")
Back to Archived Journals » Clinical Oncology in Adolescents and Young Adults » Volume 6
Treating renal cell carcinoma in young adults: challenges and solutions
Received 26 September 2015
Accepted for publication 24 February 2016
Published 1 September 2016 Volume 2016:6 Pages 31—38
DOI https://doi.org/10.2147/COAYA.S82799
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mark Kieran
Alex Dreisin,1 Marc R Matrana1,2
1The University of Queensland School of Medicine, Ochsner Clinical School, 2Ochsner Cancer Institute, Ochsner Clinic Foundation, New Orleans, LA, USA
Abstract: Renal cell carcinoma (RCC) is uncommon in adolescents and young adults, although rare variants of the disease, including Xp11.2 translocation carcinoma, collecting duct carcinoma, and renal medullary carcinoma, occur with a higher preponderance in young patients. Likewise, non-RCC kidney tumors such as those of the Ewing sarcoma family of tumors are also seen in younger patients. The mainstay therapy for most forms of localized RCC and other kidney cancer is surgical removal of the tumors. Advances in treatments for metastatic clear cell carcinoma have been made in the last decade with the development of several new targeted agents. These therapies have revolutionized the treatment of metastatic clear cell RCC but are not targeted for other types of kidney cancer that are more often found in young patients. The management of RCC in adolescents and young adults remains a challenge for clinicians, but further advances are anticipated as less selective targeted immunotherapies become more widely available.
Keywords: kidney cancer, clear cell, Xp11.2, collecting duct carcinoma, renal medullary carcinoma, adolescents, targeted therapy
Introduction
More and more data are emerging that confirm that the needs of adolescent and young adult cancer patients vary from those of pediatric and older adult patients. These needs are not just psychosocial in nature but extend to the biology of cancer in adolescents and young adults, with this population exhibiting malignancies of varying pathologies that often have different responses to treatment and outcomes compared to those of patients in other age groups.
Kidney cancers are relatively rare in adolescents and young adults. While renal cell carcinomas (RCCs) comprise the bulk of kidney cancers in older adults, they are relatively uncommon in young patients. Likewise, while the majority of older adults with RCC develop the clear cell subtype, young patients are more likely to have less common forms of RCC such as translocation carcinoma. Other non-RCCs are also more common in young patients. Wilms’ tumor is common in pediatric patients and can certainly be seen in the adolescent and young adult populations. Other rarer kidney cancers such as renal medullary carcinoma (RMC) and those of the Ewing sarcoma family of tumors (ESFT) are also seen in younger patients. The cytological features of adolescent and young adult renal malignancies are reviewed and compared in Table 1.
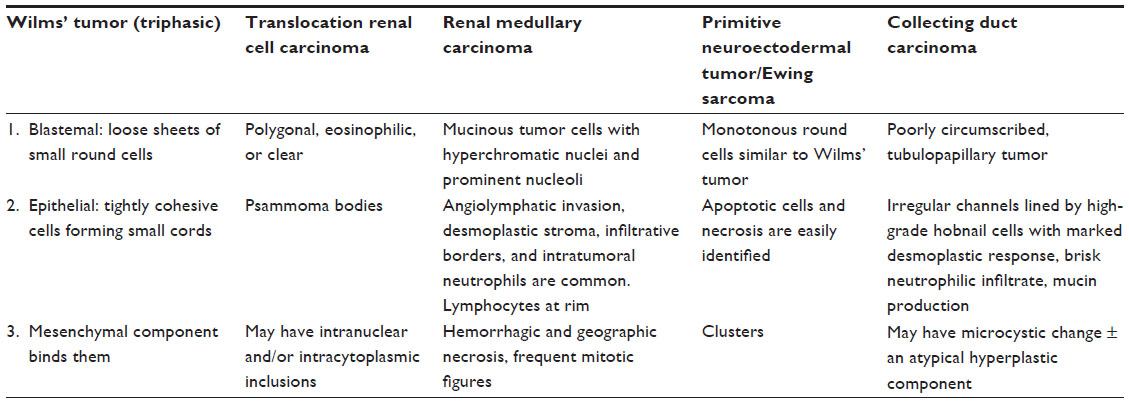 | Table 1 Cytological features of key adolescent and young adult renal malignancies |
Clear cell RCC in adolescents and young adults
Epidemiology
RCC is much more common in adults than children but does comprise 5% of pediatric renal tumors. It has an age-adjusted incidence of 0.01 per 100,000 children, with a median age of 9–12 years. There is no significant sex predilection. Outcomes worsen with age, but RCC has a 50%–60% survival rate among pediatric patients. This number rises to ~90% when confined to local lymph nodes but falls to 10%–15% when it presents with hematogenous metastasis.1
Suh et al2 reviewed 838 consecutive cases of RCC that occurred in a single tertiary hospital. They found that 44 (5.2%) cases occurred in young adults ≤40 years of age (range 24–40 years). They also found a higher incidence of non-clear cell RCC compared to older patients. For example, 12% of the younger cohort had chromophobe RCC, while only 6% of older patients exhibited this subtype. Similar results were found for collecting duct carcinoma (CDC) (5% vs 0.5%).
