")
Back to Journals » Nature and Science of Sleep » Volume 13
Transvenous Phrenic Nerve Stimulation for Treatment of Central Sleep Apnea: Five-Year Safety and Efficacy Outcomes
Authors Costanzo MR, Javaheri S, Ponikowski P, Oldenburg O, Augostini R, Goldberg LR, Stellbrink C, Fox H , Schwartz AR , Gupta S, McKane S, Meyer TE, Abraham WT
Received 8 January 2021
Accepted for publication 9 April 2021
Published 29 April 2021 Volume 2021:13 Pages 515—526
DOI https://doi.org/10.2147/NSS.S300713
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ahmed BaHammam
Maria Rosa Costanzo,1 Shahrokh Javaheri,2,3 Piotr Ponikowski,4 Olaf Oldenburg,5 Ralph Augostini,3 Lee R Goldberg,6 Christoph Stellbrink,7 Henrik Fox,8 Alan R Schwartz,6 Sanjaya Gupta,9 Scott McKane,10 Timothy E Meyer,10 William T Abraham3 for the remedē®System Pivotal Trial Study Group
1Advocate Heart Institute, Naperville, IL, USA; 2Bethesda North Hospital, Cincinnati, OH, USA; 3The Ohio State University, Columbus, OH, USA; 4Department of Heart Diseases, Medical University, Military Hospital, Wroclaw, Poland; 5Ludgerus-Kliniken Münster, Clemens Hospital, Münster, Germany; 6University of Pennsylvania Perelman School of Medicine, Philadelphia, PA, USA; 7Bielefeld Medical Center, Bielefeld, Germany; 8Clinic for General and Interventional Cardiology Angiology, Herz- und Diabeteszentrum NRW, Ruhr-Universität Bochum, Bad Oeynhausen, Germany; 9University of Missouri-Kansas City School of Medicine, Saint Luke’s Mid-America Heart Institute, Kansas City, MO, USA; 10Respicardia, Inc., Minnetonka, MN, USA
Correspondence: Maria Rosa Costanzo
Advocate Heart Institute, Naperville, IL, 60566, USA
Email [email protected]
Background: The remedē System Pivotal Trial was a prospective, multi-center, randomized trial demonstrating transvenous phrenic nerve stimulation (TPNS) therapy is safe and effectively treats central sleep apnea (CSA) and improves sleep architecture and daytime sleepiness. Subsequently, the remedē System was approved by FDA in 2017. As a condition of approval, the Post Approval Study (PAS) collected clinical evidence regarding long-term safety and effectiveness in adults with moderate to severe CSA through five years post implant.
Methods: Patients remaining in the Pivotal Trial at the time of FDA approval were invited to enroll in the PAS and consented to undergo sleep studies (scored by a central laboratory), complete the Epworth Sleepiness Scale (ESS) questionnaire to assess daytime sleepiness, and safety assessment. All subjects (treatment and former control group) receiving active therapy were pooled; data from both trials were combined for analysis.
Results: Fifty-three of the original 151 Pivotal Trial patients consented to participate in the PAS and 52 completed the 5-year visit. Following TPNS therapy, the apnea-hypopnea index (AHI), central-apnea index (CAI), arousal index, oxygen desaturation index, and sleep architecture showed sustained improvements. Comparing 5 years to baseline, AHI and CAI decreased significantly (AHI baseline median 46 events/hour vs 17 at 5 years; CAI baseline median 23 events/hour vs 1 at 5 years), though residual hypopneas were present. In parallel, the arousal index, oxygen desaturation index and sleep architecture improved. The ESS improved by a statistically significant median reduction of 3 points at 5 years. Serious adverse events related to implant procedure, device or delivered therapy were reported by 14% of patients which include 16 (9%) patients who underwent a pulse generator reposition or lead revision (primarily in the first year). None of the events caused long-term harm. No unanticipated adverse device effects or related deaths occurred through 5 years.
Conclusion: Long-term TPNS safely improves CSA, sleep architecture and daytime sleepiness through 5 years post implant.
Clinical Trial Registration: ClinicalTrials.gov Identifier: NCT01816776.
Keywords: central sleep apnea, phrenic nerve stimulation
Introduction
Central sleep apnea (CSA) is characterized by a lack of drive to breathe during sleep, leading to absent or inadequate ventilation. Although a rare disorder in the general population, CSA is frequently associated with heart failure (HF), atrial fibrillation, stroke, and opioid use.1–3 When all identifiable causes are excluded, it is called idiopathic CSA.4
Studies performed decades ago demonstrated that unilateral phrenic nerve stimulation could maintain adequate ventilation both in animals5 and humans.6 In 2017, the United States Food and Drug Administration (FDA) approved the remedē System for unilateral transvenous phrenic nerve stimulation (TPNS) for treatment of moderate to severe CSA in adults based on the results of a randomized clinical trial.7,8 Subsequent follow-up and analysis confirmed the results of the Pivotal Trial.9,10 The TPNS system (Supplement Figure 1) is an implantable neurostimulation device consisting of a pulse generator implanted in the pectoral region, a stimulation lead placed in either the left pericardiophrenic or right brachiocephalic vein, and an optional sensing lead placed in the azygos vein.11 The TPNS device automatically delivers therapy, continuously throughout the night during the scheduled time when the patient is in a sleeping position and at rest, without requiring patients to manually activate therapy.12
As a condition of FDA approval, the Post Approval Study (PAS) collected evidence regarding long-term safety and effectiveness in adult subjects with moderate to severe CSA. Here, we report the 5-year analysis of TPNS on CSA metrics, sleep architecture and safety from the PAS. Based on the data presented in this paper, the FDA recently concluded the 5-year PAS data fulfilled the post-approval requirement, providing sufficient evidence of long-term efficacy and safety.13
Methods
The remedē System Pivotal Trial (NCT01816776) was a prospective, multicenter, randomized, open-label trial in patients with moderate to severe CSA to assess TPNS versus no stimulation.14 The trial enrolled patients with any CSA etiology (except CSA induced by pain medications) who had apnea hypopnea index (AHI, the number of apnea and hypopnea events per hour of sleep) of ≥20 events/hour of sleep, with ≥30 central apneas, with central apneas accounting for ≥50% of all apneas, and the obstructive apnea index ≤20% of the AHI.
All patients were implanted and randomized to treatment or control at time of implant. The Control group had therapy activated after the primary endpoint assessment at the 6-month visit.
