significance for treatment group differences at a study visit was prospectively defined in both CAC trials as a ≥1.0 unit between-group difference in scores at the majority of observation time points at a study visit, and a ≥0.5 unit between-group difference at all post-CAC time points at a study visit.
Subgroup analyses of potential statistical or clinical significance for reduction in ocular itching or conjunctival hyperemia scores were conducted as well in subjects in the PP population with a more severe allergic response (itching score in at least one eye ≥3.0 units at one or more time points at screening visit 2), and in subjects who had complete relief of CAC-induced ocular itching (symptom score =0) in a study eye.
Secondary efficacy variables
P-values determined for all secondary efficacy variables other than allergen-induced tearing were calculated for each time point at a study visit by the Wilcoxon rank-sum test and across all time points using ANCOVA with repeated measures as described for the primary efficacy variables. Clinical significance for secondary efficacy variables other than tearing was also defined the same as for the primary efficacy variables.
P-values for treatment differences in tearing data (placebo % scored “present” less BBOS 1.5% scored “present”) were calculated at each time point using Fisher’s exact test, treating each subject as a cluster. P-values were subsequently adjusted for multiplicity by the false discovery rate method.40 Statistical and clinical significance for reduced tearing required adjusted α (two-sided) ≤0.05 at a majority of observation time points at a study visit. Clinical significance was prospectively equated to statistical significance in the absence of a validated standard for clinical relevance to describe reduced tearing.
Results
Subject characteristics
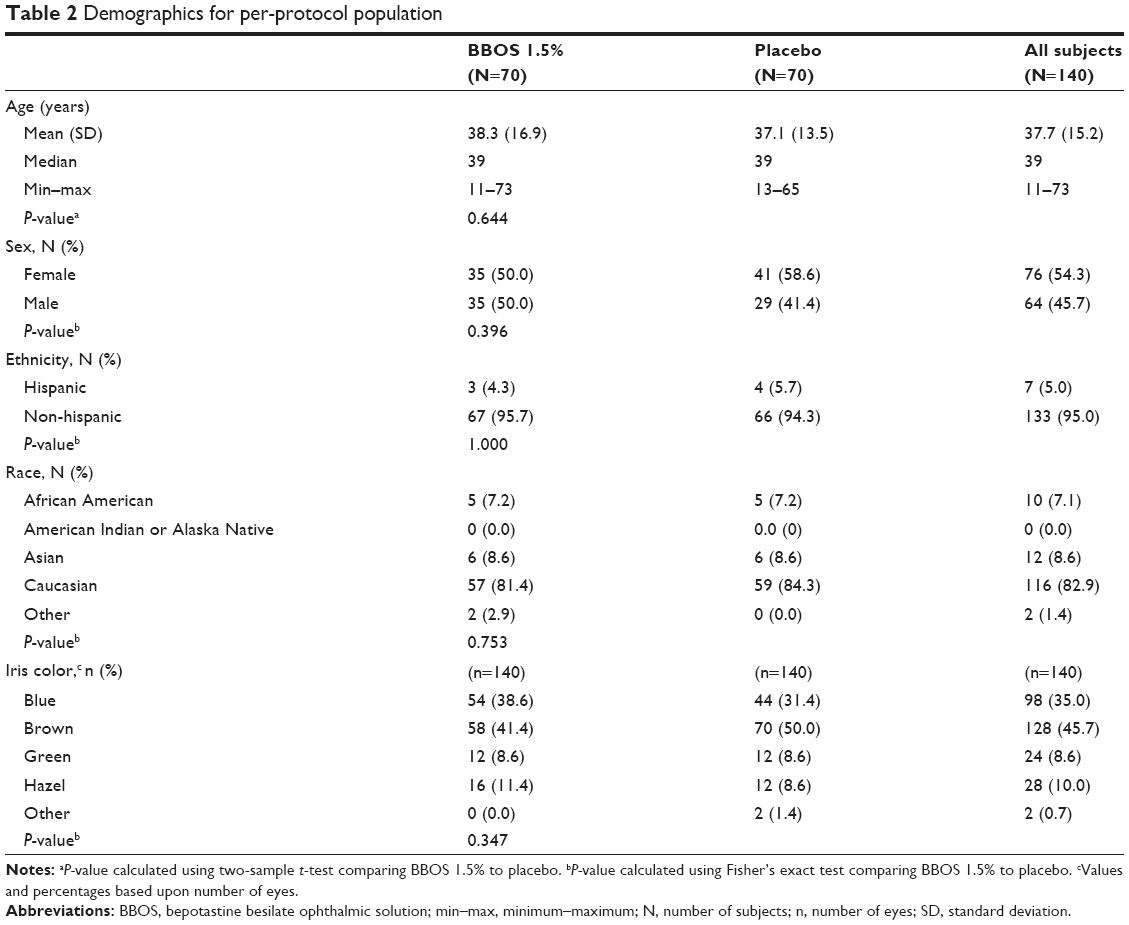
Subject demographics were well balanced between the BBOS 1.5% and placebo treatment groups (Table 2). No significant differences were found in the pair-wise comparison of the demographic characteristics between the BBOS 1.5% and placebo treatment groups.
