")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Transgenic animal models for study of the pathogenesis of Huntington’s disease and therapy
Authors Chang R, Liu X, Li S, Li X
Received 23 January 2015
Accepted for publication 10 March 2015
Published 15 April 2015 Volume 2015:9 Pages 2179—2188
DOI https://doi.org/10.2147/DDDT.S58470
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Renbao Chang,1 Xudong Liu,1 Shihua Li,2 Xiao-Jiang Li1,2
1State Key Laboratory of Molecular Developmental Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing, People’s Republic of China; 2Department of Human Genetics, Emory University School of Medicine, Atlanta, GA, USA
Abstract: Huntington’s disease (HD) is caused by a genetic mutation that results in polyglutamine expansion in the N-terminal regions of huntingtin. As a result, this polyQ expansion leads to the misfolding and aggregation of mutant huntingtin as well as age-dependent neurodegeneration. The genetic mutation in HD allows for generating a variety of animal models that express different forms of mutant huntingtin and show differential pathology. Studies of these animal models have provided an important insight into the pathogenesis of HD. Mouse models of HD include transgenic mice, which express N-terminal or full-length mutant huntingtin ubiquitously or selectively in different cell types, and knock-in mice that express full-length mutant Htt at the endogenous level. Large animals, such as pig, sheep, and monkeys, have also been used to generate animal HD models. This review focuses on the different features of commonly used transgenic HD mouse models as well as transgenic large animal models of HD, and also discusses how to use them to identify potential therapeutics. Since HD shares many pathological features with other neurodegenerative diseases, identification of therapies for HD would also help to develop effective treatment for different neurodegenerative diseases that are also caused by protein misfolding and occur in an age-dependent manner.
Keywords: transgenic animal models, Huntington’s disease, pathogenesis, therapy
Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder caused by a polymorphic trinucleotide CAG repeat expansion in exon 1 of the HD gene, and this expansion encodes the polyglutamine (polyQ) repeat in the N-terminal region of the disease protein, huntingtin (Htt).1 Expanded polyQ in Htt (>36 glutamines) results in HD, and the length of expanded polyQ is inversely correlated with the onset of the disease.2,3 The majority of HD patients carry expanded polyQ repeats in the range of 38–55 glutamines and develop late-onset neurological symptoms in mid-life, typically between the ages of 30 and 50 years,4 and longer expansions (>60 repeats) may lead to juvenile-onset HD.5 HD patients are clinically characterized by cognitive, psychiatric, and motor disturbances,6 as well as peripheral phenotypes that include weight loss and muscle wasting.7 The pathological feature in patients post mortem is a prominent neuronal loss in the striatum, especially in the caudate putamen region, which is usually accompanied by cell loss in the cerebral cortex and widespread brain atrophy in the brains of patients with grade III–V HD.8,9 The neuronal loss occurs in a cell type-specific manner, and the most vulnerable are the GABAergic medium spiny neurons (MSNs) in the striatum and cortical projection neurons (CPNs) in the deep layer cortex.10
Identification of the genetic mutation in HD lead to generation of a variety of animal models that express expanded-polyQ containing Htt. Different species from Drosophila and mice, to monkeys have been used to establish animal models of HD.9,11,12 The mouse model is by far the most commonly used mammalian genetic model because of its efficiency, economy, and ease of manipulation. These animal models have provided us with important tools to investigate the pathogenesis of the disease and develop therapeutic strategies. There have been a number of excellent reviews about behavioral phenotypes of HD mouse models and drug treatments.7,13–19 In this review, we focus on the insights into pathogenesis from HD animal models that will help us to develop therapeutic strategies. Since species-dependent pathology was also seen in large animal models of HD, we also discuss the implications of large HD animal models for future therapeutic applications.