Patients who remained in the Pivotal Trial at the time of FDA approval were identified for enrollment in the PAS. All sites were requested to enroll all eligible patients regardless of prior therapy effectiveness. All eligible patients had completed at least 2 years of follow-up in the Pivotal Trial and all subjects were to continue receiving therapy through 5 years post implant in the PAS. Subjects attended annual visits in the PAS and were contacted by phone between visits. Since the Pivotal Trial was closed prior to all subjects completing the 3-year visit and the PAS initiated as a new trial, safety data was collected throughout the period between the trials, but some 3- and 4-year visits did not occur.
Overnight, attended polysomnography (PSG) was performed at baseline and 1, 2, and 5 years; home polygraphy (PG) (NOX T3 consisting of pressure transducer, abdominal and rib cage respiratory inductance plethysmography belts and pulse oximeter; Nox Medical, Reykjavík, Iceland) was performed at 3 years, and in some subjects at 5 years due to patients declining a PSG or COVID-19 pandemic site restrictions.
The AHI, central apnea index (CAI), obstructive apnea index, mixed apnea index, hypopnea index, 4% oxygen desaturation index (ODI4), arousal index, percentage and minutes of sleep with oxygen saturation <90% and percentage of total sleep time in N1, N2, N3 and rapid eye movement (REM) were assessed for the PSGs. Sample sizes are lower for arousals and sleep stages at 5 years due to the inability to measure these parameters in patients who chose a PG rather than a PSG.
For consistency, the same central sleep core laboratory (Registered Sleepers, Winter Haven, FL, USA) used in the Pivotal Trial scored all PAS PSGs and PGs using the American Academy of Sleep Medicine guidelines detailed previously.14,15 Daytime sleepiness was assessed at baseline, 1 and 5 years using the ESS questionnaire (0=none, 24=worst).16
The study was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice, and ISO-14155:2011 and registered at ClinicalTrials.gov (Identifier: NCT01816776). The protocol was approved by local ethics or institutional review boards; all patients provided written informed consent.
Statistical Methods
Since previous Pivotal Trial publications demonstrated similar and sustained effects of therapy in the treatment and former control groups after therapy was activated,8,9,12 the groups were pooled for analysis. The effectiveness analyses continue to use the per protocol population. All originally randomized subjects were used in safety analyses. Effectiveness results were combined based on years since therapy activation for the 1-year visit; in year 2 and beyond, visit results were combined based on years post implant and represent 6 fewer months of therapy for former Control subjects than Treatment subjects. Nominal 2-sided P-values from the Wilcoxon Signed-Rank test for paired change from baseline are provided; however, the PAS was not powered for statistical tests. Imputation was not performed for missing data. Select results are also presented limiting to data for subjects with sleep study data at baseline and 5 years to assess changes in the complete data subset. Continuous variable results at each visit are displayed in text as median and interquartile range.
Freedom from related serious adverse events (SAE) through 5 years is summarized as a binomial proportion and individual events are summarized by year of onset post implant. The same independent clinical events committee that adjudicated adverse events in the Pivotal Trial was used to provide a standard and systematic adjudication of clinical adverse events in the PAS regarding relationship to device, procedure or therapy, SAE status (and reasons including hospitalization), and heart failure relationship. Overall survival (censored at last date known alive) and cardiovascular survival (censored at last date known alive or died due to non-cardiovascular cause) in the pooled group were assessed using the Kaplan–Meier method.
The subgroup of subjects with HF, defined in the Pivotal Trial as patients having a New York Heart Association functional class ≥1, was also analyzed to provide an update of previously reported results17 on sleep data, as well as Kaplan–Meier analysis of survival and freedom from HF hospitalization 5 years post implant. Sleep data results are also presented for the subgroup without HF.
SAS version 9.4 (Cary, NC) was used for analysis.
Results
Fifty-three subjects eligible at the end of the Pivotal Trial participated in the PAS and 52 completed the 5-year visit. The CONSORT diagram (Figure 1) shows reasons for patient exclusion or withdrawal from the PAS. The reasons subjects did not enroll in PAS varied, with the primary reasons being four sites declined participation for lack of resources (17 subjects) and 17 additional subjects were not interested in participating in the long-term follow-up study.
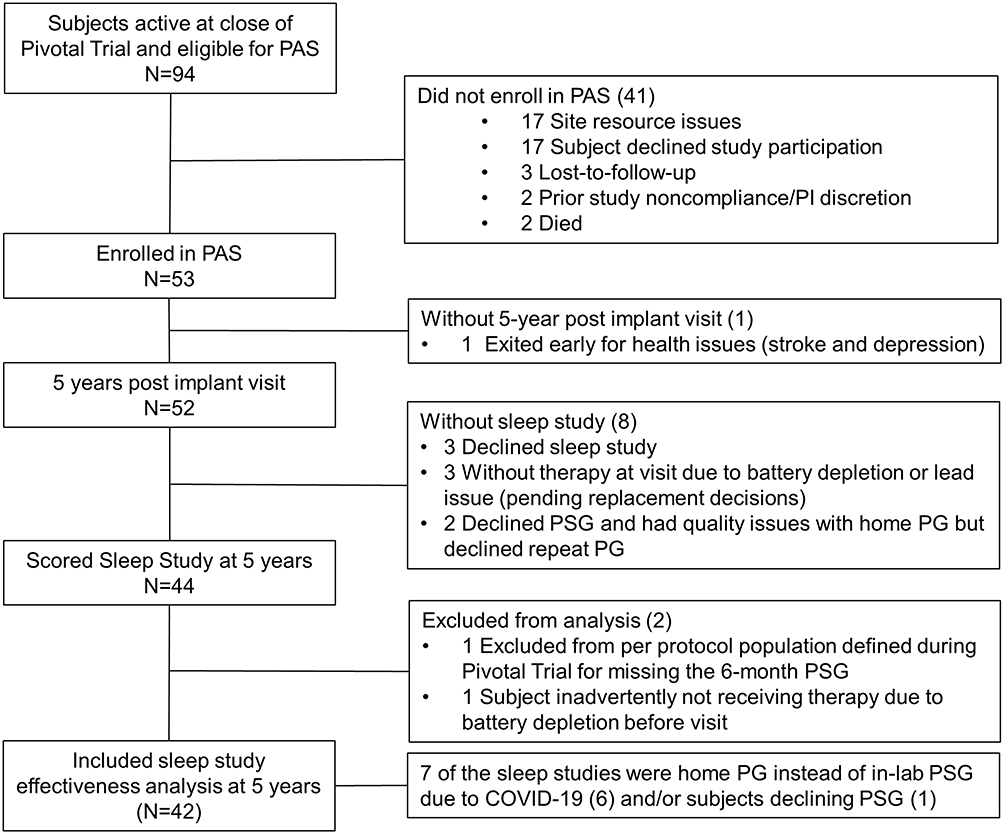 |
Figure 1 CONSORT diagram. Composition of the subjects eligible for the post approval study through the 5-year post-implant visit. |
Characteristics for pooled treatment and control patients at implant (n=151) and those enrolled in PAS are shown in Table 1. Characteristics for all PAS eligible and not enrolled subjects are displayed in Supplement Table 1 for comparison. No clinically relevant baseline characteristics differed among subjects enrolled in the pivotal, PAS or eligible but not enrolled in the PAS. Examination of subjects’ experience with therapy also did not yield differences. Two of the 94 eligible subjects had ongoing stimulation discomfort at or just prior to the Pivotal Trial closure and one of them enrolled in the PAS. Also, no signal of enrollment bias was apparent based on response to therapy during the Pivotal Trial.