Similarly, Rodríguez et al3 reviewed the medical records of 400 patients treated surgically for RCC between January 1984 and December 1999. They found that 29 (7.25%) patients were <40 years of age. When comparing patients <40 years to those >40 years, these researchers found significant differences in histologic type, with only 69% of younger patients having clear cell carcinoma compared to 91% of patients >40 years (P=0.0001). There were also differences in tumor stage at presentation (pT2 =34.5% vs 17.3%, P=0.04; pT3 =20.7% vs 42%, P=0.03).
Diagnosis
Pediatric RCC often presents with generalized symptoms of malaise and abdominal pain. When present, localized signs and symptoms include gross hematuria, flank pain, and a palpable abdominal mass. Twelve percent of pediatric RCCs are completely asymptomatic. However, pediatric RCCs are easier to identify than their adult counterparts, where 50% of cases present asymptomatically. RCC is usually first suspected on abdominal ultrasound and then better defined by computed tomography. Staging workup begins with computed tomography of the lungs, as it is the most frequent site of metastasis. Thorough investigation for disseminated metastasis is essential, as 30% of cases metastasized on initial workup.1
While histological analysis is required for definitive diagnosis, age may guide initial clinical suspicion. For example, mesoblastic nephroma and rhabdoid tumors present in patients <12 months old, while Wilms’ tumor and pediatric RCC usually present in patients ≥3 years old. Immunostaining of transcription factor E3 (TTE3) should be a part of every workup and will identify translocation RCC, the most common subtype of pediatric RCC (discussed later). Tumor–node–metastasis staging is felt to be a good prognostic indicator.
Treatment
RCC in adolescents and young adults may often represent a distinct entity from adult RCC. For instance, ~75% of adult cases are clear cell RCC, while the vast majority of pediatric cases are translocation RCC. Consequently, treatments that have been proven effective in adults are not necessarily appropriate for the younger population. So while antiangiogenic agents and tyrosine kinase inhibitors (TKIs) are the cornerstones of therapy for metastatic RCC in adults, it remains unclear how effective they are in children and adolescents. Recent investigations into the use of TKIs and antiangiogenic agents in pediatric cases have tended to disfavor their use in children because of their unreliable effectiveness and incompletely understood side-effect profile in the pediatric population.4–8 These agents should be reserved for children with unresectable metastatic or advanced-stage RCC. However, the literature does advocate continued research into the manipulation of the vascular endothelial growth factor (VEGF) and mammalian target of rapamycin (mTOR) pathways in treating younger patients. The role of newer targeted immunotherapies in this population is largely unstudied.
For localized and resectable oligometastatic RCC, the gold standard of treatment is radical nephrectomy with complete tumor resection. It is unclear whether lymph node dissection is appropriate. Children with clinical evidence of lymph node metastases seem to benefit from resection, but it is less clear whether patients without lymphadenopathy derive a similar benefit.9 In fact, it has yet to be proven that the presence of lymph node metastasis can be associated with a worse prognosis in children. Moreover, it remains unclear what the optimal preoperative tumor size needs to be prior to making an attempt at a partial nephrectomy.
The role of cytoreductive nephrectomy in patients with advanced and metastatic disease continues to be debated, although almost everyone agrees that proper patient selection is vital. Complete regression of metastatic disease following surgical removal of the primary kidney tumor has been reported but is exceedingly rare. The argument in favor of cytoreductive nephrectomy comes from two randomized controlled trials, both of which were conducted in the era of immunotherapy.
A Southwest Oncology Group study found that nephrectomy followed by interferon alpha versus interferon alpha alone was associated with a significant improvement in overall survival (OS; median 11 months vs 8 months, respectively).10 A European Organisation for Research and Treatment of Cancer study similarly found an OS benefit (median 17 months versus 7 months) also favoring cytoreductive nephrectomy prior to immunotherapy.11
However, it is unclear whether this survival benefit extends to patients who undergo treatment with molecularly targeted therapies. Small retrospective studies have suggested that there may be some OS benefit in this setting, but randomized trials are still ongoing.
Translocation RCC (Xp11.2 translocation carcinoma)
Epidemiology
Translocation RCC is the most common type of RCC in children and occurs much more frequently in adolescents and young adults compared to older adults.1,2 Although translocation RCC has an incidence of 0.9% in the general adult population, this subtype accounts for up to 50% of all pediatric RCCs and occurs predominantly in children and young adults <40 years of age.12 Xu et al13 reviewed 98 cases of RCC in young adults (≤45 years of age) at their institution and found that 16 patients had translocation RCC.
