")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Treatment discontinuation and tolerability as a function of dose and titration of duloxetine in the treatment of major depressive disorder
Authors Harada E, Shirakawa O, Satoi Y, Marangell L, Escobar R
Received 14 April 2015
Accepted for publication 2 October 2015
Published 11 January 2016 Volume 2016:12 Pages 89—97
DOI https://doi.org/10.2147/NDT.S86598
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Wai Kwong Tang
Eiji Harada,1 Osamu Shirakawa,2 Yoichi Satoi,3 Lauren B Marangell,4 Rodrigo Escobar5
1Eli Lilly Japan K.K., Medicines Development Unit Japan, Medical Science, Kobe, 2Department of Neuropsychiatry, Kinki University Faculty of Medicine, Osakasayama, 3Eli Lilly Japan K.K., Medicines Development Unit Japan, Statistical Science, Kobe, Japan; 4The University of Texas Health Science Center School of Medicine, Houston, TX, 5Eli Lilly and Company, Neuroscience, Indianapolis, IN, USA
Purpose: We sought to better understand how dose and titration with duloxetine treatment may impact tolerability and treatment discontinuation in patients with major depressive disorder.
Patients and methods: We investigated Phase III duloxetine trials. Group 1 was a single placebo-controlled study with a 20 mg initial dose and a slow titration to 40 and 60 mg. Group 2 was a single study with a 40 mg initial dose and final “active” doses of 40 and 60 mg (5 mg control group), with 1-week titration. Group 3 consisted of eight placebo-controlled studies with starting doses of 40, 60, and 80 mg/day with minimal titration (final dose 40–120 mg/day). Tolerability was measured by rate of discontinuation due to adverse events (DCAE).
Results: The DCAE in Group 1 were 3.6% in the 60 mg group, 3.3% in the 40 mg group, and 3.2% in the placebo group. In Group 2, the DCAE were 15.0% in the 60 mg group, 8.1% in the 40 mg group, and 4.9% in the 5 mg group. In Group 3, the DCAE were 9.7% and 4.2% in the duloxetine and placebo groups, respectively.
Conclusion: This study suggests that starting dose and titration may have impacted tolerability and treatment discontinuation. A lower starting dose of duloxetine and slower titration may contribute to improving treatment tolerability for patients with major depressive disorder.
Keywords: antidepressant, dose, duloxetine, major depressive disorder, titration
Introduction
Major depressive disorder (MDD) affects more than 350 million people, of all ages, worldwide1 and is a significant contributor to the global burden of disease. Depression is expected to become the second-leading cause of disability by 2020,2 and untreated depression may lead to increased morbidity and mortality.3 Globally, there is an increase in the demand to take action to curb depression.4,5
One of the difficulties of treating MDD with antidepressants is early discontinuation of treatment. Although continuing therapy for 4 to 9 months is recommended for resolution of a depressive episode,6 and patients who continue therapy with their initial antidepressant are least likely to experience relapse or recurrence of a depressive episode,7 up to 68% of patients will discontinue antidepressant treatment by 3 months.8 Hence, staying on treatment for the entire prescribed time is important to make the most of psychopharmacological treatment.
Discontinuation may be affected by a variety of reasons, one of which is the patient’s experience with side effects.9 Adverse events associated with antidepressant medications hinder the patients’ ability to continue taking pharmacological treatment.8,10 In an observational study of patients with MDD treated with a selective serotonin reuptake inhibitor, side effects were the leading cause of patients discontinuing treatment after 3 months.10 In another study, up to 62% of patients who discontinued their medication cited the medication’s side effects as one of the reasons for doing so.11 An inability to complete a treatment regimen or early treatment discontinuation leads to worsening of a patient’s condition and subsequent failure to achieve treatment goals.6 One way to reduce adverse events that can lead to discontinuation, and thereby increase the continuation rate, may be to titrate the antidepressant up to a therapeutic dose so that patients can maximize initial tolerability to the drug.12,13
Duloxetine is a serotonin and norepinephrine reuptake inhibitor that is efficacious in the treatment of MDD.14–17 In randomized, double-blind, placebo-controlled studies, duloxetine has been well tolerated, with mild-to-moderate adverse events,18,19 and has demonstrated comparable safety with other antidepressants.20,21 While a previous study evaluated the impact of starting duloxetine at either 30 or 60 mg once daily for MDD and found comparable tolerability,22 this study had the intrinsic limitations of an observational study such as the absence of patient randomization to each dose and titration, a large proportion of patients dropping out of the study, and lack of information about reasons for discontinuation. In this regard, clinical trials, which have more rigorous methods of evaluation, may provide a different lens through which to view tolerability. In addition, to the best of our knowledge, there is no publication available that has evaluated the impact of starting duloxetine at either 20 or 40 mg once daily for MDD.
In this article, we primarily examine the results of two Japanese registration clinical trials of duloxetine and aim to better understand how differences of dose and titration methods in duloxetine treatment can impact tolerability. In addition, in order to gain insights on the possible influence of ethnicity on the results, we also examine other global placebo-controlled studies of duloxetine to determine if ethnicity may have an impact on tolerability.