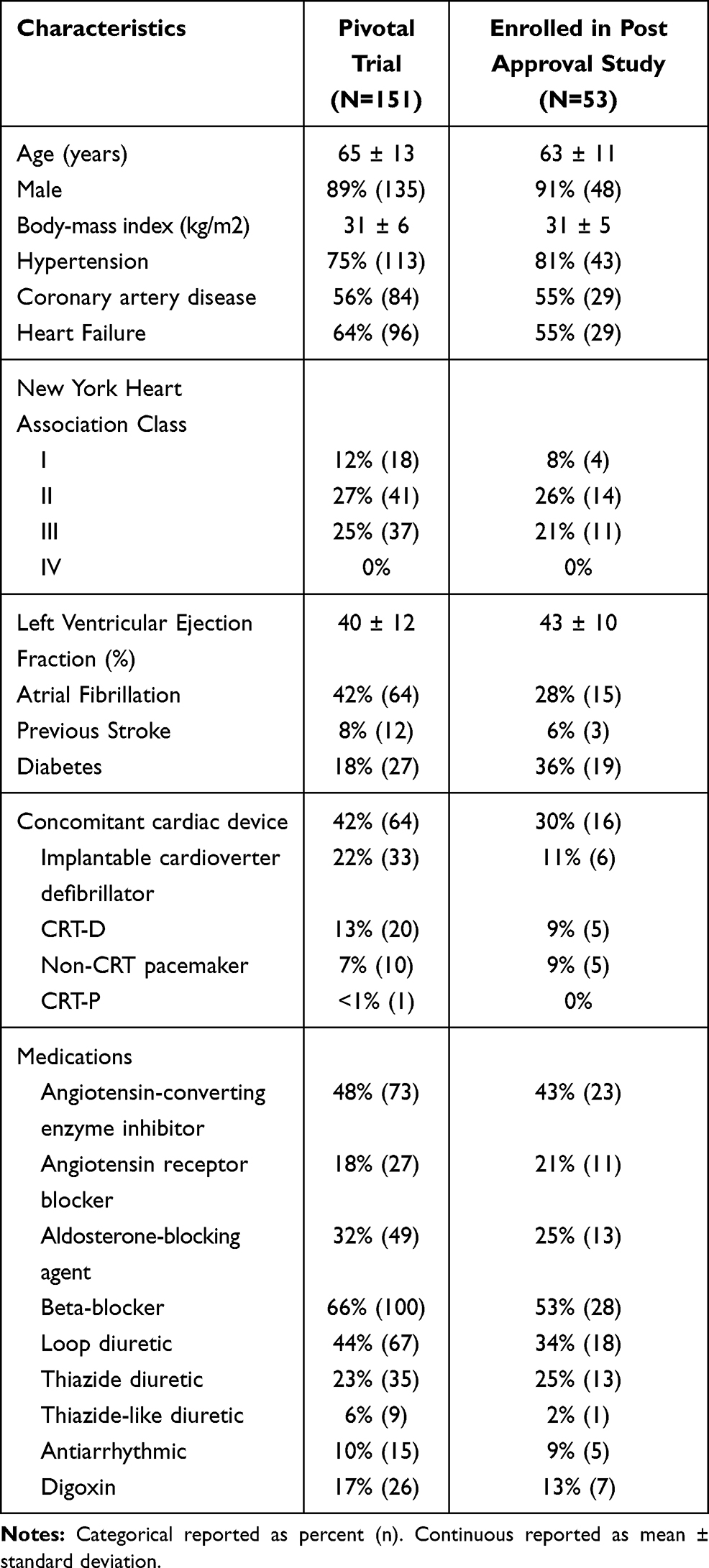 |
Table 1 Baseline Characteristics |
At the time of Pivotal Trial enrollment, 64% of subjects had HF, 42% had atrial fibrillation, and 8% previous stroke. Eleven percent of subjects had idiopathic CSA. One of the 53 enrolled subjects was not part of the per protocol population determined during the Pivotal Trial and is excluded from effectiveness analyses.
Sleep-Related Disordered Breathing Events
Subjects had severe CSA at baseline. The baseline median AHI was 46 [interquartile range 34, 60] events/hour of sleep and reduced to <20 events/hour at 1, 2 and 5 years, with median paired change from baseline to 5 years of −22 [−42, −7] events/hour (P<0.001) (Table 2, Figure 2). CAI, which comprised half of the baseline AHI, improved from a median of 23 [13, 39] events/hour at baseline to 1 at each yearly follow-up. The mixed apnea index, obstructive apnea index and hypopnea index were unchanged through 5 years. Note that 9 subjects performed a home PG instead of PSG at 5 years, and of the 9 PGs 2 could not be scored due to lack of or insufficient airflow signal quality and subjects’ refusal to repeat the study.
 |
Table 2 Sleep Study and Daytime Sleepiness Results by Visit |
 |
Figure 2 Apnea-hypopnea components by visit. Median AHI and its components (CAI, OAI, MAI and HI) are displayed by visit. A PSG was performed at baseline, 1 and 2 years. A PG was performed at 3 years (35 subjects had not reached this visit at time of study closure). aAt 5 years, 35 subjects performed a PSG and 7 a PG (due to subjects declining PSG and COVID-19 restrictions). Not all Pivotal Trial subjects enrolled in the post approval study resulting in a smaller sample size at year 5. The figure on the right shows the results when restricted to only patients with paired data at baseline and 5 years. Note the sum of the component medians does not equal the median AHI. Abbreviations: AHI, apnea hypopnea index; CAI, central apnea index; HI, hypopnea index; MAI, mixed apnea index; OAI, obstructive apnea index; PG, polygraphy; PSG, polysomnography. |
In parallel with changes in sleep-disordered breathing events, other metrics demonstrating continued improvement from baseline to 5 years include ODI4 improved from a median 39 [26, 57] events/hour to 15 [6, 26] at 5 years, the arousal index decreased from 39 [27, 57] events/hour at baseline to 23 [12, 36] events/hour at 5 years, and the percentage of sleep with oxygen saturation <90% at 5 years (4% [1, 17]) was lower than at baseline (9% [3, 21]), although the median paired change from baseline at 5 years was a 2% [−12, 6] decrease.