Transgenic mouse models of HD
R6/2 is the first and most extensively studied rodent model of HD. It was generated by expressing exon 1 of human Htt with 144 CAG repeats under the control of 1 kb human Htt promoter.20 R6/2 mice have a severe phenotype, with motor deficits at 5–6 weeks and often cannot survive more than 13 weeks20 without intervention. The early death and severe phenotypes, which make R6/2 mice a plausible model for juvenile-onset HD, indicate the toxicity of the N-terminal fragment of mutant Htt with a large polyQ repeat.21,22 Consistently, transgenic N-terminal mutant Htt forms nuclear inclusions and aggregates in R6/2 mice, which led to the discovery of similar inclusions in the post mortem brains of HD patients.23–26 Importantly, the aggregates in the brains of HD patients are only labeled by antibodies to the N-terminal region of Htt, validating the idea that only N-terminal mutant Htt is able to misfold and form aggregates.23,26 Although R6/2 mimicked human HD pathology in many aspects, it showed no apoptotic neuronal death, which is different from the profound neuronal loss in the striatum and cortex in HD patients.27 These may be due to the early death caused by weight loss or species resistance to damage of mutant Htt.9
The theory that the N-terminal mutant Htt is toxic is further proved by transgenic mice expressing different N-terminal mutant Htt fragments. For instance, N171-82Q transgenic HD mice, which express the first 171 amino acids with 82 glutamines in the polyQ domain under the control of the mouse prion promoter, also show progressive neurological phenotypes and early death, which often occurs at 4–6 months of age.28 Also, N171-82Q mice show the age-dependent formation of Htt aggregates in neuronal cells, which is consistent with the notion that N-terminal mutant Htt has altered an conformation that leads to protein aggregation.29,30
Other important evidence indicating the toxicity of N-terminal mutant Htt is derived from transgenic mice expressing full-length mutant Htt with expanded polyQ repeats. BAC and YAC mouse models express full-length human mutant Htt under the human Htt promoter and regulatory elements that provide relatively intact human genomic regulatory elements and protein context within the span of the transgene. Of these HD mice, the most extensively studied are BACHD with 97 CAG/CAA mixed repeats and YAC128 with 128 CAG pure repeats in human Htt.31,32 BAC and YAC HD mice show selective atrophy in the striatum and cortex along with progressive motor deficits, thus recapitulating to some extent the regional selectivity of adult-onset HD. These mice present electrophysiological abnormalities suggestive of an alteration at glutamatergic synapses.33 However, unlike YAC128 mice, BACHD mice express a higher level of Htt but exhibit fewer aggregates. As mixed CAG/CAA repeats lead to more stable polyQ proteins in the rodent brain, the instability of mutant Htt may play a role in aggregate or inclusion formation. A shared phenotype commonly observed in human Htt genomic transgene mice is body weight gain, which is not observed in HD patients and is probably due to the Htt dosage effect, as YAC or BAC mice overexpressing wild-type Htt also show weight gain.34 Another possibility, which remains to be ruled out, is that additional gene expression from the large genomic DNA in BAC and YAC vectors could contribute to the obesity phenotype.
HD knock-in mouse models
Transgenic mouse models of HD using exogenous promoters often raise a concern about the effects of overexpression of the transgene and multiple copies of integrated transgenes.35 In theory, knock-in models should be optimal to reproduce human pathology because they are the most faithful reproduction of the genetic mutations.9 A number of knock-in models with expanded CAG repeats or human mutant Htt exon 1 replacing the corresponding sequences in the endogenous murine htt gene locus generated.13,36–39 Also, a series of mutant Htt-KI models with increasing polyQ length repeats (111,40 140,39 150,38,41,42 and 17543,44) are available for studying HD pathogenesis and therapies. However, all these HD KI mice showed late-onset of phenotype and progressive but mild pathology.36,37 Many behavioral abnormalities in this KI model were similar to transgenic mouse models but much milder,42 which also supports the idea that N-terminal mutant Htt could be more toxic. Indeed, mutant Htt also forms aggregates in KI mice, and these aggregates are only labeled by the antibody to N-terminal region of Htt.45 In addition, Western blotting of HD KI mouse brain tissue demonstrates the presence of a number of degraded N-terminal mutant Htt that carry an expanded polyQ repeat.45 All these observations lead to the theory that proteolysis of full-length mutant Htt is critical for accumulation of N-terminal mutant Htt, which then causes age-dependent neuropathology in HD.24,46