The majority of cases are of the microphthalmia transcription factor (MiT)-TFE3 family of translocation carcinomas that involve the TFE3 gene on chromosome Xp11.2 or, less often, the TFE3 gene on chromosome 6p21. Immunohistochemical staining for TFE3 protein is the best diagnostic tool available to identify cases of translocation RCC. In a retrospective analysis of eight cases of translocation RCC, the most common presenting complaints were abdominal pain and gross hematuria. The right kidney was involved in 75% of cases.12
Diagnosis
Translocation RCCs have their own distinct morphological and immunohistological phenotypes that distinguish them from other types of RCC, although they can sometimes be difficult to distinguish from clear cell RCC and papillary RCC due to pathological heterogeneity classically found in these tumors. They are often papillary, high-grade tumors with bulky, oxyphilic cells. Immunohistology reveals low or absent expression of CAM5.2, keratin 7, and epithelial membrane antigen (EMA), but samples stain positive for CD10, cathepsin-K, and racemase expression. TFE3 immunohistochemistry (IHC) can be used to help distinguish translocation RCC from other subtypes, but IHC sometimes shows low sensitivity. Florescent in situ hybridization assays may be more accurate.
They are almost always primary tumors but can occasionally present as secondary malignancies in children who have been previously treated with chemotherapy.12 These children typically present 4–13 years postchemotherapy.14–18 Argani et al17 analyzed six cases of secondary RCC. They were all translocation type and occurred in patients who had been treated with topoisomerase II inhibitors and/or alkylating agents, both drugs that cause chromosomal destabilization.
Classification
Historically, translocation RCC has been described as an aggressive pediatric tumor. In the study by Su et al,12 62.5% of patients presented with stage III or IV disease; however, there was no statistically significant correlation between cancer characteristics and patient survival. A more recent analysis of pediatric cases reveals that certain subclasses are more indolent.19 The two most prominent subclasses involve either the ASPL-TFE3 or the PRCC-TFE3 translocation. They can be reliably differentiated by their variable expression of cathepsin-K; PRCC is usually positive, while ASPL always stains negative.20 A retrospective review by Ellis et al20 yielded the following key points:
- ASPL tended to present at a more advanced stage and was more likely to present as N1 or M1.
- Among patients with N1 stage III disease, outcomes tended to be worse in PRCC (although the difference was not statistically significant).
- ASPL follows a relatively indolent course. It has a low proliferation rate and often recurs late. Consequently, long-term follow-up is advised. This finding stands in contrast to other forms of translocation RCC that typically follow an extremely aggressive course.
- According to a multivariate analysis by Malouf et al,21 advanced age was associated with poorer outcomes in both subclasses.
Of note, per the International Society of Urological Pathology Vancouver Classification of renal neoplasia, translocation RCCs have been classified under the MiT family and contain either a TFE3 or a TFEB translocation. TFE3 translocations, such as the ASPL-TFE3 or PRCC-TFE3 translocations discussed earlier, are significantly more common. However, TFEB translocations may also occur and are the result of the fusion of TFEB on chromosome 6p21 with the alpha gene on chromosome 11q12. Regardless of subtype, both members of the MiT family respond similarly to treatment, although distinctions between subtypes may be more important in the future as more targeted therapies are developed.22
Treatment
The literature has failed to reveal any consistent guidelines for surgical or medical intervention.1 In the review by Ellis et al,20 one patient with the ASPL subtype had a complete response to sorafenib, and in another study, one patient with the PRCC had a complete response to sunitinib.23,24 While the use of TKIs shows some promise, many other patients have failed to respond to TKIs. Similarly, interleukin-2 and vaccine tumor lysate-pulsed dendritic cell therapy worked in one patient but failed in two others. In summary, there is no single effective treatment at this time for metastatic disease, and further research into potential targets is needed.