Sleep Architecture
The distribution of sleep stages as a percentage of total sleep time at baseline was N1=31% [19, 47], N2=47% [40, 58], N3=2% [0, 9] and REM sleep=10% [6, 16]. In parallel with changes in AHI and CAI, sleep architecture improved (Table 2, Figure 3). At 5 years, median paired absolute changes from baseline revealed N1 decreased by 19 [−23, −9] absolute percentage points, whereas N2 increased by 9 [−2, 23], and REM by 5 [0, 13]. N3 did not change (0 [−3, 4]).
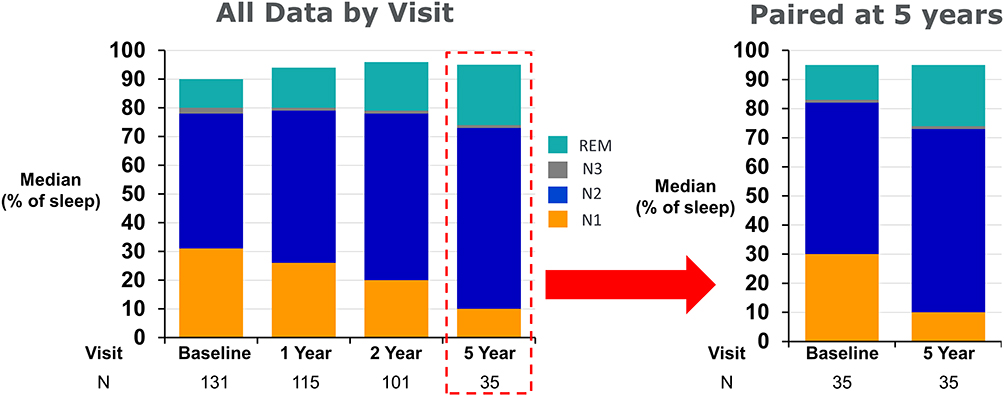 |
Figure 3 Sleep stages by visit. Median percentage of total sleep time in N1, N2, N3, and REM. Light sleep (N1) decreases over time in favor of deeper stages of sleep (N2, N3, REM). Not all Pivotal Trial subjects enrolled in the post approval study resulting in a smaller sample size at year 5. The figure on the right shows the results when restricted to only patients with paired data at baseline and 5 years. N1 decreased 19 absolute percentage points and deep stages of sleep increased 14 percentage points. Note the sum of the medians of the sleep stages does not add to 100%. Abbreviation: REM, rapid eye movement. |
Daytime Sleepiness
The median ESS at baseline and 5-years were 9 [5, 14] and 6 [4, 11], respectively (Table 2), demonstrating a consistent and clinically meaningful ≥3 point reduction18,19 in daytime sleepiness with therapy at each visit. In the subset of subjects with excessive daytime sleepiness (ESS>10) at baseline who had 5-year data, 62% (16/26) of subjects shifted to an ESS ≤10. Examination of the percentage of subjects with a clinically meaningful improvement from baseline at 5 years, 74% (37/50) improved by ≥2 points and 62% (31/50) improved by ≥3 points. Analysis restricted to subjects with baseline ESS>10 showed that 96% (25/26) improved by ≥2 points and 88% (23/26) by ≥3 points.
Safety
SAEs adjudicated as related to the implant procedure, device or delivered therapy occurred in 14% (n=21) of subjects through 5 years (Table 3), with the majority occurring in the first year after implant. The related SAEs are displayed by year in Table 3. Between years 2 and 3, no related SAEs occurred. Also, no related concomitant cardiac device interaction adverse events were observed after 2 years. Four subjects reported related SAEs in years 3 to 5:
- One subject experienced a stimulation lead dislocation after a fall, followed by multiple procedures with hospitalization to extract the lead and place a new lead, which subsequently dislodged and required another replacement procedure.
- Two subjects experienced a stimulation lead component failure (high lead impedance on all electrode pairs used to deliver therapy) that involved a hospital stay for a lead replacement.
- One subject experienced an implant site infection after device replacement requiring device explant and hospital stay.
 |
Table 3 Implant Procedure, Device or Delivered Therapy-Related Serious Adverse Events Through 5 Years of Follow-Up |
The events all resolved with remedē System revisions and routine care. For completeness, all SAEs adjudicated as not related to the implant procedure, device, or delivered therapy are shown in Supplement Table 2.
No deaths occurred in patients enrolled in the PAS. The 5-year survival estimate from Kaplan–Meier analysis was 78% (Supplement Table 3). The causes of death occurring during the Pivotal Trial have been described previously (restated in Supplement Table 4 for reference) and no death during the Pivotal Trial was adjudicated by the Clinical Events Committee as being related to the implant procedure, device, or delivered therapy.12
Battery longevity estimates were 47 months for a left stimulation lead (N=92) and 35 months for a right stimulation lead (N=55), for a combined estimate of 41 months for all subjects who had a successful implant in the Pivotal Trial. Of the 53 subjects participating in the PAS, 51 had their IPG replaced prior to completing 5 years of follow-up.
Heart Failure Subgroup Analysis
Twenty-nine subjects identified at the baseline visit as having HF participated in the PAS. Subject characteristics for the HF subgroup enrolled in the PAS are displayed in Supplement Table 5 (average age of 62 years, 97% male, average left ventricular ejection fraction 38%, 41% having atrial fibrillation).