It is now known that full-length Htt is proteolytically cleaved to N-terminal fragments, and these fragments are ubiquitinated and cleared by the proteasome and autophagy.24,46–48 Shorter N-terminal fragments are more stable and prone to aggregation, suggesting that specific proteolysis of full-length Htt into smaller fragments might be one of the initial steps in the disease process.9 A number of N-terminal fragments and a subset of the enzymes that generate N-terminal Htt fragments of varying sizes have been determined.24 These include caspase-3, caspase-6,49–51 calpain,52 matrix metalloproteinase 10,53 and an undefined aspartyl protease.54 The presence of caspase cleavage sites in Htt makes the caspase inhibitors promising drugs for HD therapy. YAC transgenic mice expressing human full-length mutant Htt carrying a mutation that blocks caspase-6 cleavage did not show neuropathology or phenotypes when compared with the YAC-128Q model.55 However, later studies did not find prevention of generation of the toxic fragment when caspase-6 was knocked out in mice.56
Although HD KI mice do not develop phenotypes as robust as those in transgenic mice expressing N-terminal mutant Htt, they recapitulate an important pathological change seen in the brain of the HD patient, which is the preferential accumulation of mutant Htt in striatal neurons that are mostly affected in HD.37,38,57 Such important features of HD KI mice provide an avenue to dissect out the mechanism underlying the selective neurodegeneration and early pathology seen in HD.
Conditional HD mouse models expressing N-terminal mutant Htt
The above transgenic and KI HD mice ubiquitously express mutant Htt in both neuronal and non-neuronal tissues, which is also distributed in various subcellular locations, making it difficult to determine the cell type-specific toxicity of mutant Htt. Conditional HD mouse models that express mutant Htt in specific types of cells have been established to address the specific effects of mutant Htt in a cell type-dependent manner.
In brains from HD patients, striatal and cortical neurons are mainly deteriorated. In the striatum, MSNs comprise more than 90% of the total neuronal population,58 and in the cortex the large pyramidal neurons in layers III, V, and VI are predominantly affected in HD.10 Conditional HD mouse models with mutant Htt expression restricted to the striatum or cortex were established.59 In conditional BACHD mice, LoxP sequences were inserted into the 5′-untranslated region of mutant Htt and in intron 1 flanking mutant Htt exon 1. Therefore, these two LoxP do not interfere with expression of mutant Htt but do allow BACHD mice to be a conditional inactivation model in which Cre expression can switch off mutant Htt expression. Wang et al crossed BACHD mice with Emx1-Cre (cortex CPN-specific Cre), Rgs9-Cre (striatal MSN-specific Cre), or Emx1-Cre; Rgs9-Cre double-transgenic mice to genetically reduce mutant Htt expression in neuronal populations in the cortex, striatum, or both.59 They showed that reduction of cortical mutant Htt expression in BACHD mice partially ameliorates motor and psychiatric-like behavioral deficits, but does not improve brain atrophy, whereas reduction of mutant Htt expression in both cortical and striatal neurons results in the most robust amelioration of all behavioral deficits and selective brain atrophy in BACHD mice.59 This study suggests that the optimal therapeutic strategy may require targeting mutant Htt in both cortical and striatal neurons in HD.
The animal brain consists of neurons and glial cells, which comprise more than 90% of the total cell population. Glia can maintain homeostasis, form myelin, and provide support and protection for neurons in the brain. Although mutant Htt is ubiquitously expressed in neuronal and non-neuronal cells, the role of mutant Htt in glial cells remains elusive, despite the fact that white matter degeneration is found in the brains of HD patient.60–65 A mouse model (GFAP [glial fibrillary acidic protein]-Htt), which expresses N-terminal mutant Htt in astrocytes, the major type of glial cell, under the control of the human GFAP promoter, also shows age-dependent neurological phenotypes.66 These phenotypes are characterized by body weight loss, motor function deficits, and earlier death than wild-type or control transgenic mice. The GFAP-Htt model revealed that mutant Htt binding to Sp1 reduces the associating of Sp1 with the promoter of glutamate transporter, thus resulting in decreased expression of GLT-1 and impaired glutamate uptake.66 The findings provide a new mechanistic insight into the excitotoxic damage in HD. Recently, transgenic HD mice expressing mutant Htt in microglial cells are also reported and show some pathological changes.67,68 All these findings imply an important role for non-neuronal mutant Htt in HD pathology.