 | Table 2 Demographics for per-protocol population |
Subject disposition
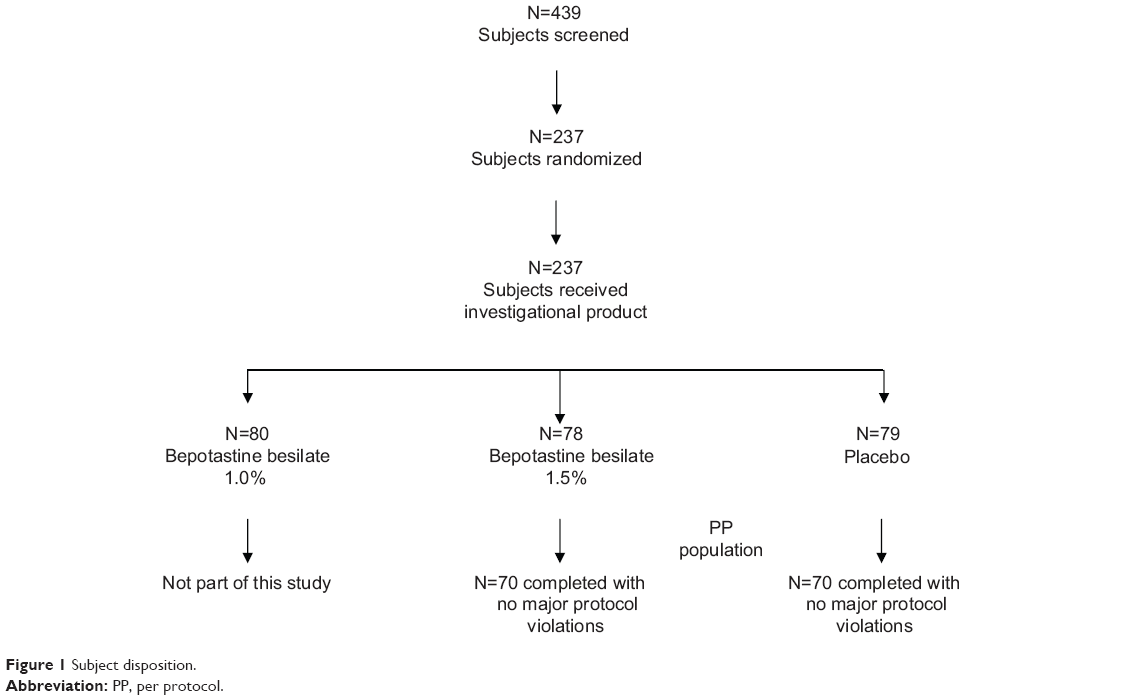
A total of 439 subjects were screened as part of either the single-site or the multicenter trials. Of those, 202 subjects were excluded at visit 1 or visit 2 because they did not meet all of the inclusion criteria (Figure 1). A total of 237 patients were subsequently enrolled and assigned to treatment groups according to a computer-generated randomization code at visit 3A. A third treatment arm that was not included in this analysis of pooled data, BBOS 1.0%, accounted for 80 of these subjects and has been previously discussed.33,34 The remaining 157 subjects comprised the intent-to-treat population. Of these, 140 subjects completed the study without a significant protocol deviation, and thus this subgroup constituted the per protocol population.
 | Figure 1 Subject disposition. |
Efficacy
Ocular itching
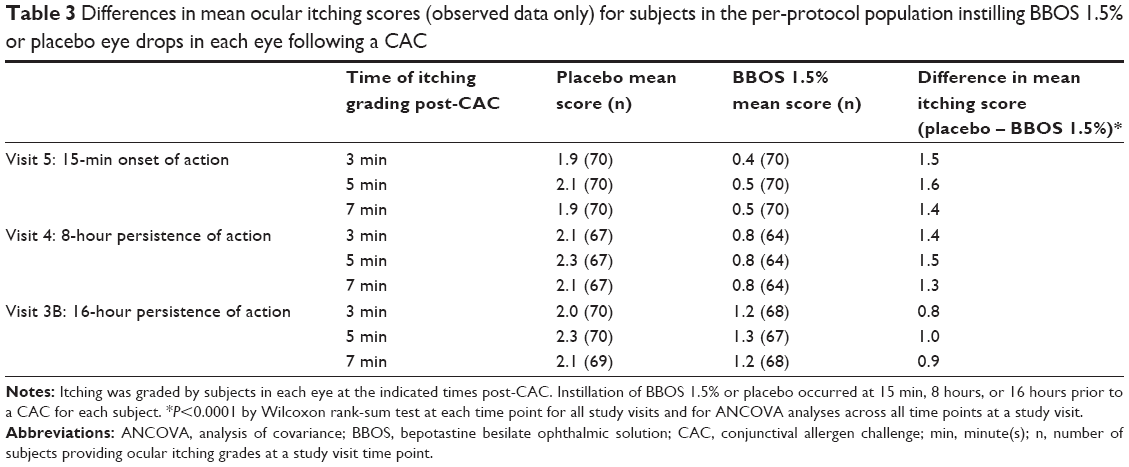
For the PP population, the differences in mean ocular itching scores between the BBOS 1.5% and the placebo group were found to be statistically significant (P<0.001) at all visits and time points by the Wilcoxon rank-sum test and across all time points at a study visit by ANCOVA assessments (Table 3). In addition, the onset and 8-hour persistence-of-action measures were also clinically significant at all time points. A comparison of the difference between onset and 8-hour persistence-of-action scores shows that the treatment effect remained relatively constant over this time period (differences of 1.4–1.6 units at onset, and 1.3–1.5 units at 8 hours). Differences in mean ocular itching scores for BBOS 1.5% compared to placebo for the 16-hour persistence-of-action measures were also statistically significant (P<0.001) at all time points; however, the range of differences did not reach ≥1.0 unit difference at a majority of time points, the standard prospectively designated as clinically significant.