Similar to the full population, improvements in sleep disordered breathing events, sleep architecture, and daytime sleepiness were sustained throughout 5 years (Table 4). AHI was reduced by a median of 25 events/hour (n=22), the median residual CAI at 5 years was 2 events/hour, the median ODI4 improved from 41 events/hour at baseline to 20 events/hour at 5 years, and the median arousal index decreased from 41 events/hour at baseline to 19 (n=17). The ESS improved from 8 at baseline to 4 at 5 years (n=27, paired change from baseline −3 [−7, −1], P value=0.002).
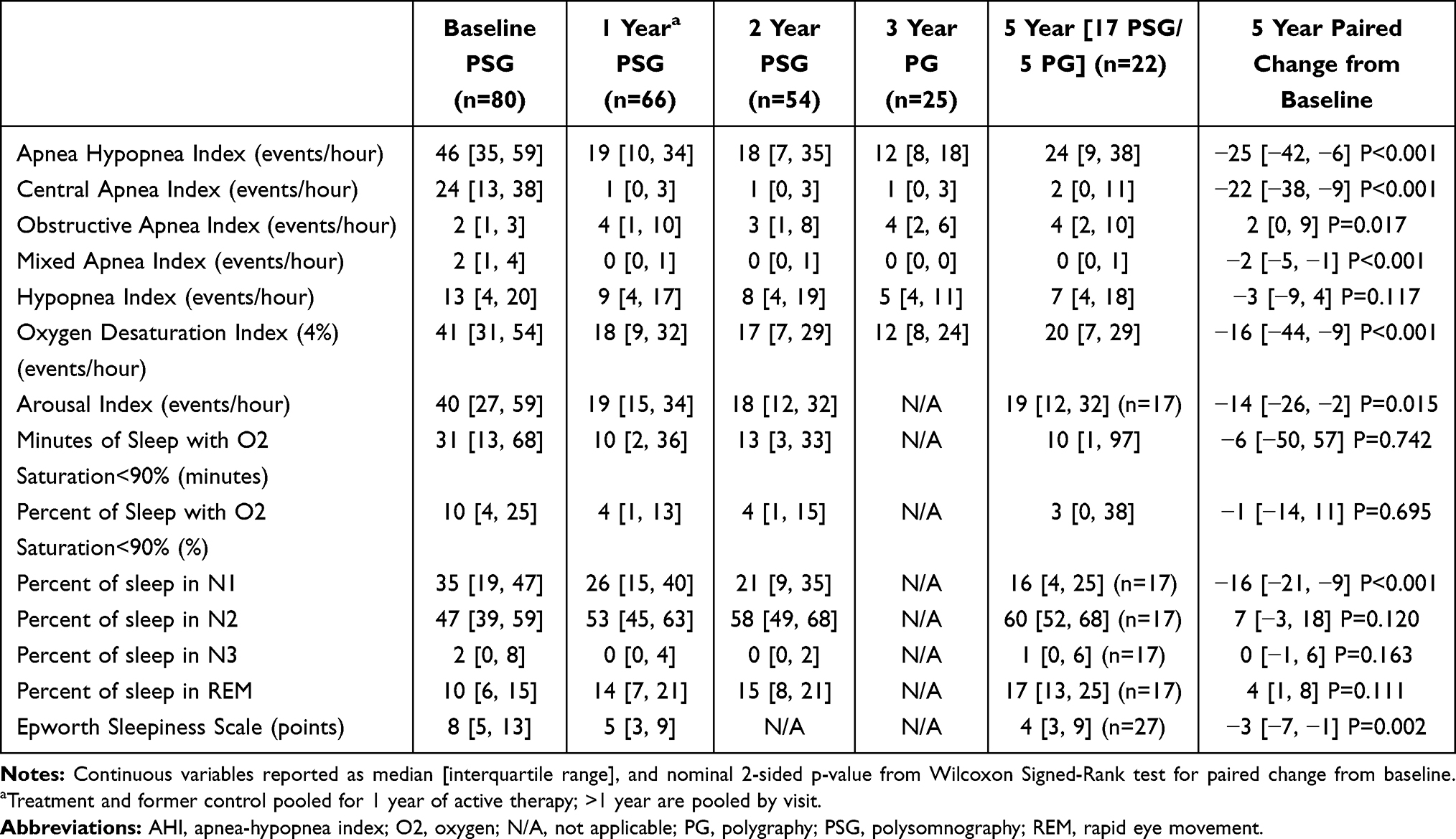 |
Table 4 Sleep Study and Daytime Sleepiness Results by Visit for Heart Failure Subgroup |
The estimated Kaplan–Meier 5-year overall survival rate of the 96 subjects with HF enrolled in the Pivotal Trial and including follow-up data from the cohort enrolled in the PAS was 68% and the cardiovascular survival rate was 78% (subjects censored at time of non-cardiovascular death or end of follow-up if still alive). The estimated 5-year post-therapy activation rate of freedom from HF-related hospitalization by Kaplan–Meier analysis was 55%. See Supplement Table 3 for details of these time to event analyses.
Non-Heart Failure Subgroup Analysis
Twenty-four Pivotal Trial subjects identified at the baseline visit as not having HF participated in the PAS. Subject characteristics for the non-HF subgroup enrolled in the PAS are displayed in Supplement Table 6 (average age of 63 years, 83% male, 67% having hypertension, 13% having atrial fibrillation).
Similar to the overall population, improvements in sleep disordered breathing events, sleep architecture, and daytime sleepiness were sustained throughout 5 years (Table 5). AHI was reduced by a median of 21 events/hour (n=20), the median residual CAI at 5 years was 1 event/hour, the median ODI4 improved from 37 events/hour at baseline to 10 events/hour at 5 years, and the median arousal index decreased from 37 events/hour at baseline to 23 (n=18). The ESS improved from 13 at baseline to 7 at 5 years (n=23, paired change from baseline −5 [−8, 0], P value=0.008).