The unique structure for neuronal interactions is the synapse. However, how synaptic mutant Htt directly mediates the neuropathology remains unclear. A transgenic HD mouse model was also generated, which selectively expresses mutant Htt in the presynaptic nerve terminal by fusing mutant Htt to synaptosomal-associated protein 25.69 Although transgenic Htt is expressed at a lower level than control Htt with a normal polyQ repeat, it causes progressive and age-dependent neurological symptoms and early death. This interaction of mutant Htt with the C-terminal proline-rich domain of synapsin-1 in this mouse model caused a decrease of glutamate release from synapses.69 Thus, the transgenic HD mouse models that selectively express mutant Htt in selective types of cells or subcellular regions have provided us with new insight into the pathogenesis of HD (Figure 1).
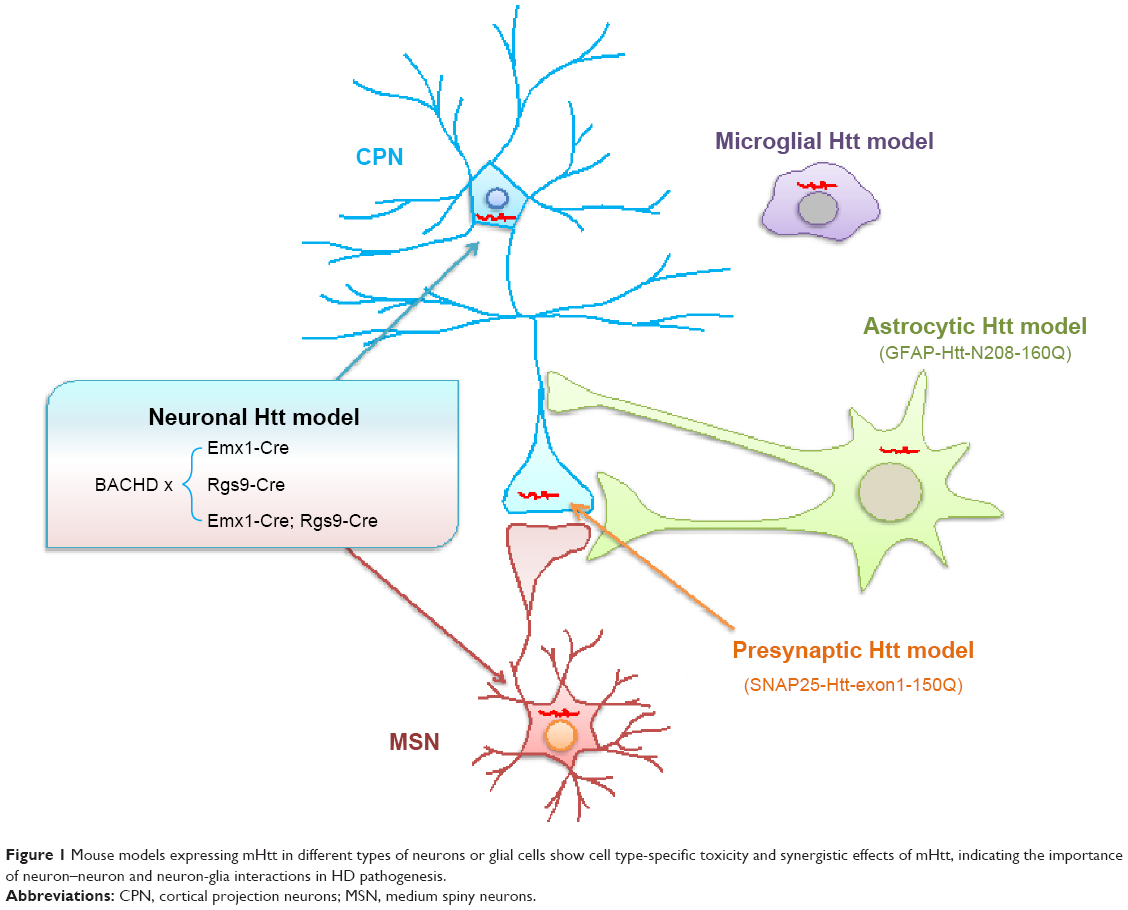 | Figure 1 Mouse models expressing mHtt in different types of neurons or glial cells show cell type-specific toxicity and synergistic effects of mHtt, indicating the importance of neuron–neuron and neuron-glia interactions in HD pathogenesis. |
Transgenic large animal models of HD
Although a variety of mouse models of HD have been extensively studied, none of them show robust neurodegeneration like that seen in the brains of HD patients. This scenario is similar to the transgenic mouse models of Alzheimer’s disease and Parkinson’s disease, in which there is also a lack of overt neurodegeneration.70,71 The failure of transgenic mouse models to replicate striking neurodegeneration in patient brains could be due to biological differences between humans and mice.72 Since the aging process is quite different between small and large animals, mutant Htt may accumulate or be cleared differentially in the brains of different species, which could also contribute to differential pathologies in rodents and large animals. Whether larger transgenic animal models can mimic important neurodegenerative features caused by misfolded proteins remains to be rigorously tested. Transgenic non-human primate models expressing the disease genes were established.12,73 Of these large animal models, transgenic HD rhesus monkeys express exon 1 mutant Htt with 84Q under the control of the human ubiquitin promoter.12 These HD monkeys were generated by injecting lentiviruses into fertilized oocytes to express mutant Htt. Unlike