 | Table 3 Differences in mean ocular itching scores (observed data only) for subjects in the per-protocol population instilling BBOS 1.5% or placebo eye drops in each eye following a CAC |
Figure 2A shows the data for ocular itching scores by degree of resolution of itching (itching score =0), and demonstrates that more subjects in the BBOS 1.5% group compared to those in the placebo group experienced complete relief of CAC-induced ocular itching. BBOS 1.5% was statistically superior to placebo at all CAC visits. In addition, the majority of BBOS 1.5% subjects reported zero itching at the onset of action as early as 3 minutes after a CAC. Total itching relief was sustained at the 8-hour persistence-of-action visit for one-third of all subjects. In addition, a sizable number of subjects experienced complete relief from ocular itch even at the 16-hour persistence-of-action visit.
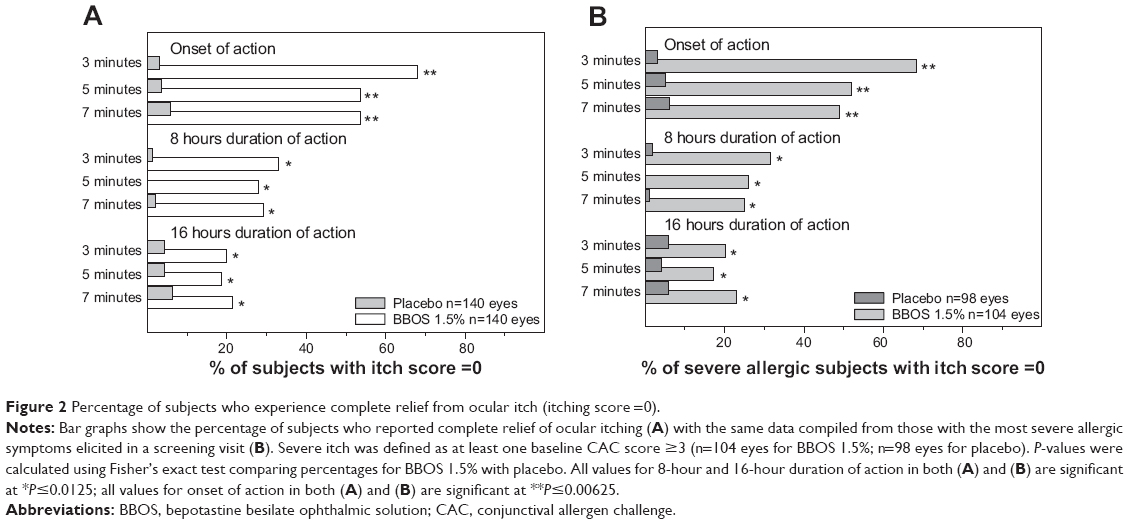 | Figure 2 Percentage of subjects who experience complete relief from ocular itch (itching score =0). |
Figure 2B shows the proportion of severely allergic subjects (baseline [visit 2] ocular itching scores ≥3) who experienced complete resolution of CAC-induced ocular itching. The data demonstrate that the extent of complete relief was significantly better for BBOS 1.5% than for placebo at all study visits and was comparable to the results of the entire PP population, as seen in Figure 2A.
Conjunctival hyperemia
Table 4 shows the differences in mean conjunctival hyperemia scores for the PP population. Numerical reduction in hyperemia scores was superior for BBOS 1.5% compared to placebo at almost all time points. Statistically significant differences were seen at a majority of time points for the onset-of-action CAC test. However, the criterion for clinical significance (≥1.0 unit reduction) was not met at any study visit.