Materials and methods
Data were examined from an integrated database of Phase III clinical trials for registration submission in the following country/region: two Japan registration trials for duloxetine in the treatment of MDD, and eight double-blind, placebo-controlled trials that formed the basis of the US New Drug Application or the European Union (EU) submission package. The protocols for the individual studies were reviewed and approved by the applicable organizational ethical review boards. The studies were conducted in accordance with the ethical principles of the Declaration of Helsinki, Good Clinical Practice, and applicable laws and regulations.
The first data source, Group 1, was the Japan registration trial (F1J-MC-HMFH) for duloxetine in the treatment of MDD. The dose and titration used in this study formed the basis of the approved labeling in Japan. As detailed in Higuchi et al,23 this Phase III, randomized, double-blind, parallel-group, comparative study was conducted in patients with MDD. This study consisted of a duloxetine 60 mg group (n=84), duloxetine 40 mg group (n=91), paroxetine 20 to 40 mg group (n=164), and the placebo group (n=156). This study had a 1- to 2-week pretreatment observation period, followed by a 7-week treatment period (duration: 9 weeks [1-week placebo lead-in, 6-week drug exposure, 2-week gradual reduction period]). The study drug was administered once daily for a total of 9 weeks including the treatment period and gradual reduction period. After 1-week administration of placebo, patients were randomly assigned to one of four treatment groups. In the duloxetine 60 mg group, 20 mg of duloxetine was administered as the initial dose for 1 week, and the dose was increased by forced dose escalation to 40 mg, and again to 60 mg after 1 week of 40 mg treatment. The dose of 60 mg was fixed as the maintenance dose for the subsequent treatment. In the duloxetine 40 mg group, 20 mg of duloxetine was administered as the initial dose for 1 week, and the dose was increased to 40 mg by forced dose escalation. The 40 mg dose was fixed as the maintenance dose for the subsequent treatment.
The second data source, Group 2, was the other Japan registration trial (F1J-JE-HMEC) for duloxetine in the treatment of MDD. As detailed in Higuchi et al,24 this Phase III, randomized, double-blind, parallel-group, comparative study was conducted in patients with MDD. The patients were administered an ineffective, 5 mg dose of duloxetine as a pseudo-placebo control to verify the superiority of duloxetine 40 and 60 mg dose regimens. All patients started treatment at 40 mg/day except patients who were administered a 5 mg dose of duloxetine. Duloxetine was administered once daily for 8 weeks in these patients. For patients assigned to the 60 mg group, duloxetine was administered at 40 mg/day for 1 week and at 60 mg/day from weeks 2 to 8. For patients assigned to the 5 and 40 mg groups, the doses were fixed at 5 and 40 mg/day, respectively, for the entire 8-week treatment period.
The third data source, Group 3, was from an integrated data base that included patient-level data from eight double-blind, placebo-controlled trials (F1J-MC-HMBHa, F1J-MC-HMBHb, F1J-MC-HMAQa, F1J-MC-HMAQb, F1J-MC-HMATa, F1J-MC-HMATb, F1J-MC-HMAYa, F1J-MC-HMATb) in which patients with MDD were randomized to either duloxetine (maximum dose of 40–120 mg/day; n=1,139) or placebo (n=777) for up to 9 weeks.25 These eight studies constituted all acute- and continuation-phase, placebo-controlled trials that were included in either the US New Drug Application or the EU submission package for duloxetine in the treatment of MDD. Six of the studies were conducted in the USA and two were conducted in Eastern Europe. The doses and titration used in several of the studies differ from the approved labeling in the USA and EU. As detailed in Hudson et al,25 in study 1 (duloxetine, n=123; placebo, n=122) and study 2 (duloxetine, n=128; placebo, n=139), duloxetine was administered at 60 mg once daily. In study 3 (duloxetine, n=70; placebo, n=70) and study 4 (duloxetine, n=82; placebo, n=75), duloxetine was administered as a forced titration from 20 to 60 mg twice daily over a period of 3 weeks. In study 5 (duloxetine 80 mg, n=84; duloxetine 40 mg, n=91; placebo, n=90) and study 6 (duloxetine 80 mg, n=91; duloxetine 40 mg, n=86; placebo, n=89), duloxetine was administered at 20 or 40 mg twice daily (no titration). In study 7 (duloxetine 120 mg, n=93; duloxetine 80 mg, n=95; placebo, n=93) and study 8 (duloxetine 120 mg, n=103; duloxetine 80 mg, n=93; placebo, n=99), duloxetine 120 mg/day was administered as a titration from 20 mg twice daily (3 days) to 40 mg twice daily (3 days) to 60 mg twice daily, and duloxetine 80 mg/day was administered as a titration from 20 mg twice daily (3 days) to 40 mg twice daily. In summary, the initial dose was 40 mg/day for 63% of the patients and 60 or 80 mg/day for 37% of the patients. A total of 53% of the patients received no titration period, 34% received 3 days titration, and only 13% of the patients received slower titration. The maximum fixed dose was 40 mg/day for 16%, 60 mg/day for 22%, 80 mg/day for 32%, and 120 mg/day for 31% of the patients in Group 3. The doses for the data sources are shown in Table 1.