Current guidelines generally support the use of partial nephrectomy over total nephrectomy when resecting smaller renal masses because partial nephrectomy has a lower incidence of surgically induced chronic kidney disease. However, this guideline is not uniformly appropriate. For example, the ASPL subtype involves lymph nodes 75% of the time. Because partial nephrectomies do not sample regional lymph nodes, a total nephrectomy is indicated if the ASPL subtype is identified preoperatively. If an ASPL sample is identified postoperatively, a completion nephrectomy should be performed. Moreover, partial nephrectomy should only be considered if clear surgical margins can be achieved; otherwise, total nephrectomy is indicated.25
Collecting duct carcinoma (of Bellini)
Background
As its name implies, CDC originates from the distal collecting ducts of the kidneys. It accounts for ~1%–3% of all renal neoplasms and tends to occur in young adults, with a male predominance of 2:1. It is centrally located near the renal pelvis.26 Grossly, it is gray/white in color and devoid of any hemorrhagic or necrotic features.27,28 However, the absence of necrosis does not preclude the diagnosis of a CDC as anecdotal cases of necrotic features in diagnosed CDC exist.29 Microscopically, CDC displays a tubulopapillary growth pattern associated with a prominent stromal reaction.30–32 Although it is cited as one of the classic pediatric renal tumors, CDC actually has a mean age of onset beyond the sixth decade of life.29 When it presents in children, it exhibits a predilection for the right kidney at a ratio of 2:1.33
Treatment
CDC is an extremely aggressive tumor and almost always fails to respond to systemic therapy. The literature rarely describes some success using TKIs such as sorafenib. TKIs have shown great promise in the treatment of clear cell RCC, but aside from some cases of CDC that have responded to TKI therapy, the evidence supporting its efficacy is sporadic and inconsistent.29,33
The following diagnostic guidelines have been adopted by the International Society of Urological Pathology:33
- Involvement of at least some of the lesions with the renal medulla.
- Prominent tubule formation.
- Presence of desmoplastic stromal reaction.
- High-grade cytological features.
- Infiltrative growth pattern.
- Absence of other RCC subtypes.
A number of similarities exist between CDC and RMC, and one can often be misdiagnosed as the other. Some have postulated that both tumors may in fact represent the same family of cancers that exist on different ends of a single spectrum of renal malignancies.33
Renal medullary carcinoma
Background
RMC is a very rare and extremely aggressive neoplasm with an often dismal prognosis that presents almost exclusively in young patients with sickle cell trait (SCT).34 RMC was first identified by the Genitourinary Pathology Department, Armed Forces Institute of Pathology, Washington, DC, USA in 1995, where it was recognized as “the seventh sickle cell nephropathy”, adding to a list that includes gross hematuria, papillary necrosis, nephrotic syndrome, renal infarction, the inability to concentrate urine, and pyelonephritis.35 The original study reported a mean patient age of 22 years, with a male predominance of 2:1. Presenting symptoms are highly variable, but in a study by Swartz et al,36 >60% of cases presented with at least one component of the classic renal tumor triad: flank pain, hematuria, and/or palpable mass. Seven of the 40 cases analyzed had clinical suspicion for urinary tract infection or renal abscess. There was also a striking predilection for involvement of the right kidney with a right–left ratio of 3:1. Most cases of RMC have proved resistant to standard treatment modalities, including various combinations of surgery, chemotherapy, immunotherapy, and radiation.37–40 The average life expectancy from the time of diagnosis is 4 months.41
Pathogenesis
The pathogenesis of RMC remains poorly understood, although one prominent theory has gained some momentum in recent literature. It proposes that the hypoxic environment created in the renal medulla of patients with SCT interacts with hypoxia-inducible factor (HIF). In their study, Swartz et al36 stained eight RMC samples for HIF, VEGF, and p53 and noted increased expression of all three markers in every sample they analyzed. In normal cells, HIF induces p53 that activates cell death via apoptosis. In hypoxic cells, there is an absence of p53, and HIF instead induces VEGF that contributes to the angiogenic environment necessary for tumor progression. This theory is bolstered by a study conducted by Gatalica et al42 that analyzed two cases of RMC in patients without SCT and noted that they both contained mutations that promoted hypoxia, suggesting that the hypoxic environment is responsible for tumor proliferation.
Genetics
Genetic analyses have thus far been inconsistent in yielding potential therapeutic targets. A study by Stahlschmidt et al43 demonstrated evidence of a (9,22) translocation, but the tumor did not respond to imatinib. In nine tumors analyzed by Swartz et al,36 only one case had any aneuploidy. The single aneuploidy was a loss in chromosome 22. The lack of significant aneuploidy seems at odds with the extremely malignant nature of this tumor. A separate study identified the absence of SMARCB1/INI1 with loss of heterozygosity at the SMARCB1/INI1 gene locus on chromosome 22.44 SMARCB1 is a tumor suppressor gene involved in chromatin remodeling, cell cycle control, and regulation of cytoskeletal dynamics. Finally, a study by Schaeffer et al45 revealed high expression of topoisomerase II in a patient who achieved 9 months of complete remission with doxorubicin.