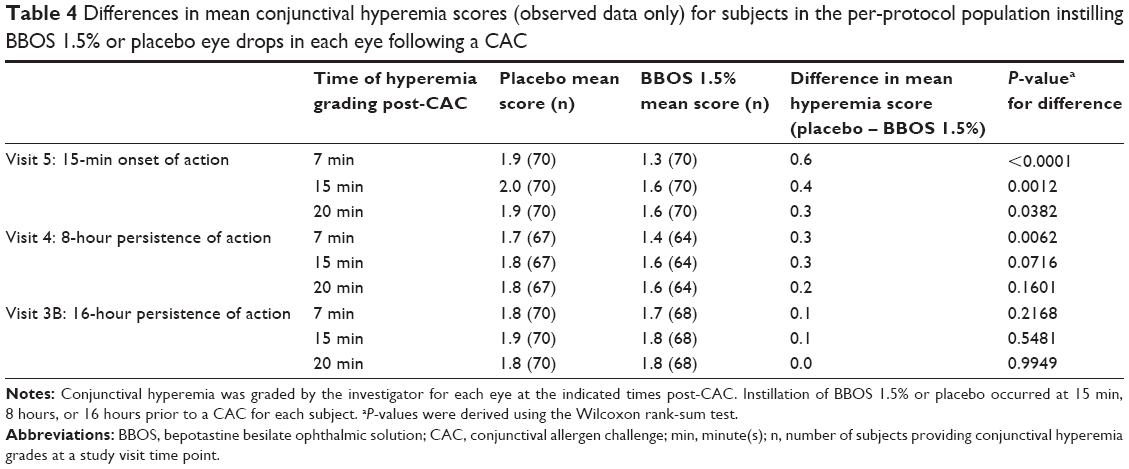 | Table 4 Differences in mean conjunctival hyperemia scores (observed data only) for subjects in the per-protocol population instilling BBOS 1.5% or placebo eye drops in each eye following a CAC |
Secondary efficacy variables
Consistent improvements in several secondary efficacy variables were seen with BBOS 1.5% compared to placebo. Data for ocular tearing were notable and are summarized in Table 5. The percentage of subjects exhibiting tearing in response to a CAC ranged from 23%–39% from onset of action to 16 hours after drug instillation in placebo-treated eyes compared to 6%–15% from onset of action up to 16 hours after drug instillation in BBOS 1.5%-treated eyes. This reduction was both statistically and clinically significant for all time points measured.
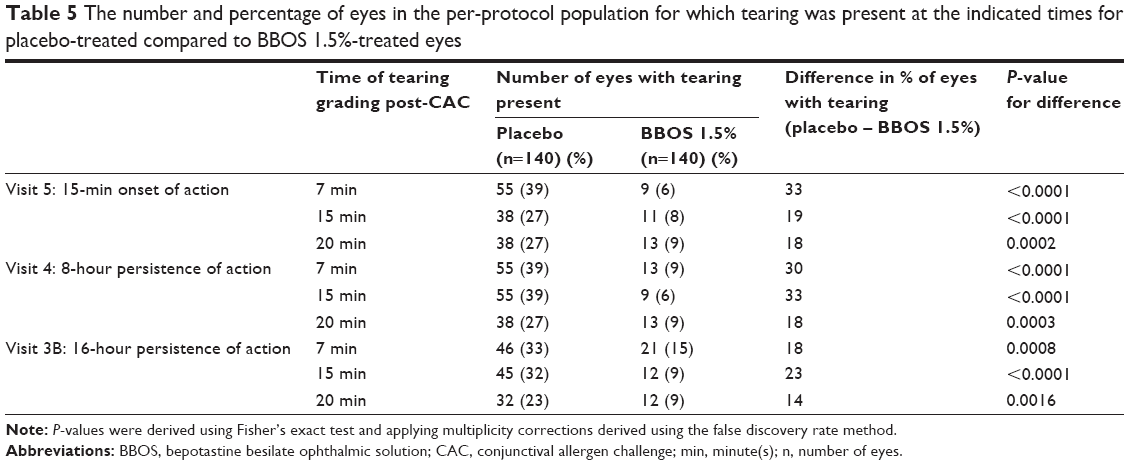 | Table 5 The number and percentage of eyes in the per-protocol population for which tearing was present at the indicated times for placebo-treated compared to BBOS 1.5%-treated eyes |
Additional end points, including ciliary and episcleral hyperemia, chemosis, and eyelid swelling, are summarized in Table 6. All measures showed improvement for BBOS 1.5% compared to placebo based upon reduced mean scores. In particular, all of the end points showed statistically significant reductions by BBOS 1.5% at the onset-of-action CAC test for either all time points (chemosis and eyelid swelling) or the majority of the assessed time points (ciliary and episcleral hyperemia), and the results for ciliary and episcleral hyperemia were very similar to those seen for conjunctival hyperemia. Eyelid swelling was significantly reduced by BBOS 1.5% at all of the 8-hour persistence-of-action time points.