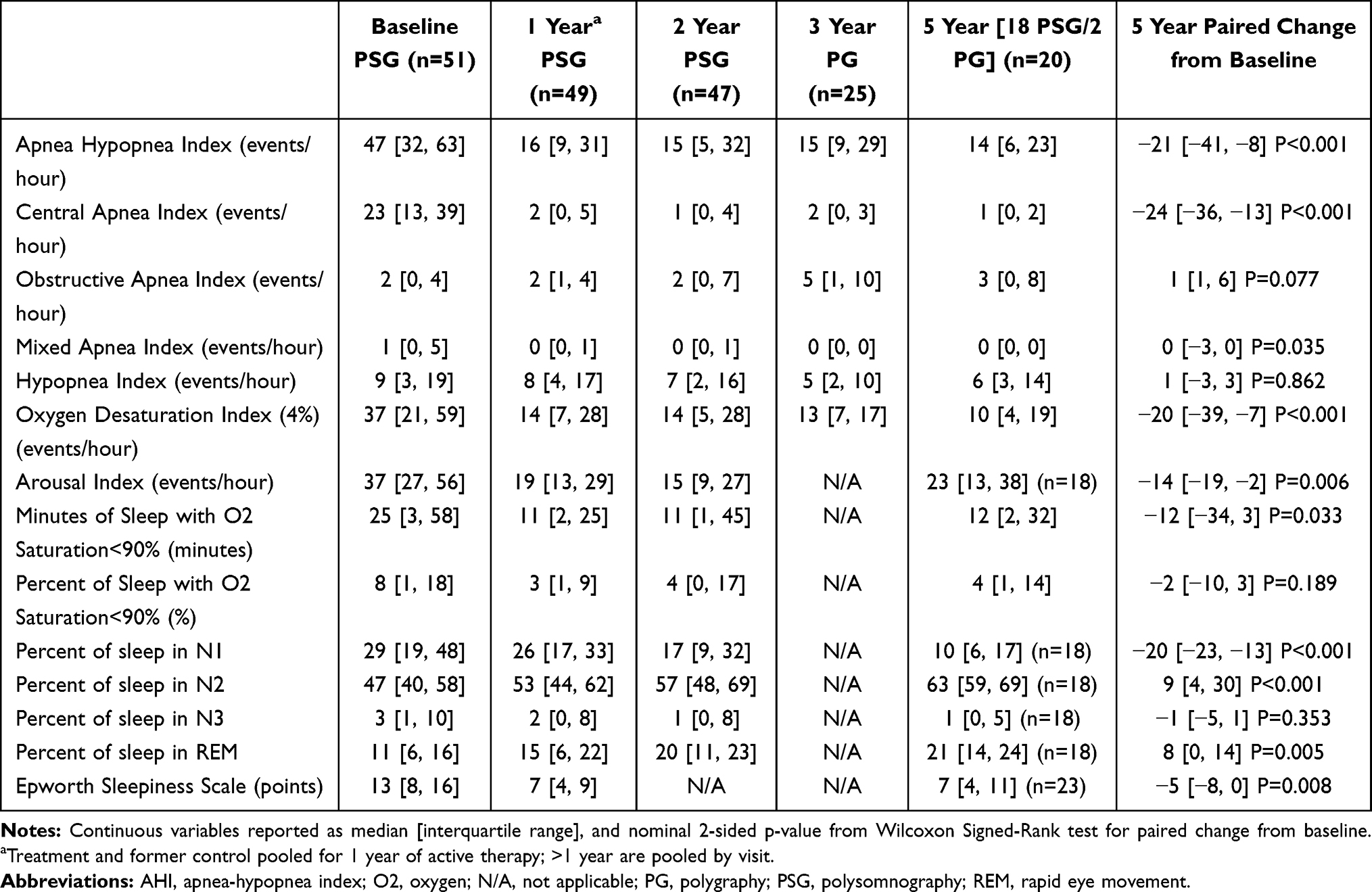 |
Table 5 Sleep Study and Daytime Sleepiness Results by Visit for Non-Heart Failure Subgroup |
Discussion
Following the randomized controlled Pivotal Trial, in 2017 the FDA approved the use of TPNS for the treatment of moderate to severe CSA in adults. As a condition of approval, the PAS collected clinical evidence regarding the long-term safety and effectiveness of TPNS in subjects from the Pivotal Trial. Importantly, the FDA considered the results of the PAS report as providing sufficient evidence of long-term effectiveness and safety of TPNS.13
The subjects enrolled in the trial had severe CSA as evidenced by baseline median AHI of 46 events/hour of sleep and CAI accounted for most of the events. The TPNS treatment, designed to specifically target CSA, nearly eliminated central apneas in subjects during the entire 5-year follow-up. The reduction is similar to that seen with continuous positive airway pressure (CPAP),20 although the automaticity of TPNS ensures that duration of therapy throughout the night is independent of patients’ adherence. Similar to trials using theophylline,21 acetazolamide22 CPAP20 and adaptive servoventilation,23,24 residual events were present with TPNS. At this point in time, no single therapy has been capable of completely eliminating sleep-related disordered breathing events. Several reasons exist for residual events observed in the aforementioned therapeutic trials. Specifically, regarding TPNS, hypopneas may have occurred at sub-therapeutic stimulation levels following resumption of sleeping position after the subject may have gotten out of bed, sat up, or rolled to a different position; this is related to the device algorithm which is programmed to gradually ramp back up to the therapeutic stimulation level over several minutes to allow the patient to fall back to and remain asleep, an algorithm similar to some automatic positive airway pressure devices. Furthermore, some hypopneas may have been obstructive, and therefore remained untreated by TPNS. Consistent with our previous studies,21,22 hypopneas were not differentiated as central versus obstructive by the Sleep core laboratory. However, if hypopneas were obstructive in nature, we suggest that TPNS did not cause the hypopneas as the median obstructive apnea index did not significantly change over 5 years.
Consequent to a reduction in central sleep apnea events, sleep architecture improved considerably and significantly, reflected by a reduction in arousals and N1 sleep stage, and increased percentage of N2 and REM sleep stages. The improved sleep architecture following TPNS could partially account for the improvement in ESS, which is the longest sustained improvement seen in randomized trials of CSA therapies. In addition, overall hypoxemic burden including ODI4 and time with oxygen saturation<90% remained improved through 5 years with TPNS.