transgenic mice, which can survive after birth when expressing the same exon 1 mutant Htt with an even longer polyQ repeat (150Q),30,74 HD transgenic monkeys with 84Q die postnatally, and this early death is associated with the levels of mutant Htt.12 Despite their early death, some transgenic monkeys developed key clinical HD features including dystonia, chorea, and seizure,12 which have not been replicated by mouse models or other small animal models. Like the brains of HD mouse models and patients, HD monkey brains also show abundant Htt aggregates in the neuronal nuclei and neuronal processes. More importantly, transgenic HD monkeys display degeneration of axons and neuronal processes in the absence of obvious cell body degeneration,45 suggesting that neuronal degeneration in HD may initiate from neuronal processes.
Similarly, transgenic HD pigs that express N-terminal mutant Htt consisting of the first 208 amino acids with 105Q (N208–105Q) were also generated.75 The transgenes were expressed under the control of the cytomegalovirus enhancer and chicken beta-actin (CAG) promoter to allow the ubiquitous expression of transgenes in all tissues. Primary porcine fetal fibroblast cells expressing this mutant Htt fragment were used to generate transgenic HD pigs via nuclear transfer. Six early pregnancies were established, and four of them went to term, with five live births. Like transgenic monkey models of HD, most of these transgenic HD piglets die postnatally, and some transgenic HD pigs show a severe chorea phenotype before death. Thus, the postnatal death of transgenic HD piglets also suggests that mutant Htt is more toxic to larger animals. More importantly, in all transgenic pig brains examined, there were apoptotic cells, which have not been reported in any HD mouse models.
Studies of transgenic large animals also show that smaller N-terminal mutant Htt is more toxic. This is because transgenic pigs76 and sheep77 that express much large Htt fragments develop non-detectable or very mild phenotypes. Thus, even in large animals, expression of small N-terminal mutant Htt fragments appears to be necessary to facilitate disease progression.
Current therapeutic strategies
Based on the findings from animal models of HD, it is clear that continuous accumulation of the misfolded mutant Htt is key to developing HD pathology. Accordingly, several therapeutic strategies have been developed.