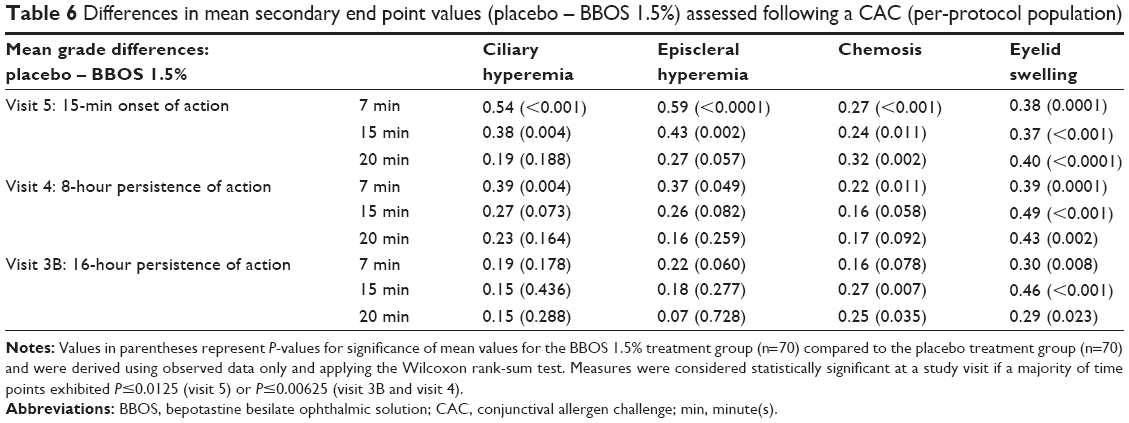 | Table 6 Differences in mean secondary end point values (placebo – BBOS 1.5%) assessed following a CAC (per-protocol population) |
Safety
The AEs from these studies were based upon a safety population of all subjects who received at least one instillation of BBOS 1.5% or placebo. Overall, there were a total of 24 AEs in the PP population reported for the BBOS 1.5% treatment group and ten AEs in the placebo treatment group (Table 7). AEs in the PP population were generally non-ocular and not considered related to the test agent. The most common nonocular AEs included nasopharyngitis (n=3 for both treatment groups) and a mild taste upon instillation for the BBOS 1.5% treatment group (n=4). Among the few AEs that were ocular, the most commonly reported was eye irritation, which was seen in both the placebo (n=2) and BBOS 1.5% (n=3) treatment groups. In addition, visits throughout both CAC trials included an ocular examination by slit-lamp biomicroscopy for each subject, and there were no clinically significant findings reported in the course of these examinations.
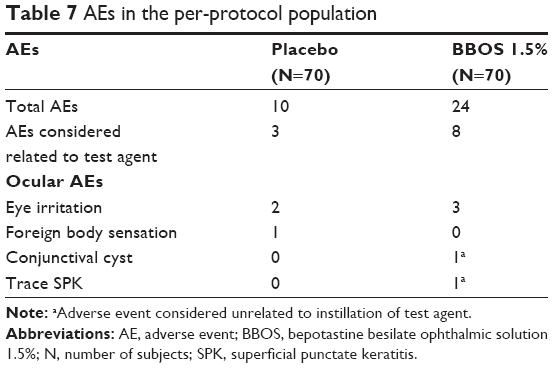 | Table 7 AEs in the per-protocol population |
Discussion
The analysis of this pooled data from two CAC clinical trials was conducted to extend our ability to characterize the efficacy of BBOS 1.5% for the treatment of AC. When analyzed as a single integrated dataset, BBOS 1.5% provided clinically and statistically significant reduction of ocular itching for up to 8 hours post-dosing, as previously reported for the individual CAC trials.33,34 Subjects experienced rapid onset (within 15 minutes) of clinical relief following instillation of drug, as shown by the onset-of-action CACs. The clinical relief was sustained even at 16 hours after dosing, at which the reduction in ocular itching was significantly better and approached a clinically meaningful 1.0 unit difference at all time points (Δ =0.81–0.96 units) with BBOS 1.5% compared to placebo. Ocular itch relief provided by BBOS 1.5% was also sustained at the prospectively defined clinically significant level for at least 8 hours. Overall, these data for the PP population demonstrate that BBOS 1.5% can provide ocular itching relief for up to 16 hours for many allergy sufferers.
To assess the extent to which subjects experience complete relief from ocular itching when exposed to allergens, we examined the percentage of subjects with itch scores of zero at each time point following BBOS 1.5% or placebo instillation. Over one-half (54%–68%) of the subjects in the BBOS 1.5% group reported “complete relief” (no itch) at the onset-of-action visit compared to 3%–6% of those in the placebo group. In the BBOS 1.5%-treated group, 19%–21% of subjects experienced no CAC-induced itch symptoms even 16 hours post-instillation compared to only 4%–6% of the placebo-treated subjects, thus demonstrating the long, persistent effectiveness of BBOS 1.5%. This zero-itch, complete-relief response to BBOS 1.5% was also seen in the more severely allergic subjects. In this group, 68% of the subjects receiving BBOS 1.5% reported zero itching at 3 minutes post-CAC in an onset-of-action CAC test, while 25% of BBOS 1.5% subjects continued to report zero itch 7 minutes post-CAC at 8 hours post-dose; <2% of severely allergic subjects in the placebo treatment group reported complete relief.