Interestingly, ESS improvements observed at 5 years were similar to those occurring at one year of TPNS therapy when there was also amelioration of quality of life. At the one-year visit (the last visit collecting quality of life data), 60% of subjects reported marked or moderate improvement in patient global assessment compared to before device implantation.9
The Pivotal Trial population experienced CSA more severe than that occurring in other randomized trials of CSA in HF patients (CANPAP20 and SERVE-HF23). In the positive airway pressure trials, the fact that, despite reduction in CAI, sleep architecture and arousal index failed to improve, possibly explaining why in said trials only minimal improvement in ESS was shown. In the SERVE-HF trial, ESS improved by 1 point through 3 years and the improvement began to dissipate at 4 years.23 Similarly, in the CANPAP trial, arousal index did not change significantly, even in those whose CSA was suppressed by CPAP.25 Reduced arousals with TPNS may result in both ESS improvement and reduction in the detrimental surges of sympathetic activity triggered by arousals.26,27
The safety profile through 5 years did not change significantly compared to prior years.8–10 No unanticipated adverse device effects occurred, and all related SAEs through five years were expected for this type of device. The related SAEs occurring after 2 years were either related to lead component failures, lead dislocation, or were perioperative events, such as infection or lead dislodgment, following battery or lead replacement procedures. The 2 additional lead component failures bring the 5-year rate to 3%, which is favorable compared to published rates for other neurostimulation devices.28–30
Importantly, no signal of increased mortality has been observed during long-term use of TPNS. While the trial lacks a comparator group at 5 years, the overall 5-year survival rates were 78% for the entire population and 68% for the HF cohort. This is reassuring because typically subjects with severe CSA experience excess mortality in most studies.31,32 In addition, no publications on survival in a CSA population which included those without HF are available to compare overall survival rates. In the SERVE-HF trial,23 the 5-year survival rate was approximately 60% in the control group and 55% in the adaptive servo-ventilation arm. However, SERVE-HF only included patients with CSA and symptomatic HF with reduced ejection fraction. Moreover, the SERVE-HF participants had an average AHI of about 30/hour compared to an average of 47/hour in the current study. In the study by Oldenburg et al,32 the 5-year survival of subjects with HF with reduced ejection fraction and moderate to severe CSA was just above 50%. However, the results of these studies may not be directly comparable because they included HF populations different from the HF cohort in our study.
Currently, a large, prospective single-arm post-market study (The remedē System Therapy [rēST] Study) of TPNS is collecting real-world experience in adult patients with moderate to severe CSA (NCT03884660).
This long-term, single-arm follow-up study has limitations that need to be mentioned. First, it is an observational study that followed the randomized, controlled Pivotal Trial and all patients received active therapy, so the study lacked a control group or external control similar enough to meaningfully assess impact of long-term outcomes such as survival. However, change from baseline assessments were able to be analyzed for some endpoints. Another limitation is the lack of availability of data for some eligible participants who completed the Pivotal Trial. This was due to the interval between closure of the Pivotal Trial and initiation of the 5-year PAS as a separate study. In addition, not all Pivotal Trial sites and patients chose to participate in the PAS, a fact which may have led to underestimation of adverse events. However, the decision to participate in the PAS study was independent of and unrelated to issues with stimulation discomfort, or therapy effectiveness.
Conclusion
Results of this prospective long-term 5-year study suggest TPNS is a safe and effective therapy, resulting in clinically meaningful improvements in sleep and excessive daytime sleepiness for patients with CSA. The consistent safety and effectiveness of the remedē System through 5 years provides further evidence that TPNS is a viable therapy for adult patients with moderate to severe CSA and particularly those lacking alternative treatment options or unable to tolerate mask-based therapies.
Data Sharing Statement
The de-identified dataset used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Committee/Institutional Review Board Approvals
The protocol was approved by the following local ethics or institutional review boards: Quorum Review IRB, Western Institutional Review Board, Cleveland Clinic Institutional Review Board, The Cooper Health System Institutional Review Board, University of Pennsylvania Office of Regulatory Affairs, Office of Human Subjects Research Institutional Review Board Johns Hopkins University, Lancaster General Hospital Institutional Review Board, Marshfield Clinic Research Foundation, Methodist Healthcare Institutional Review Board, Schulman IRB, Presbyterian Healthcare Institutional Review Board, Spectrum Health Human Research Protection Program, University of Maryland-Baltimore Institutional Review Board, University of Southern California Health Sciences Campus Institutional Review Board, Washington University St. Louis Institutional Review Board, Henry Ford Health System Research Administration, Christ Hospital Institutional Review Board, Baptist Institutional Review Board, Forsyth Medical Center Institutional Review Board, Edward Hospital & Health Services Institutional Review Board, Komisja Bioetyczna przy Dolnoślaskiej Izbie Lekarskiej, and Vorsitzender Ethikkommission Der Medizinischen Fakultät Der Ruhr-Universität Bochum Sitz Bad Oeynhausen.
Acknowledgments
The authors wish to acknowledge the technical expertise of the sleep core laboratory, Registered Sleepers, Inc., supervised by Tim Winchester (Winter Haven, FL, USA).
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
Respicardia, Inc. funded the study.
Disclosure
Dr Maria Rosa Costanzo report grants, personal fees from Respicardia, during the conduct of the study; Dr Shahrokh Javaheri report personal fees from Respicardia, during the conduct of the study; Dr Piotr Ponikowski report personal fees from Respicardia, during the conduct of the study; personal fees from Vifor Pharma, personal fees from Amgen, personal fees from Servier, personal fees from Novartis, personal fees from Berlin Chemie, personal fees from Bayer, personal fees from Pfizer, personal fees from Cibiem, personal fees from Coridea, personal fees from Impulse Dynamics, personal fees from Renal Guard Solutions, personal fees from Boehringer Ingelheim, personal fees from AstraZeneca, personal fees from BMS, personal fees from AbbottVascular, personal fees from Radcliffe Group, personal fees from Ono Pharmaceutical, outside the submitted work; Dr Ralph Augostini report personal fees from Respicardia, outside the submitted work; Dr Lee R Goldberg report grants, personal fees from Respicardia, during the conduct of the study; personal fees from Abbott, outside the submitted work; Dr Christoph Stellbrink report grants from Respicardia, during the conduct of the study; Dr Alan R Schwartz report grants from NIH, personal fees from Respicardia, during the conduct of the study; personal fees from Respicardia, outside the submitted work; Mr Scott McKane and Dr Timothy E. Meyer are employees of Respicardia so receive salary and have ownership in the company during the conduct of the study. Dr William T Abraham report personal fees from Respicardia, during the conduct of the study; Dr Costanzo (study principal investigator for remedē® System Pivotal Trial). The authors report no other conflicts of interest in this work.
References
1. Javaheri S, Dempsey JA. Central sleep apnea. Compr Physiol. 2013;3:141–163. doi:10.1002/cphy.c110057
2. Hernandez AB, Patil SP. Pathophysiology of central sleep apneas. Sleep Breath. 2016;20(2):467–482. doi:10.1007/s11325-015-1290-z
3. Javaheri S, Elliott M. Central sleep apnoea. In: Elliott M, Nava S, Schönhofer B, editors. Non-Invasive Ventilation and Weaning: Principles and Practice.