Decrease production of mutant Htt
Gene suppression methods provide us with a great tool in HD therapy, and the most popular approach is to use antisense oligonucleotide and RNA interference (RNAi).78,79 This approach has been successfully used in HD mouse models and demonstrates favorable effects in alleviating neurological symptoms in HD mice.80–83 Because the function of normal Htt is essential for early embryonic development,84–86 great effort has been made to develop specific RNAi that only suppresses the expression of mutant Htt.87,88 It has been reported that partial lowering of mutant Htt over months in adult rodent models of HD is effective in reducing neuropathology, improving motor behavior, and prolonging survival.89–93 However, allele-specific RNAi has to be designed based on polymorphic differences in Htt genes between individuals, and an efficient tool to deliver RNAi and antisense oligonucleotide into human brains remains to be established before their clinical application to suppress expression of mutant Htt in patients.83,94
Enhance clearance of mutant Htt
Misfolded and degraded proteins can be cleared by the ubiquitin-proteasome system (UPS). It has been reported that the activity of the UPS declines with age,95–97 which correlates with age-dependent accumulation of mutant Htt in the mouse brain. Also, there is less UPS activity in neuronal cells than in glial cells, suggesting that differential UPS activity contributes to the preferential accumulation of mutant Htt in neuronal cells.98 Many in vitro studies have demonstrated that inhibiting UPS activity can promote accumulation of mutant Htt in cultured cells.99,100 Normally the N-terminal of mutant Htt follows a K48-mediated ubiquitination, which is required for degradation by the proteasome. Degradation of ubiquitinated proteins by the proteasomes requires ubiquitin-conjugating enzymes and ubiquitin-protein ligase E3A. While mutant Htt can be ubiquitinated via K48 in ubiquitin and then targeted via ubiquitin-ligating (E3) enzymes to the proteasome for degradation, the ubiquitin-conjugating (E2) enzyme promotes ubiquitination of mutant Htt via K63 in ubiquitin, resulting in more aggregation of mutant Htt. Reducing ubiquitin-conjugating (E2) or overexpressing ubiquitin-ligating (E3) in the HD mouse brain attenuates Htt aggregation in HD mouse models.100,101 These findings suggest that drugs that activate the UPS would be likely to help remove mutant Htt.
The autophagy-lysosomal pathway is another way to degrade mutant Htt.47,48 The lipophilic macrolide antibiotic rapamycin was reported to inactivate mammalian target of rapamycin, and this inhibition can induce autophagy to reduce HD symptoms in fly and mouse models.102,103 However, rapamycin has broad effects, so its protection is likely due to the combination of its effects on different cellular functions. A more specific drug that only acts on autophagy remains to be developed and may show more specific and efficient clearance of mutant Htt, with less off-target effects.
Reduce gain of toxicity of mutant Htt
PolyQ expansion confers abnormal conformation of Htt, which alters its interactions with other proteins. For example, mutant Htt binds more tightly to Hap1 and Sp1 to alter protein trafficking and transcriptional function.104–106 Drugs or small peptides that can prevent this abnormal interaction would possibly reduce the toxicity of mutant Htt. This possibility is supported by expressing engineered intracellular antibodies, known as intrabodies, which were designed to selectively bind to mutant Htt exon 1 in HD mouse brains. The expression of such intrabodies can ameliorate motor deficit symptoms in multiple HD mouse models.107–109 Similarly, overexpression of Q-rich prion-like proteins suppresses polyQ cytotoxicity via altering the interactions of polyQ proteins with other interactors.110
Improve specific cellular function that can be affected by mutant Htt
Accumulation of mutant Htt can affect a variety of cellular functions. Mutant Htt in the nucleus can affect gene transcription. Cytoplasmic mutant Htt associates with a number of proteins to affect intracellular trafficking, mitochondrial function, and synaptic transmission to induce excitotoxicity, inflammation, and cell death (Figure 2).