As with earlier analyses of conjunctival hyperemia data in CAC trials with BBOS 1.5%,33,34 the assessment of pooled data shows that BBOS 1.5% treatment consistently reduced the extent of hyperemia, and that these reductions were statistically significant at the earliest time points. Overall, BBOS 1.5%-induced hyperemia reductions did not reach the level designated for clinical significance. This result has been seen with other ophthalmic antihistamines using the CAC model41–44 and may be due to the timing of the efficacy assessments as well as a differential response to H1 receptor-specific antihistamines in dilated vascular tissue and ocular nerve endings responsible for itching sensation following a CAC.
The pooled analysis of secondary efficacy variables provides additional power to the assessments, allowing several measures that were not significant in the earlier analyses to reach the level of statistical significance. For example, reductions in lid swelling at 8 hours were significant at all time points in the pooled dataset. Similarly, BBOS 1.5% significantly reduced chemosis and ciliary hyperemia for the onset-of-action CAC tests for the pooled data analyses.
The CAC is a model designed to replicate the accompanying signs and symptoms of AC in a way that is reproducible, quantifiable, and standardized among subject populations that may have differing levels of allergen sensitivity.35 In contrast, studies that depend upon daily fluctuations in environmental allergens are hampered by high variability and the resulting need for a large number of subjects.45 A limitation of the CAC model is that it reproduces neither the exact manner in which patients are typically exposed to allergens in the environment nor the degree or constancy of exposure to environmental allergens. Despite this, the CAC model is able to accurately measure the ability of investigational products to prevent or treat both the early and the late phases of AC36 and has been used to support approval of a number of ophthalmic allergy medications for over 20 years.41–44 The CAC-based trials of BBOS 1.5% provided the basis for the US Food and Drug Administration approval of this ophthalmic solution for treatment of itching associated with AC in September 2009.46
Conclusion
The pooled CAC trial data analysis affirms the usefulness of BBOS 1.5% for treating ocular itching associated with AC in a more substantially sized sample of allergy subjects. These results show that BBOS 1.5% is an effective anti-allergic medication that provides rapid and sustained reduction of ocular itching, even for the most severe AC sufferers. These post hoc results also show that BBOS 1.5% provides statistically significant relief for other allergy-associated ocular symptoms, including tearing, eyelid swelling, and conjunctival hyperemia. In keeping with the statistically significant findings discussed in this paper, additional clinical trials may identify optimal use patterns for improving allergen-induced ocular signs or symptoms with BBOS 1.5%, in addition to ocular itching, and should be considered.
Acknowledgments
The authors would like to acknowledge the following individuals for their assistance in preparation of this manuscript: James T McLaughlin, of Ora (Andover, MA, USA) for manuscript preparation; Kathryn S Kennedy of Statistics and Data Corporation (Tempe, AZ, USA) for statistical analyses; and Mauricio Muñoz and Karen Gertz of Bausch + Lomb Pharmaceuticals (Irvine, CA, USA) for data review and verification. Portions of the data presented here have previously been presented at annual meetings of the American Academy of Ophthalmology, the American Academy of Allergy, Asthma and Immunology, and the Association for Research in Vision and Ophthalmology. The Bepotastine Besilate Ophthalmic Solutions Clinical Study Group comprises: Stacey L Ackerman, MD, Philadelphia Eye Associates, Philadelphia, PA, USA; Mark T Bergmann, MD, Eye Care Associates of Greater Cincinnati, Inc., Cincinnati, OH, USA; Jung T Dao, MD, Cornea Consultants of Arizona, Phoenix, AZ, USA; Fred K Kurata, MD, East West Eye Institute, Los Angeles, CA, USA; Thomas T Macejko, MD, Eye Care Associates of Greater Cincinnati, Inc., Fairfield, OH, USA; Eugene B McLaurin, MD, Total Eye Care, Memphis, TN, USA; Edward J Meier, MD, Eye Care Associates of Greater Cincinnati, Inc., Mason, OH, USA; Clifford M Michaelson, MD, Andover Eye Associates, Andover, MA, USA; Thomas K Mundorf, MD, Mundorf Eye Center, Charlotte, NC, USA; Eugene E Protzko, MD, Seidenberg Protzko Eye Associates, Bel Air, MD, USA; Tushina A Reddy, MD, Ophthalmic Associates, Las Vegas, NV, USA; and Gail L Torkildsen, MD, Andover Eye Associates, Andover, MA, USA.
Disclosure
JI Williams is an employee of Bausch + Lomb. PJ Gomes is an employee of Ora, Inc. The authors have no other conflicts of interest in this work.