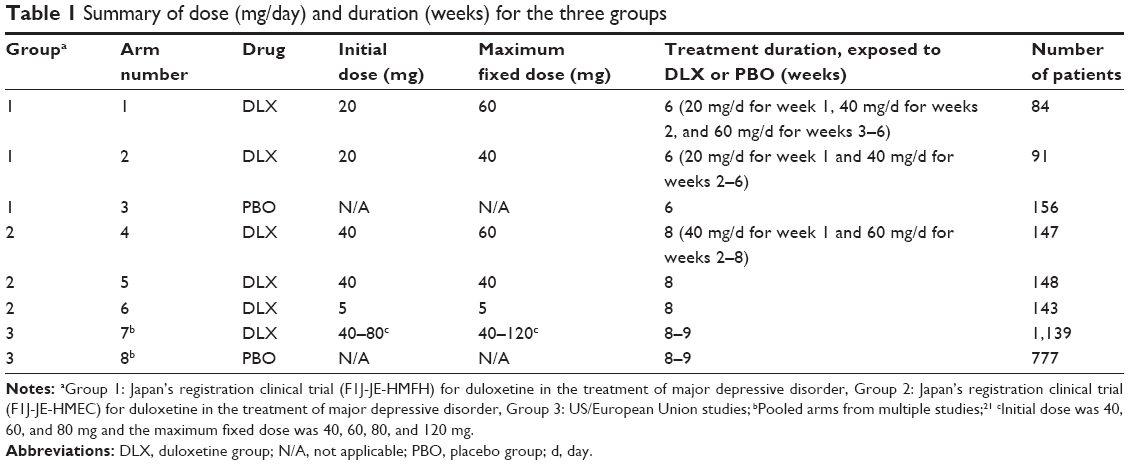 | Table 1 Summary of dose (mg/day) and duration (weeks) for the three groups |
In Groups 1 and 2, enrolled patients had to meet the criteria for depression according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) classification26 for a single episode of MDD or recurrent MDD, which was further classified as mild, moderate, severe without psychotic features, or severe with psychotic features. In both studies, patients had to have a 17-item Hamilton Rating Scale for Depression (HAMD17)27 total score ≥19 and a HAMD17 score for depressed mood ≥2 points at baseline or enrollment. In addition, patients had to be ≥20 to <65 years of age in Group 1 and ≥20 to <70 years of age in Group 2.
In Group 3, enrolled patients had to be at least 18 years of age, have a current primary diagnosis of MDD as defined by the DSM-IV, and have an HAMD17 total score of ≥15 and a Clinical Global Impression of severity28 score ≥4 at the screening and baseline study visits. The maximum age of patients in studies 3 and 4 was 65 years, whereas the other studies had no restriction on maximum age.
Patient demographics were examined for each treatment group in all of the data sources. Safety was defined by the rate of adverse events, and tolerability was defined by the rate of discontinuation due to adverse events (DCAE). An adverse event was defined as any untoward medical occurrence that did not necessarily have a causal relationship with the treatment. A treatment-emergent adverse event (TEAE) was an adverse event that occurred, was newly developed, or when a precondition worsened. The adverse events were coded using Medical Dictionary for Regulatory Activities version 10.0, 8.1, and 5.0 in Groups 1, 2, and 3, respectively.
Statistical methods
Analyses were performed according to the intent-to-treat principle. In Group 1, the incidence of adverse events was compared between each duloxetine dose and the placebo group among the treatment groups. In Group 2, the incidence of adverse events was compared between the 5 and 60 mg groups and between the 5 and 40 mg groups. In Group 3, the incidence of adverse events was compared between duloxetine and placebo.
Case numbers were adopted for the safety analysis population defined in each study. The incidence of adverse events was defined as the proportion of patients with adverse events in the safety analysis population; the number of patients with adverse events was tabulated by the event type, and the percentage of patients with a specific adverse event was calculated. Only adverse events spontaneously reported by the patients were used in the analyses. Fisher’s exact test was used for comparison. Statistical tests were performed only for comparisons between treatment arms within each group. All comparisons were made by using two-sided tests at α=0.05 level.
Results
Patient demographics
Patient demographics for the studies are summarized in Table 2. In Groups 1 and 2, all patients were East/Southeast Asian, had a mean age of 38 to 39 years in both groups, and approximately 44% to 51% were female. In Group 3, most patients were Caucasian, there were a higher proportion of females than males, and patients had a mean age of 42.5 years. There were no significant differences between treatment arms in each group for demographics and patient baseline characteristics.