Histopathology and IHC
RMC is histologically distinct from RCC.42 RMC demonstrates a distinct reticular appearance with sheets of poorly differentiated tumor cells.35 According to Swartz et al,36 stromal dysplasia and acute and chronic inflammatory infiltrate are a consistent finding, distinguishing RMC from RCC. Swartz et al36 analyzed 28 tumors, and they all stained positive for CAM 5.2, EMA, and vimentin. All tumors showed diffuse expression of both HIF and VEGF. Eighty-two percent had rhabdoid features, and only 11% were cystic. Of note, RMC is often described as a subset of CDC in many texts despite their pathological differences. The mean age of onset for CDC is 53 years, whereas RMC presents in early adolescence. Moreover, CDC is characteristic for being cytokeratin 34βE12 positive, but all RMC samples in the study of Swartz et al36 were negative. Limited data show similarity to transitional cell carcinoma.46
Treatment
While most cases of RMC have proved resistant to traditional chemotherapy regimens, scattered cases exist that report a period of disease-free survival extending beyond the 4-month life expectancy of RMC. Several studies looking at the use of platinum-based chemotherapy regimens have yielded incremental improvements in life expectancy. In a retrospective review of nine patients treated for RMC, the regimen associated with the longest survival consisted of doxorubicin and gemcitabine followed by carboplatin and paclitaxel.40 In 2010, a regimen of carboplatin, gemcitabine, and paclitaxel produced a complete radiological response, with a progression-free survival of 24 months.47 In 2006, a case report described a complete radiological response to a 7-month course of bortezomib.48 At the time of publication, the patient had remained without radiological evidence of disease for 27 months. Moreover, bortezomib may exhibit a synergistic effect with doxorubicin.49 In 2011, a case report combined the use of bortezomib with carboplatin/paclitaxel/gemcitabine, and the patient was alive with no signs of disease 24 months after diagnosis.50 Finally, Lipkin et al51 started a patient on everolimus maintenance therapy after having achieved remission with platinum-based induction therapy, and the patient was still alive with no signs of disease 14 months later. Everolimus, an mTOR inhibitor, interferes with the PI3K–AKT pathway, which is one proposed mechanism of RMC proliferation. Given the similarities of RMC to transitional cell carcinoma, methotrexate, vinblastine, doxorubicin, and cisplatin has also been investigated as a promising chemotherapy regimen. A recent RMC case report showed 16 months of disease-free survival using methotrexate, vinblastine, doxorubicin, and cisplatin.34
Ewing sarcoma family of tumors
Background
The ESFT is a group of malignancies that includes Ewing sarcoma, primitive neuroectodermal tumors, and Askin tumor. They are poorly differentiated and quite aggressive.52 Although ESFT is not commonly considered a primary renal tumor, the kidney is being increasingly recognized as an unexpected primary ESFT site. The tumors tend to occur in young adult patients. Although there is a general lack of high quality studies into primary renal ESFT, a retrospective analysis of ten cases of primary renal ESFT revealed the following:52
- Mean age of diagnosis was 24.1 years (range 16–58 years).
- All ten tumors were positive for CD99, and 7/7 tumors had rearrangement of the EWSR1 locus.
- Gross morphology shows small, round, blue cell tumors. They are often misdiagnosed as Wilms’ tumor because both Wilms’ tumor and ESFT are monotonous round cell tumors.
- Forty percent had metastasized at presentation. All metastatic cases involved the lungs. Two cases also presented with bone metastasis.
A review of the literature conducted by Rowe et al52 identified 97 relevant cases and revealed the following background information:
- Only one of three patients with primary ESFT underwent diagnostic biopsy before surgery.
- An EWS–FLI1 fusion was noted in 33 of 35 tumors analyzed.
- In >95% of cases (25), t(11;22)(q24;q12) was observed.
- Forty-four percent had evidence of distant metastasis at diagnosis. Of note, this is a significantly greater rate than is typical for bone or soft tissue ESFT that presents with metastatic spread only 25% of the time.
- The tumors often grew to large volume before detection, usually due to their occult intra-abdominal location.
These last two points are associated with unfavorable outcomes.53,54
Treatment
Primary renal manifestations of ESFT are treated in much the same way as those originating in bone or soft tissue. Aggressive chemotherapy regimens include cycles of vincristine, adriamycin, and cyclophosphamide, dactinomycin, or ifosfamide and etoposide for 8–12 weeks. Once the tumor has shrunk to an operable size, local control (nephrectomy) can be attempted and followed up with postoperative chemotherapy if possible. The rationale behind postponing surgical intervention is that many first-line chemotherapy agents – such as ifosfamide – are nephrotoxic, and a prior nephrectomy would preclude their use. However, while this remains the current standard of treatment, there is no reliable evidence to suggest that it is superior to local control with only postoperative chemotherapy, and more data are needed before any conclusion can be drawn.52
Wilms’ tumor
Wilms’ tumor, the most common pediatric renal malignancy among younger pediatric patients, is relatively rare among young adults and adolescents. The median age at diagnosis of Wilms’ tumor is ~3.5 years.55 The principles of treatment involve both surgical and neoadjuvant therapies, with radiation therapy reserved for patients with advanced disease. With the current multimodal therapy, >90% of children diagnosed with Wilms’ tumor are expected to be long-term survivors.56
Future directions
During the past decade, major advances in the understanding of the underlying biology of clear cell RCC have led to the approval of several targeted agents (VEGF-targeted TKIs and mTOR inhibitors) for the treatment of unresectable or metastatic clear cell RCC. These therapies improve the quality of life and, when used sequentially, are thought to improve survival, but they are primarily targeted to the molecular drivers of clear cell carcinoma. Despite their efficacy, they do not elicit cures. The efficacy of these therapies in adolescent and young adult patients is not fully understood.