4. Javaheri S, McKane S. Transvenous phrenic nerve stimulation to treat idiopathic central sleep apnea. J Clin Sleep Med. 2020. doi:10.5664/jcsm.8802
5. Sarnoff SJ, Gaensler EA, Maloney JV. The effectiveness of contralateral ventilation during activity of one phrenic nerve. J Thorac Surg. 1949:929–937.
6. Whittenberger JL, Sarnoff SJ, Hardenbergh E. Electrophrenic respiration. II. Its use in man. J Clin Invest. 1949;28(1):124–128. doi:10.1172/JCI102040
7. U.S. Food & Drug Administration. remedē® System – P160039. Available from: https://www.fda.gov/medical-devices/recently-approved-devices/remeder-system-p160039.
8. Costanzo MR, Ponikowski P, Javaheri S, et al. Randomised Controlled Trial of transvenous neurostimulation for central sleep apnoea. Lancet. 2016;388(10048):974–982. doi:10.1016/S0140-6736(16)30961-8
9. Costanzo MR, Ponikowski P, Javaheri S, et al. Sustained 12 month benefit of phrenic nerve stimulation for central sleep apnea. Am J Cardiol. 2018;121(11):1400–1408. doi:10.1016/j.amjcard.2018.02.022
10. Fox H, Oldenburg O, Javaheri S, et al. Long-term efficacy and safety of phrenic nerve stimulation for the treatment of central sleep apnea. Sleep. 2019;42(11). doi:10.1093/sleep/zsz158
11. Augostini RS, Afzal MR, Costanzo MR, et al. How to implant a phrenic nerve stimulator for treatment of central sleep apnea? J Cardiovasc Electrophysiol. 2019;30(5):792–799. doi:10.1111/jce.13898
12. Schwartz AR, Sgambati FP, James KJ, et al. Novel phrenic nerve stimulator treats Cheyne-Stokes respiration: polysomnographic insights. J Clin Sleep Med. 2020;16(5):817–820. doi:10.5664/jcsm.8328
13. U.S. Food & Drug Administration [internet]. Post-approval studies (PAS) database. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma_pas.cfm?t_id=602722&c_id=4561.
14. Costanzo MR, Augostini R, Goldberg LR, et al. Design of the remedē® System Pivotal Trial: a Prospective, Randomized Study in the use of respiratory rhythm management to treat central sleep apnea. J Card Fail. 2015;21(11):892–902. doi:10.1016/j.cardfail.2015.08.344
15. Iber C, Ancoli-Israel S, Chesson AL
16. Johns MW. A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep. 1991;14(6):540–545. doi:10.1093/sleep/14.6.540
17. Costanzo MR, Ponikowski P, Coats A, et al. Phrenic nerve stimulation to treat patients with central sleep apnoea and heart failure. Eur J Heart Fail. 2018;20(12):1746–1754. doi:10.1002/ejhf.1312
18. Patel S, Kon SSC, Nolan CM, et al. The Epworth Sleepiness Scale: minimum clinically important difference in obstructive sleep apnea. Am J Respir Crit Care Med. 2018;197(7):961–963. doi:10.1164/rccm.201704-0672LE
19. Patil SP, Ayappa IA, Caples SM, Kimoff RJ, Patel SR, Harrod CG. Treatment of adult obstructive sleep apnea with positive airway pressure: an American Academy of Sleep Medicine systematic review, meta-analysis, and GRADE assessment. J Clin Sleep Med. 2019;15(2):301–334. doi:10.5664/jcsm.7638
20. Bradley TD, Logan AG, Kimoff RJ, et al. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med. 2005;353(19):2025–2033. doi:10.1056/NEJMoa051001
21. Javaheri S, Parker TJ, Wexler L, Liming JD, Lindower P, Roselle GA. Effects of theophylline on sleep disordered breathing in stable heart failure: a prospective, double-blind, placebo controlled, and crossover study. N Engl J Med. 1996;335:562–567. doi:10.1056/NEJM199608223350805
22. Javaheri S. Acetazolamide improves central sleep apnea in heart failure: a double-blind prospective study. Am J Respir Crit Care Med. 2006;173:234–237. doi:10.1164/rccm.200507-1035OC
23. Cowie MR, Hoehrle H, Wegscheider K, et al. Adaptive servo-ventilation for central sleep apnea in systolic heart failure. New Engl J Med. 2015;373:1095–1105. doi:10.1056/NEJMoa1506459
24. Javaheri S, Brown LK, Randerath W, Khayat R. SERVE-HF: more questions than answers. Chest. 2016;149(4):900–904. doi:10.1016/j.chest.2015.12.021
25. Ruttanaumpawan P, Logan AG, Floras JS, Bradley TD. Effect of continuous positive airway pressure on sleep structure in heart failure patients with central sleep apnea. Sleep. 2009;32(1):91–98. doi:10.5665/sleep/32.1.91
26. Javaheri S, Gay P. To die, to sleep, perchance to dream … without hypertension: dreams of the visionary Christian Guilleminault revisited. J Clin Sleep Med. 2019;15(9):1261–1270. doi:10.5664/jcsm.7952
27. Cohn JN, Levine TB, Olivari MT, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311(13):819–823. doi:10.1056/NEJM198409273111303
28. Morales-Conde S, Del Agua IA, Busetto L, et al. Implanted closed-loop gastric electrical stimulation (CLGES) system with sensor-based feedback safely limits weight regain at 24 months. Obes Surg. 2018;1–9.
29. Deckers K, De Smedt K, Mitchell B, et al. New therapy for refractory chronic mechanical low back pain-restorative neurostimulation to activate the lumbar multifidus: one year results of a Prospective Multicenter Clinical Trial. Neuromodulation. 2018;21(1):48–55. doi:10.1111/ner.12741
30. Ostrem JL, Ziman N, Galifianakis NB, et al. Clinical outcomes using ClearPoint interventional MRI for deep brain stimulation lead placement in Parkinson’s disease. J Neurosurg. 2016;124(4):908–916. doi:10.3171/2015.4.JNS15173
31. Javaheri S, Barbe F, Campos-Rodriguez F, et al. State-of-the-Art Review: sleep apnea types, mechanisms, and clinical cardiovascular consequences. J Am Coll Cardiol. 2017;69(7):841–858. doi:10.1016/j.jacc.2016.11.069
32. Oldenburg O, Wellmann B, Buchholz A, et al. Nocturnal hypoxaemia is associated with increased mortality in stable heart failure patients. Eur Heart J. 2016;37(21):1695–1703. doi:10.1093/eurheartj/ehv624
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.