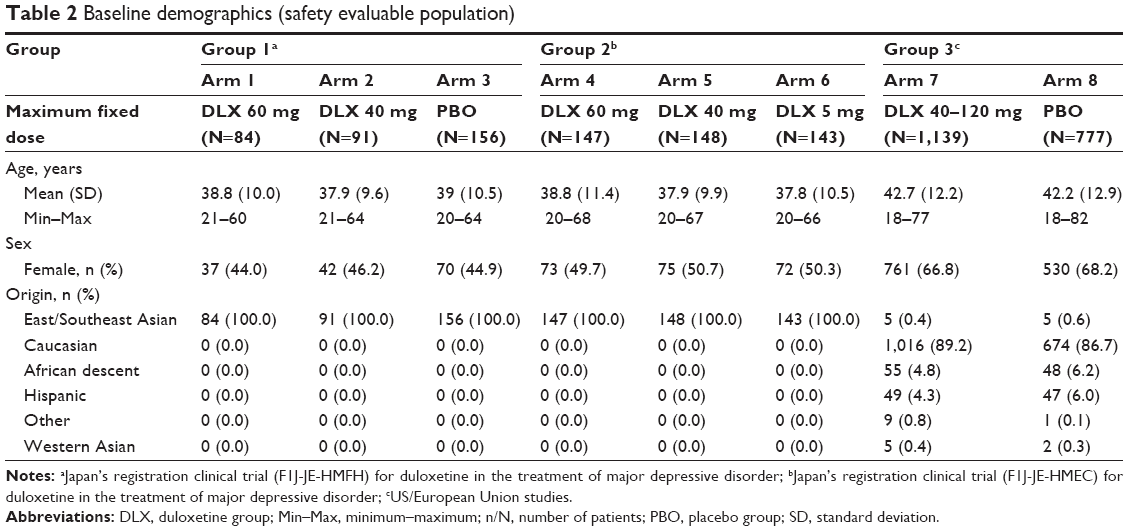 | Table 2 Baseline demographics (safety evaluable population) |
Treatment-emergent adverse events
The incidence of TEAEs that occurred in ≥5% of duloxetine-treated patients and at a rate greater than placebo is summarized in Table S1. In Group 1, the percentage of patients who experienced ≥1 TEAE was 86.9% in the 60 mg group, 87.9% in the 40 mg group, and 78.2% in the placebo group. Nausea was the most frequently reported adverse event in the 60 mg group, occurring in 34.5% (P≤0.001 vs placebo group) of patients compared with 19.8% of patients receiving duloxetine 40 mg (P=0.0321 vs placebo group) and 9.6% of patients receiving placebo. Other adverse events occurring in ≥5% of the 60 mg group and at a significantly higher rate than in the placebo group were somnolence, constipation, insomnia, thirst, and decreased appetite; adverse events occurring in ≥5% of the 40 mg group and at a significantly higher rate than in the placebo group were somnolence, nausea, thirst, dizziness, stomach discomfort, postural dizziness, and pharyngolaryngeal pain.
In Group 2, the percentage of patients who experienced ≥1 TEAE was 96.6% in the 60 mg group, 95.3% in the 40 mg group, and 88.1% in the 5 mg group. Nausea was the most frequently reported adverse event in both groups (38.1% of patients in the 60 mg group and 48.0% of patients in the 40 mg group). Other adverse events occurring in ≥5% in both duloxetine 60 and 40 mg groups were thirst, somnolence, headache, constipation, nasopharyngitis, diarrhea, decreased appetite, dizziness, anorexia, and malaise.
In Group 3, 73.4% of duloxetine-treated patients and 65.1% of placebo-treated patients experienced ≥1 TEAE. Nausea was the most frequently reported TEAE that occurred in ≥5% of duloxetine-treated patients (19.9% vs 6.9% in the placebo group; P≤0.001). Other adverse events occurring in duloxetine-treated patients that were significantly higher than those reported in placebo-treated patients were dry mouth, constipation, insomnia, dizziness, fatigue, somnolence, hyperhidrosis (increased sweating), and decreased appetite.
Rate of discontinuation due to adverse events
In Group 1, DCAE were 3.6% of patients in the 60 mg group, 3.3% in the 40 mg group, and 3.2% in the placebo group. In the 40 and 60 mg pooled groups, DCAE were 3.4%. In Group 2, DCAE were 15.0% in the 60 mg group, 8.1% in the 40 mg group, and 4.9% in the 5 mg group; the DCAE were significantly higher in the 60 mg group than in the 5 mg group (P=0.011). In the 40 and 60 mg pooled groups, DCAE were 11.5%. In Group 3, DCAE were 9.7% in the duloxetine group and 4.2% in the placebo group (P<0.001) (Figure 1).
 | Figure 1 Rate of discontinuation due to adverse events. |
Rate of discontinuation due to specific adverse events
Nausea was the adverse event that most frequently resulted in discontinuation before the end of the preplanned treatment among duloxetine-treated patients in Group 2 (60 mg group =6.0%, 40 mg group =4.7%, 5 mg group =0.7%; P<0.05 for 60 mg group vs 5 mg group) and Group 3 (duloxetine group =1.4%, placebo group =0.1%; P<0.01). In Group 1, no significant differences were found in the incidence of adverse events leading to discontinuation between the placebo group and the 60 or 40 mg group. Additionally, in Group 1, there were no significant differences found in the incidence of nausea leading to discontinuation between the placebo group (0.0%) and the duloxetine 60 mg (0.0%) or 40 mg (1.1%) group.