Tremendous efforts are underway to develop new and innovative therapies for RCC and other kidney tumors. While none of these efforts specifically targets patients who are adolescents or young adults, patients in this unique age group may benefit from these potential future therapy options. Newer therapies that block the interaction of the PD-1 receptor and PD-L1/PD-L2 ligands have gained US Food and Drug Administration approval for metastatic melanoma and unresectable lung cancer. These therapies work by blocking inhibitory signals that suppress T-cell-mediated antitumor immune responses, essentially upregulating T-cell immunity against cancer cells. Such targeted immunotherapy has shown promise in RCC in early studies and at least theoretically would not be specifically targeted against clear cell RCC only, instead potentially having broad implications in the management of many subtypes of RCC and other forms of kidney cancer. Likewise, several autologous vaccine technologies are being studied for RCC and may have therapeutic utility in various types of renal malignancies seen in younger patients. Certainly, wider studies are needed to determine how these new targeted immunotherapies may aid in the management of kidney cancers in adolescents and young adults.
Conclusion
RCC is uncommon in adolescents and young adults, although some subtypes of kidney cancers do occur at higher rates in younger patients. The mainstay therapy for most forms of pediatric renal cancer, similar to their adult counterparts, is surgical tumor removal. While significant advances in treatments for metastatic clear cell carcinoma have been made in the last decade, these therapies have been studied much more extensively in the adult population, and more data are needed to ascertain their effectiveness and toxicities in the adolescent and young adult population. As less selective targeted immunotherapies are developed and more information is ascertained regarding their application to children and young adults, we anticipate an imminent expansion in the treatment options available to treat renal cancer in younger patients.
Disclosure
The authors report no conflicts of interest in this work.
References
Spreafico F, Collini P, Terenziani M, Marchianò A, Piva L. Renal cell carcinoma in children and adolescents. Expert Rev Anticancer Ther. 2010;10(12):1967–1978. | |
Suh JH, Oak T, Ro JY, Truong LD, Ayala AG, Shen SS. Clinicopathologic features of renal cell carcinoma in young adults: a comparison study with renal cell darcinoma in older patients. Int J Clin Exp Pathol. 2009;2(5):489–493. | |
Rodríguez A, Patard JJ, Lobel B. Renal cell carcinoma in young adults: incidence, disease outcome and review of the literature. Arch Esp Urol. 2002;55(8):969–975. | |
Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet. 2009;373(9669):1119–1132. | |
Sun M, Lughezzani G, Perrotte P, Karakiewicz PI. Treatment of metastatic renal cell carcinoma. Nat Rev Urol. 2010;7(6):327–338. | |
Bellmunt J, Guix M. The medical management of metastatic renal cell carcinoma: integrating new guidelines and recommendations. BJU Int. 2009;103(5):572–577. | |
Soulières D. Review of guidelines on the treatment of metastatic renal cell carcinoma. Curr Oncol. 2009;16(suppl 1):S67–S70. | |
Escudier B, Kataja V, ESMO Guidelines Working Group. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(suppl 5):v137–v139. | |
Pace K. Determining the best treatment for renal cell carcinoma in young patients. Can Urol Assoc J. 2008;2(6):618. | |
Flanigan RC, Salmon SE, Blumenstein BA, et al. Nephrectomy followed by interferon alfa-2b compared with interferon alfa-2b alone for metastatic renal-cell cancer. N Engl J Med. 2001;345(23):1655. | |
Mickisch GH, Garin A, van Poppel H, de Prijck L, Sylvester R; European Organisation for Research and Treatment of Cancer (EORTC) Genitourinary Group. Radical nephrectomy plus interferon-alfa-based immunotherapy compared with interferon alfa alone in metastatic renal-cell carcinoma: a randomised trial. Lancet. 2001;358(9286):966. | |