References
Collum LMT, Kilmartin DJ. Acute allergic conjunctivitis. In: MB Abelson, editor. Allergic Diseases of the Eye. Philadelphia, PA: WB Saunders Co.; 2000:108–132. | ||
Uchio E, Kimura R, Migita H, Kozawa M, Kadonosono K. Demographic aspects of allergic ocular diseases and evaluation of new criteria for clinical assessment of ocular allergy. Graefes Arch Clin Exp Ophthalmol. 2008;246:291–296. | ||
Abelson MB, Smith L, Chapin M. Ocular allergic disease: mechanisms, disease sub-types, treatment. Ocul Surf. 2003;1:127–149. | ||
Abelson MB, Schaefer K. Conjunctivitis of allergic origin: immunologic mechanisms and current approaches to therapy. Surv Ophthalmol. 1993;38 Suppl:115–132. | ||
Butrus S, Portela R. Ocular allergy: diagnosis and treatment. Ophthalmol Clin North Am. 2005;18:485–492. | ||
Kari O, Salo OP, Halmepuro L, Suvilehto K. Tear histamine during allergic conjunctivitis challenge. Graefe’s Arch Clin Exp Ophthalmol. 1985;223:60–62. | ||
Abelson MB, Udell IJ. H2-receptors in the human ocular surface. Arch Ophthalmol. 1981;99:302–304. | ||
Simons FE. Advances in H1-antihistamines. N Engl J Med. 2004;351: 2203–2217. | ||
Abelson MB, McLaughlin JT, Gomes PJ. Antihistamines in ocular allergy: are they all created equal? Curr Allergy Asthma Rep. 2011;11: 205–211. | ||
Narita H, Kikuchi M, Asahi T, et al. [General pharmacology of betotastine besilate (TAU-284), a novel antiallergic agent]. Jpn Pharmacol Ther. 1997;25:907–924. Japanese. | ||
Tashiro M, Duan X, Kato M, et al. Brain histamine H1 receptor occupancy of orally administered antihistamines, bepotastine and diphenhydramine, measured by PET with 11C-doxepin. Br J Clin Pharmacol. 2008;65:811–821. | ||
Yato N, Murata T, Saito N, et al. [Anti-allergic activity of betotastine besilate (TAU-284), a new anti-allergic drug]. Nihon Yakurigaku Zasshi. 1997;110:19–29. Japanese. | ||
Sakai A, Kikuchi M, Yato N, Kikkawa S, Saito N, Narita H. Inhibitory effect of betotastine besilate on antigen-induced airway eosinophil infiltration and peripheral blood eosinophilia in mice. Arzneimittelforschung. 1997;47:954–958. | ||
Ukai K, Takeuchi M, Masuda S, et al. [Clinical pharmacological study of anti-allergic agent TAU-184 (bepotastine besilate) – the effect on counting of eosinophils in nasal discharge, and the patency improvement of nasal cavity]. J Clin Ther Med. 1997;13:1401–1412. Japanese. | ||
Ueno M, Inagaki N, Nagai H, Koda A. Antiallergic action of betotastine besilate (TAU-284) in animal models: a comparison with ketotifen. Pharmacology. 1998;57:206–214. | ||
Honda H, Murata K, Hamazaki A, et al. [Inhibitory effect on anaphylactic reaction and histamine antagonizing action [in guinea pigs] of betotastine besilate (TAU-284), a novel anti-allergic drug]. Jpn Pharmacol Ther. 1997;25:879–888. Japanese. | ||
Sakamoto O, Okanari E, Ueno H. [Suppression effects of the novel drug bepotastine besilate (TAU-284) on experimental asthmatic reactions in guinea pigs]. Jpn Pharmacol Ther. 1997;25:889–894. Japanese. | ||
Andoh T, Kuraishi Y. Suppression by bepotastine besilate of substance P-induced itch-associated responses through the inhibition of the leukotriene B4 action in mice. Eur J Pharmacol. 2006;547:59–64. | ||
Kida T, Fujii A, Sakai O, et al. Bepotastine besilate, a highly selective histamine H(1) receptor antagonist, suppresses vascular hyperpermeability and eosinophil recruitment in in vitro and in vivo experimental allergic conjunctivitis models. Exp Eye Res. 2010;91:85–91. | ||
Ohashi Y. [Immunological study of a second-generation antihistamine – the effects of bepotastine besilate on cytokine production from the peripheral blood mononuclear cells in cedar pollen allergy patients]. Prog Med. 2004;24:157–159. Japanese. | ||
Ishibashi Y, Harada S, Niimura M, et al. [Early phase II study of TAU- 284 (betotastine besilate) on chronic urticaria]. J Clin Ther Med. 1997;13:1199–1215. Japanese. | ||
Ishibashi Y, Harada S, Niimura M, et al. [Late phase II study of TAU-284 (betotastine besilate) on chronic urticaria – optimal dose finding study by double-blind technique]. J Clin Ther Med. 1997;13:1237–1257. Japanese. | ||
Ishibashi Y, Harada S, Niimura M, et al. [Clinical evaluation of TAU- 284 (betotastine besilate) on eczema/dermatitis, prurigo, and pruritus cutaneus]. J Clin Ther Med. 1997;13:1383–1400. Japanese. | ||
Adachi J. [Investigation of the clinical effects and safety of bepotastine besilate (Talion tablets) on patients with chronic hives]. Prog Med. 2004;24:151–155. Japanese. | ||