Dizziness was the adverse event leading to discontinuation that occurred in ≥0.5% of duloxetine-treated patients in Group 1 (in at least one of the 40 or 60 mg arm), Group 2 (in at least one of the 40 or 60 mg arm), and Group 3. Other than nausea and dizziness, patterns of adverse events leading to discontinuation varied from group to group and there was no clear trend identified.
Discussion
Our results show that different starting doses and titration methods for duloxetine may result in different tolerability profiles. We explored this relationship using individual patient data collected from clinical trials. Among these three data sources, there were several similarities (eg, clinical trial phase, target disease and its severity, age, and sex) and differences (eg, initial dose, titration, and ethnicities) in their study design and target population. Here, we aimed to understand how dose, titration, and ethnicity may impact tolerability as measured by DCAE.
First, we compared arms within Groups 1 and 2 to assess the impact of starting dose on DCAE. In Group 1, all duloxetine-treated patients started at 20 mg/day and increased to 40 mg/day 1 week later, and approximately half of the patients were titrated up to 60 mg/day after another week. The DCAE in the duloxetine 60 or 40 mg arm were comparable with the DCAE in the placebo arm (3.6%, 3.3%, and 3.2%, respectively). In contrast, in Group 2, all patients (except the duloxetine 5 mg group) started at 40 mg/day, and approximately half of the patient doses were titrated up to 60 mg/day 1 week later. The DCAE in the duloxetine 60 or 40 mg arm indicated a higher trend than the DCAE in the pseudo-placebo arm (15.0%, 8.1%, and 4.9%, respectively). These results suggest that a low, subtherapeutic starting dose (ie, 20 mg/day) with a slow titration schedule (ie, weekly by 20 mg) may minimize the DCAE to a level similar to placebo, whereas a relatively high starting dose (ie, 40 mg/day) may fail to do so, regardless of its titration schedule.
Second, we compared arms across Groups 1 and 2 to assess the impact of maximum dose on DCAE. These two studies had many similar factors that may justify our comparison: clinical trial phase, target disease, and its severity, sex, age, and ethnicity (ie, East/Southeast Asian population). The DCAE in the patients titrated to 60 mg/day were 3.6% and 15.0% for Groups 1 and 2, respectively, and the DCAE in the patients titrated to 40 mg/day were 3.3% and 8.1% for Groups 1 and 2, respectively. Overall, patient groups treated with the same maximum dose but lower starting dose and slower titration resulted in lower DCAE. Thus, we may conclude that starting dose and titration, rather than the final maximum dose, may attribute more to determining treatment tolerability.
Previous studies12,13 reported that starting duloxetine treatment at 30 mg/day was associated with a reduction in treatment-emergent nausea in patients taking duloxetine when compared with patients starting duloxetine treatment at 60 mg/day. In contrast, Wilhelm et al22 concluded that initiation of duloxetine treatment with 60 mg/day was not associated with poor tolerability when compared with 30 mg/day of duloxetine. However, in this observational study, a considerable proportion of patients dropped out and their information regarding the reason for discontinuation was not available.
Third, we compared arms across Groups 2 and 3 to gain insight into how difference in ethnicity may impact DCAE. These two studies had similarities in some factors (eg, clinical trial phase, target disease and its severity, age) but had a major difference in ethnicity; all the patients were East/Southeast Asian in Group 2, but the patients were primarily Caucasian in Group 3. Dose and titration were similar to some extent; all patients in the duloxetine 40 and 60 mg groups in Group 2 started duloxetine treatment at 40 mg and half of them were titrated up to 60 mg 1 week later, whereas most patients in Group 3 started duloxetine treatment at 40 mg/day and approximately half of the patients underwent a titration period. As a result, the DCAE were similar; 11.5% for the 60 and 40 mg pooled groups in Group 2 and 9.7% in Group 3. Moreover, the DCAE in the placebo (or “pseudo-placebo”) arms were similar: 4.9% in Group 2 and 4.2% in Group 3. Overall, studies of totally different ethnicity resulted in a similar DCAE. This may indicate that ethnicity does not solely attribute to determination of tolerability of antidepressants in these trials.
Previous studies reported limited impact of ethnicity on tolerability to antidepressants: DCAE did not differ significantly between Caucasian (17.0%) and Hispanic (14.0%) patients29 or Caucasians (17.0%) and African Americans (13%).30 One study did report higher rates of some adverse events in Caucasian patients than non-Caucasian patients taking duloxetine.31 In our research, it is difficult to determine the extent to which ethnicity played a role in tolerability. Therefore, further research will be needed to provide clarification on this point in a quantitative manner.