Su HH, Sung MT, Chiang PH, Cheng YT, Chen YT. The preliminary experiences of translocation renal cell carcinoma and literature review. Kaohsiung J Med Sci. 2014;30(8):402–408. | |
Xu L, Yang R, Gan W, et al. Xp11.2 translocation renal cell carcinomas in young adults. BMC Urol. 2015;15(1):57. | |
Selle B, Furtwängler R, Graf N, Kaatsch P, Bruder E, Leuschner I. Population-based study of renal cell carcinoma in children in Germany, 1980-2005: more frequently localized tumors and underlying disorders compared with adult counterparts. Cancer. 2006;107(12):2906–2914. | |
Argani P, Ladanyi M. Recent advances in pediatric renal neoplasia. Adv Anat Pathol. 2003;10(5):243–260. | |
Ross H, Argani P. Xp11 translocation renal cell carcinoma. Pathology. 2010;42(4):369–373. | |
Argani P, Laé M, Ballard ET, et al. Translocation carcinomas of the kidney after chemotherapy in childhood. J Clin Oncol. 2006;24(10):1529–1534. | |
Schafernak KT, Yang XJ, Hsueh W, Leestma JL, Stagl J, Goldman S. Pediatric renal cell carcinoma as second malignancy: reports of two cases and review of the literature. Can J Urol. 2007;14(6):3739–3744. | |
de Jong B, Molenaar IM, Leeuw JA, Idenberg VJ, Oosterhuis JW. Cytogenetics of a renal adenocarcinoma in a 2-year-old child. Cancer Genet Cytogenet. 1986;21(2):165–169. | |
Ellis CL, Eble JN, Subhawong AP, et al. Clinical heterogeneity of Xp11 translocation renal cell carcinoma: impact of fusion subtype, age, and stage. Mod Pathol. 2014;27(6):875–876. | |
Malouf GG, Monzon FA, Couturier J, et al. Genomic heterogeneity of translocation renal cell carcinoma. Clin Cancer Res. 2013;19(17):4673–4684. | |
Magers MJ, Udager AM, Mehra R. MiT family translocation-associated renal cell carcinoma: a contemporary update with emphasis on morphologic, immunophenotypic, and molecular mimics. Arch Pathol Lab Med. 2015;139(10):1224–1233. | |
Numakura K, Tsuchiya N, Yuasa T, et al. A case study of metastatic Xp11.2 translocation renal cell carcinoma effectively treated with sunitinib. Int J Clin Oncol. 2011;16(5):577–580. | |
Choueiri TK, Mosquera JM, Hirsch MS. A case of adult metastatic Xp11 translocation renal cell carcinoma treated successfully with sunitinib. Clin Genitourin Cancer. 2009;7(3):E93–E94. | |
Gorin MA, Ball MW, Pierorazio PM, Argani P, Allaf ME. Partial nephrectomy for the treatment of translocation renal cell carcinoma. Clin Genitourin Cancer. 2015;13(3):e199–e201. | |
Zhao RR, Nie LH, Gong R, et al. Active targeted therapy for metastatic collecting duct carcinoma of the kidney: a case report and review of the literature. Int Urol Nephrol. 2013;45(4):1017–1021. | |
Bejar J, Szvalb S, Maly B, Munichor M, Cohen H. Collecting duct carcinoma of kidney: a cytological study and case report. Diagn Cytopathol. 1996;15(2):136–138. | |
Gupta R, Billis A, Shah RB, et al. Carcinoma of the collecting ducts of Bellini and renal medullary carcinoma: clinicopathologic analysis of 52 cases of rare aggressive subtypes of renal cell carcinoma with a focus on their interrelationship. Am J Surg Pathol. 2012;36(9):1265–1278. | |
Xu Q, Cao Q, Liu N, Fang Z, Ye Z, Peng T. Renal collecting duct carcinoma with extensive coagulative necrosis mimicking anemic infarct: report of a case and the literature review. Diagn Pathol. 2013;8:119. | |
Amin MB, Varma MD, Tickoo SK, Ro JY. Collecting duct carcinoma of the kidney. Adv Anat Pathol. 1997;4:85–94. | |
Srigley JR, Eble JN. Collecting duct carcinoma of kidney. Semin Diagn Pathol. 1998;15(1):54–67. | |
Eble JN, Sauter G, Epstein JI, Sesterhenn IA, editors. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. Lyon, France: IARC Press; 2004. | |
Amin MB, Smith SC, Agaimy A, et al. Collecting duct carcinoma versus renal medullary carcinoma: an appeal for nosologic and biological clarity. Am J Surg Pathol. 2014;38(7):871–874. | |
Amjad AI, Ali H, Appleman LJ, et al. Renal medullary carcinoma: case report of an aggressive malignancy with near-complete response to dose-dense methotrexate, vinblastine, doxorubicin, and cisplatin chemotherapy. Case Rep Oncol Med. 2014;2014:615895. | |