Shikanai K. [Investigation of effectiveness and safety of bepotastine besilate (Talion tablets) in cedar pollen allergy]. Prog Med. 2002;22:2472–2477. Japanese. | ||
Kawana S, Niimi Y, Higashi N, et al. [Efficacy and safety investigation of bepotastine besilate (Talion tablets) in patients with atopic dermatitis]. J New Rem Clin. 2005;54:1325–1331. Japanese. | ||
Talion® (bepotastine besilate) tablets. Osaka: Mitsubishi Tanabe Pharma Corporation (formerly Tanabe Seiyaku Co., Ltd.); 2009. | ||
Ohta N, Akatsuka N, Saito F, et al. [The effect of prophylactic treatment with bepotastine in patients with Japanese cedar pollinosis]. Practica Otologica. 2002;95:531–537. Japanese. | ||
Baba S, Sakakura Y, Iwata S, et al. [Early phase II study of TAU-284 (betotastine besilate) on perennial allergic rhinitis]. J Clin Ther Med. 1997;13:1217–1235. Japanese. | ||
Baba S, Takasaki T, Baba K, et al. [Late phase II clinical study of TAU-284 for perennial allergic rhinitis – dose finding study by the double-blind method]. J Clin Ther Med. 1997;13:1259–1286. Japanese. | ||
Ito M, Akaogi K, Shimizu KA, et al. [A clinical study of anti-allergy drugs in relation to allergic rhinitis]. Allergol Immunol. 2003;10:1328–1337. Japanese. | ||
Baba S. [Post-marketing surveillance study of Talion tablets on pediatric allergic rhinitis]. Rinsho Igaku. 2002;18:1371–1387. Japanese. | ||
Abelson MB, Torkildsen GL, Williams JI, Gow JA, Gomes PJ, McNamara TR; Bepotastine Besilate Ophthalmic Solutions Clinical Study Group. Time to onset and duration of action of the antihistamine bepotastine besilate ophthalmic solutions 1.0% and 1.5% in allergic conjunctivitis: a phase III, single-center, prospective, randomized, double-masked, placebo-controlled, conjunctival allergen challenge assessment in adults and children. Clin Ther. 2009;31:1908–1921. | ||
Macejko TT, Bergmann MT, Williams JI, et al; Bepotastine Besilate Ophthalmic Solutions Clinical Study Group. Multicenter clinical evaluation of bepotastine besilate ophthalmic solutions 1.0% and 1.5% to treat allergic conjunctivitis. Am J Ophthalmol. 2010;150:122–127. | ||
Abelson MB, Chambers WA, Smith LM. Conjunctival allergen challenge. A clinical approach to studying allergic conjunctivitis. Arch Ophthalmol. 1990;108:84–88. | ||
Abelson MB, Loeffler O. Conjunctival allergen challenge: models in the investigation of ocular allergy. Curr Allergy Asthma Rep. 2003;3:363–368. | ||
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonized tripartite guideline. Guideline for good clinical practice. Available from http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf. | ||
World Medical Association Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. 1989 Version. http://www.wma.net/en/30publications/10policies/b3/17c.pdf. Accessed 2 July 2014. | ||
International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) MedDRA® the Medical Dictionary for Regulatory Activities. A registered trademark of the International Federation of Pharmaceutical Manufacturers and Associations (IFPMA). Chantilly, VA: Northrop Grumman MSSO (distributors); 2009. Available from http://www.meddra.org/how-to-use/support-documentation/english. Accessed 2 July 2014. | ||
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. JR Statist Soc B. 1995;57:289–300. | ||
Abelson M, Howes J, George M. The conjunctival provocation test model of ocular allergy: utility for assessment of an ocular corticosteroid, loteprednol etabonate. J Ocul Pharmacol Ther. 1998;14:533–542. | ||
Abelson MB, Spitalny L. Combined analysis of two studies using the conjunctival allergen challenge model to evaluate olopatadine hydrochloride, a new ophthalmic antiallergic agent with dual activity. Am J Ophthalmol. 1998;125:797–804. | ||
Abelson MB, Chapin MJ, Kapik BM, Shams NB. Efficacy of ketotifen fumarate 0.025% ophthalmic solution compared with placebo in the conjunctival allergen challenge model. Arch Ophthalmol. 2003;121: 626–630. | ||
Abelson MB, Gomes P, Crampton HJ, Schiffman RM, Bradford RR, Whitcup SM. Efficacy and tolerability of ophthalmic epinastine assessed using the conjunctival antigen challenge model in patients with a history of allergic conjunctivitis. Clin Ther. 2004;26:35–47. | ||
Abelson MB. Comparison of the conjunctival allergen challenge model with the environmental model of allergic conjunctivitis. Acta Ophthalmol Scand. 1999;228(Suppl):43–46. | ||
Bepreve® (bepotastine besilate ophthalmic solution) 1.5% [package insert]. Irvine, CA: Bausch + Lomb Pharmaceuticals, Inc; 2012. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.