Careful consideration should be given when we interpret study results of which dose and titrations employed are different from our clinical practice, and when we apply the study results to daily clinical practice in the treatment of MDD. Meta-analyses that compared the efficacy and acceptability of 12 new-generation antidepressants concluded that the acceptability profile of duloxetine was inferior to that of escitalopram and sertraline.32 However, that meta-analysis used studies performed in Europe and North America with duloxetine doses ranging from 40 to 120 mg/day14,17,18,33–36 and with little consideration to dose titration, whereas starting with a subtherapeutic dose (ie, 30 mg/day) followed by escalation to therapeutic dose has become a standardized procedure in most recent clinical trials.22,37,38 A difference in starting dose and titration method may result in different tolerability, and this needs to be kept in mind when interpreting the study results. It should be noted that by modifying the dose and titration according to patient conditions, clinicians may be able to better manage adverse events experienced by a patient taking duloxetine, which may lead to more tolerable pharmacological treatment of MDD.
The strength of this study was that each study was a Phase III registration clinical trial. These trials were performed under close observation and the obtained information was well documented, which enables us to provide clinical implication to daily practice. However, this study is limited mainly by a small number of studies and sample size according to the study inclusion criteria; the main conclusion of this manuscript was drawn by comparing the results of two separate clinical trials that had similar factors, such as the clinical trial phase, target disease and disease severity, sex, age, and ethnicity. In addition, this study focused primarily on treatment discontinuation; time to discontinue the treatment or treatment efficacy was not compared. Furthermore, statistical tests were performed only for comparisons between treatment groups within each study and statistical significance across studies remains unknown. On another note, though we focused on dose and titration as a main driver for tolerability, it is possible that the differences in study designs beyond dose and titration (eg, pretreatment observation, placebo lead-in) may have impacted tolerability.
Conclusion
In conclusion, this study showed that starting dose and titration may have impacted tolerability and treatment discontinuation. It is suggested that a lower starting dose and a slower dose titration schedule with duloxetine may contribute to improving the treatment tolerability for patients with MDD.
Acknowledgments
This work was sponsored by Eli Lilly and Company. The authors would like to thank Deborah D’Souza, PhD, of inVentiv Health Clinical, LLC, who provided medical writing support on behalf of Eli Lilly and Company.
Disclosure
Dr Harada contributed to this work as a former full-time employee of Eli Lilly Japan K.K. The opinions expressed in this work are solely his and do not represent his current affiliation, the Japanese Ministry of Health, Labour and Welfare. Mr Satoi is an employee of Eli Lilly Japan K.K., and stock holder of Eli Lilly and Company. Dr Escobar is an employee and stock holder of Eli Lilly and Company. Dr Shirakawa has received honoraria from Otsuka Pharmaceutical Co., Ltd., Tanabe Mitsubishi Pharma Corporation, Eli Lilly Japan K.K., Shionogi & Co., Ltd., and GlaxoSmithKline K.K., and has received donations for research from Tanabe Mitsubishi Pharma Corporation. Dr Marangell is a former employee of Eli Lilly and Company. The authors report no further conflicts of interest in this work.
References