Davis CJ Jr, Mostofi FK, Sesterhenn IA. Renal medullary carcinoma: the seventh sickle cell nephropathy. Am J Surg Pathol. 1995;19(1):1–11. | |
Swartz MA, Karth J, Schneider DT, Rodriguez R, Beckwith JB, Perlman EJ. Renal medullary carcinoma: clinical, pathologic, immunohistochemical, and genetic analysis with pathogenetic implications. Urology. 2002;60(6):1083–1089. | |
Watanabe IC, Billis A, Guimarães MS, et al. Renal medullary carcinoma: report of seven cases from Brazil. Mod Pathol. 2007;20(9):914–920. | |
Coogan CL, McKiel CF Jr, Flanagan MJ, Bormes TP, Matkov TG. Renal medullary carcinoma in patients with sickle cell trait. Urology. 1998;51(6):1049–1050. | |
Noguera-Irizarry WG, Hibshoosh H, Papadopoulos KP. Renal medullary carcinoma: case report and review of the literature. Am J Clin Oncol. 2003;26(5):489–492. | |
Hakimi AA, Koi PT, Milhoua PM, et al. Renal medullary carcinoma: the Bronx experience. Urology. 2007;70(5):878–882. | |
Daher P, Bourgi A, Riachy E, Khoury A, Rehayem C, Sader-Ghorra C. Renal medullary carcinoma in a white adolescent with sickle cell trait. J Pediatr Hematol Oncol. 2014;36(5):e285–e289. | |
Gatalica Z, Lilleberg SL, Monzon FA, et al. Renal medullary carcinomas: histopathologic phenotype associated with diverse genotypes. Hum Pathol. 2011;42(12):1979–1988. | |
Stahlschmidt J, Cullinane C, Roberts P, Picton SV. Renal medullary carcinoma: prolonged remission with chemotherapy, immunohistochemical characterisation and evidence of bcr/abl rearrangement. Med Pediatr Oncol. 1999;33(6):551–557. | |
Calderaro J, Moroch J, Pierron G, et al. SMARCB1/INI1 inactivation in renal medullary carcinoma. Histopathology. 2012;61(3):428–435. | |
Schaeffer EM, Guzzo TJ, Furge KA, et al. Renal medullary carcinoma: molecular, pathological and clinical evidence for treatment with topoisomerase-inhibiting therapy. BJU Int. 2010;106(1):62–65. | |
Yang XJ, Sugimura J, Tretiakova MS, et al. Gene expression profiling of renal medullary carcinoma: potential clinical relevance. Cancer. 2004;100(5):976–985. | |
Walsh A, Kelly DR, Vaid YN, Hilliard LM, Friedman GK. Complete response to carboplatin, gemcitabine, and paclitaxel in a patient with advanced metastatic renal medullary carcinoma. Pediatr Blood Cancer. 2010;55(6):1217–1220. | |
Ronnen EA, Kondagunta GV, Motzer RJ. Medullary renal cell carcinoma and response to therapy with bortezomib. J Clin Oncol. 2006;24(9):e14. | |
Kondagunta GV, Drucker B, Schwartz L, et al. Phase II trial of bortezomib for patients with advanced renal cell carcinoma. J Clin Oncol. 2004;22(18):3720–3725. | |
Al-Rahawan MM, Rapkin L, Smith SD. Active combination of platins/ paclitaxel/gemcitabine/bortezomib in a child with metastatic renal medullary carcinoma (RMC). J Clin Oncol. 2011;29(15 suppl):e20002. | |
Lipkin JS, Rizvi SM, Gatalica Z, et al. Therapeutic approach guided by genetic alteration: use of MTOR inhibitor in renal medullary carcinoma with loss of PTEN expression. Cancer Biol Ther. 2015;16(1):28–33. | |
Rowe RG, Thomas DG, Schuetze SM, Hafez KS, Lawlor ER, Chugh R. Ewing sarcoma of the kidney: case series and literature review of an often overlooked entity in the diagnosis of primary renal tumors. Urology. 2013;81(2):347–353. | |
Rodriguez-Galindo C, Liu T, Krasin MJ, et al. Analysis of prognostic factors in Ewing sarcoma family of tumors: review of St. Jude Children’s Research Hospital studies. Cancer. 2007;110(2):375–384. | |
Bacci G, Longhi A, Ferrari S, Mercuri M, Versari M, Bertoni F. Prognostic factors in non-metastatic Ewing’s sarcoma tumor of bone: an analysis of 579 patients treated at a single institution with adjuvant or neoadjuvant chemotherapy between 1972 and 1998. Acta Oncol. 2006;45(4):469–475. | |
Breslow N, Olshan A, Beckwith JB, Green DM. Epidemiology of Wilms tumor. Med Pediatr Oncol. 1993;21(3):172–181. | |
Cotton CA, Peterson S, Norkool PA, et al. Early and late mortality after diagnosis of Wilms tumor. J Clin Oncol. 2009;27:1304–1309. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.