Marcus M, Yasany MT, van Ommeren M, Chisholm D, Saxena S. Depression: A global public health concern [Internet]. c.2012 [cited July 1, 2013]. Available from: http://www.who.int/mental_health/management/depression/who_paper_depression_wfmh_2012.pdf. Accessed March 23, 2015. | ||
World Health Organization. The World Health Report 2001 – Mental Health: New Understanding, New Hope. Geneva: World Health Organization; 2001. | ||
Sartorius N, Angstt J. Suicide in population subgroups. Int Clin Psychopharmacol. 2001;16(Suppl 2):2p preceding S1. | ||
World Health Organization. Sixty-fifth world health assembly 2012: Daily notes on proceedings [Internet]. [Updated May 22, 2012; cited July 1, 2013]. Available from: http://www.who.int/mediacentre/events/2012/wha65/journal/en/index4.html. Accessed March 23, 2015. | ||
World Health Organization. Depression. What is depression [Internet]? [Updated 2013; cited July 1, 2013]. Available from: http://www.who.int/mental_health/management/depression/definition/en/print.html. Accessed March 23, 2015. | ||
Qaseem A, Snow V, Denberg TD, et al. Using second-generation antidepressants to treat depressive disorders: a clinical practice guideline from the American College of Physicians. Ann Intern Med. 2008;149:725–733. | ||
Melfi CA, Chawla AJ, Croghan TW, Hanna MP, Kennedy S, Sredl K. The effects of adherence to antidepressant treatment guidelines on relapse and recurrence of depression. Arch Gen Psychiatry. 1998;55(12):1128–1132. | ||
Bull SA, Hu XH, Hunkeler EM, et al. Discontinuation of use and switching of antidepressants: influence of patient-physician communication. JAMA. 2002;288(11):1403–1409. | ||
Masand PS. Tolerability and adherence issues in antidepressant therapy. Clin Ther. 2003;25(8):2289–2304. | ||
Goethe JW, Woolley SB, Cardoni AA, Woznicki BA, Piez DA. Selective serotonin reuptake inhibitor discontinuation: side effects and other factors that influence medication adherence. J Clin Psychopharmacol. 2007;27(5):451–458. | ||
Lin EH, Von Korff M, Katon W, et al. The role of the primary care physician in patients’ adherence to antidepressant therapy. Med Care. 1995;33(1):67–74. | ||
Whitmyer VG, Dunner DL, Kornstein SG, et al. A comparison of initial duloxetine dosing strategies in patients with depressive disorder. J Clin Psychiatry. 2007;68(12):1921–1930. | ||
Dunner DL, Wohlreich MM, Mallinckrodt CG, Watkin JG, Fava M. Clinical consequences of initial duloxetine dosing strategies: comparison of 30 and 60 mg QD starting doses. Curr Ther Res. 2005;66(6):522–540. | ||
Goldstein DJ, Mallinckrodt C, Lu Y, Demitrack MA. Duloxetine in the treatment of major depressive disorder: a double-blind clinical trial. J Clin Psychiatry. 2002;63(3):225–231. | ||
Goldstein DJ, Lu Y, Detke MJ, Hudson J, Iyengar S, Demitrack MA. Effects of duloxetine on painful physical symptoms associated with depression. Psychosomatics. 2004;45(1):17–28. | ||
Detke MJ, Lu Y, Goldstein DJ, Hayes JR, Demitrack MA. Duloxetine, 60 mg once daily, for major depressive disorder: a randomized double-blind placebo-controlled trial. J Clin Psychiatry. 2002;63(4):308–315. | ||
Detke MJ, Wiltse CG, Mallinckrodt CH, McNamara RK, Demitrack MA, Bitter I. Duloxetine in the acute and long-term treatment of major depressive disorder: a placebo- and paroxetine-controlled trial. Eur Neuropsychopharmacol. 2004;14(6):457–470. | ||
Goldstein DJ, Lu Y, Detke MJ, Wiltse C, Mallinckrodt C, Demitrack MA. Duloxetine in the treatment of depression: a double-blind placebo-controlled comparison with paroxetine. J Clin Psychopharmacol. 2004;24(4):389–399. | ||
Detke MJ, Lu Y, Goldstein DJ, McNamara RK, Demitrack MA. Duloxetine 60 mg once daily dosing versus placebo in the acute treatment of major depression. J Psychiatr Res. 2002;36(6):383–390. | ||
Perahia DG, Pritchett YL, Kajdasz DK, et al. A randomized, double-blind comparison of duloxetine and venlafaxine in the treatment of patients with major depressive disorder. J Psychiatr Res. 2008;41(1):22–34. | ||
Badyal DK, Khosla PP, Deswal RS, Matreja PS. Safety and efficacy of duloxetine versus venlafaxine in major depression in Indian patients. JK Sci. 2006;8(4):195–199. | ||
Wilhelm S, Boess FG, Hegerl U, et al. Tolerability aspects in duloxetine-treated patients with depression: should one use a lower starting dose in clinical practice? Expert Opin Drug Saf. 2012;11(5):699–711. | ||
Higuchi T, Murasaki M, Kamijima K. [Clinical evaluation of duloxetine in the treatment of major depressive disorder – Placebo- and paroxetine-controlled double-blind comparative study]. Jpn J Clin Psychopharmacol. 2009;12:1613–1634. Japanese. | ||
Higuchi T, Murasaki M, Kamijima K. [Clinical evaluation of duloxetine in the treatment of major depressive disorder – Superiority study of 40 mg and 60 mg versus 5 mg]. Jpn J Clin Pscyhopharmacol. 2009;12:1595–1612. Japanese. | ||
Hudson JI, Wohlreich MM, Kajdasz DK, Mallinckrodt CH, Watkin JG, Martynov OV. Safety and tolerability of duloxetine in the treatment of major depressive disorder: analysis of pooled data from eight placebo-controlled clinical trials. Hum Psychopharmacol. 2005;20:327–341. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th ed. Washington, DC: American Psychiatric Association; 1994. | ||
Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. | ||
Guy W. ECDEU Assessment Manual for Psychopharmacology, Revised 1976. Rockville, MD: National Institutes of Mental Health; 1976. | ||
Lewis-Fernandez R, Blanco C, Mallinckrodt CH, Wohlreich MM, Watkin JG, Plewes JM. Duloxetine in the treatment of major depressive disorder: comparisons of safety and efficacy in U.S. Hispanic and majority Caucasian patients. J Clin Psychiatry. 2006;67(9):1379–1390. | ||
Bailey RK, Mallinckrodt CH, Wohlreich MM, Watkin JG, Plewes JM. Duloxetine in the treatment of major depressive disorder: comparisons of safety and efficacy. J Natl Med Assoc. 2006;98(3):437–447. | ||
Brunton S, Wang F, Edwards SB, et al. Profile of adverse events with duloxetine treatment. Drug Saf. 2010:33(5):393–407. | ||
Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373(9665):746–758. | ||
Khan A, Bose A, Alexopoulos GS, Gommoll C, Li D, Gandhi C. Double-blind comparison of escitalopram and duloxetine in the acute treatment of major depressive disorder. Clin Drug Investig. 2007;27:481–492. | ||
Nierenberg AA, Greist JH, Mallinckrodt CH, et al. Duloxetine versus escitalopram and placebo in the treatment of patients with major depressive disorder: onset of antidepressant action, a non-inferiority study. Curr Med Res Opin. 2007;23:401–416. | ||
Perahia DG, Wang F, Mallinckrodt CH, Walker DJ, Detke MJ. Duloxetine in the treatment of major depressive disorder: a placebo- and paroxetine-controlled trial. Eur Psychiatry. 2006;21:367–378. | ||
Wade A, Gembert K, Florea I. A comparative study of the efficacy of acute and continuation treatment with escitalopram versus duloxetine in patients with major depressive disorder. Curr Med Res Opin. 2007;23:1605–1614. | ||
Perahia DG, Maina G, Thase M, et al. Duloxetine in the prevention of depressive recurrences: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(5):706–716. | ||
Gaynor PJ, Gopal M, Zheng W, et al. A randomized placebo-controlled trial of duloxetine in patients with major depressive disorder and associated painful physical symptoms. Curr Med Res Opin. 2011;27(10):1849–1858. |
Supplementary material
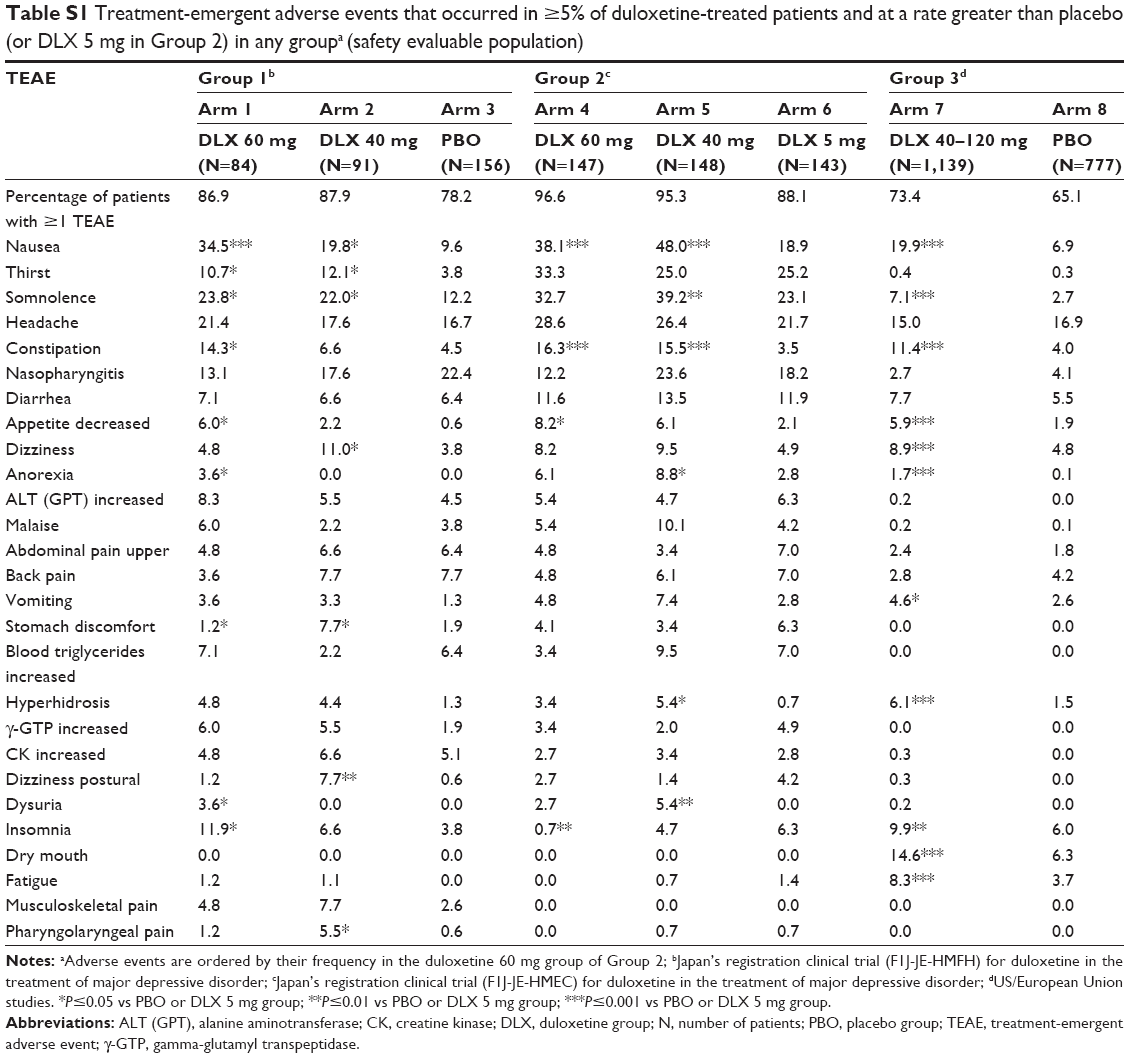 | Table S1 Treatment-emergent adverse events that occurred in ≥5% of duloxetine-treated patients and at a rate greater than placebo (or DLX 5 mg in Group 2) in any groupa (safety evaluable population) |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.