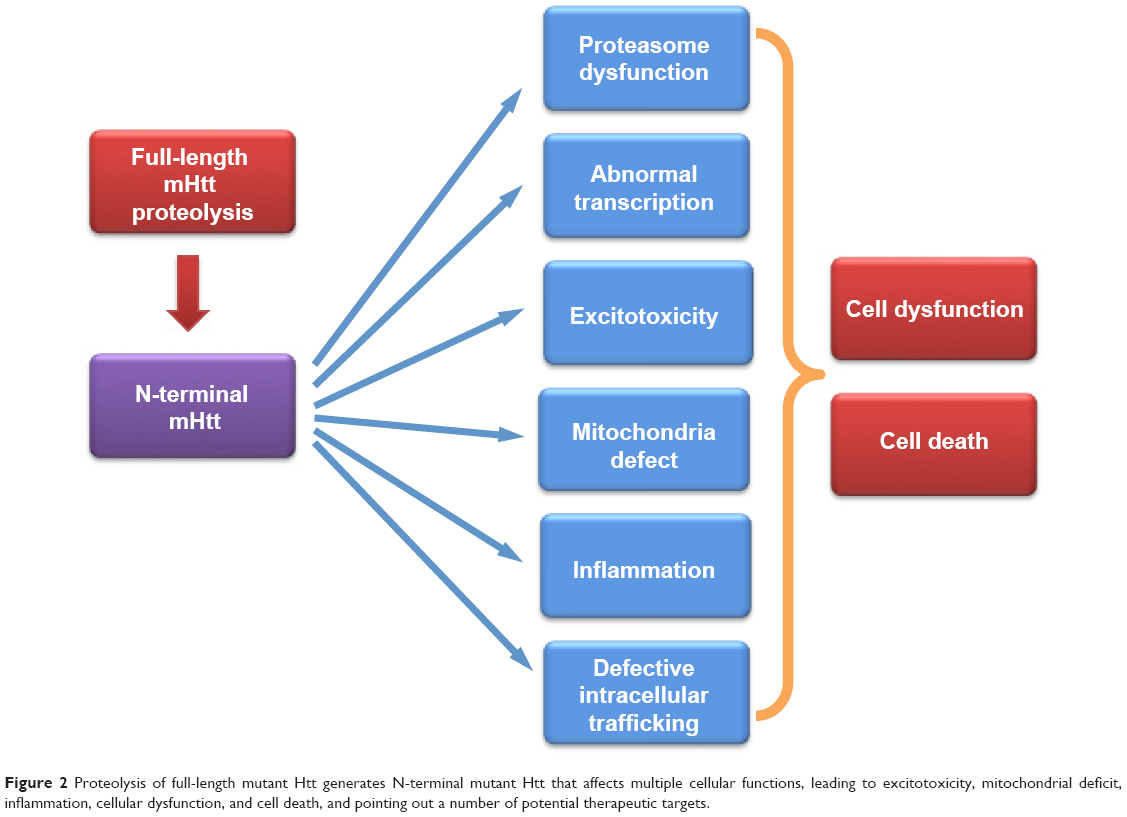 | Figure 2 Proteolysis of full-length mutant Htt generates N-terminal mutant Htt that affects multiple cellular functions, leading to excitotoxicity, mitochondrial deficit, inflammation, cellular dysfunction, and cell death, and pointing out a number of potential therapeutic targets. |
Drugs that selectively improve one or more of the above defects caused by mutant Htt have been tested in HD mouse models. For example, histone deacetylase inhibitors, which are known to improve transcriptional activity, have been used in HD mice and show beneficial effects.111–113 Neurotrophic factors that can prevent neuronal cells also show protective effects in HD mouse models.114–117 Several drugs, such as coenzyme Q10, which can improve mitochondrial function, have been used clinically.118,119 However, to date, there is no effective drug that can be used clinically to stop or reverse progression of HD.
Use of HD animal models to identify therapeutics
The established animal models of HD provide us with important tools to identify therapies for HD. However, these animal models express different forms of mutant Htt and show different neurological phenotypes in various time frames. Thus, it would be important to utilize the unique features of these HD animal models when choosing them for drug studies. Transgenic HD mice, such as R6/2 and N171-82Q, develop severe phenotypes with rapid disease progression. The pathological alterations in these mice are not restricted to neuronal cells in the central nervous system, and may also occur in the peripheral tissues. Thus, drugs that can significantly delay disease progression in these mice are likely to improve the function in the brain and peripheral tissues as well. However, overexpression of small truncated N-terminal mutant Htt may generate pathological events that do not happen when full-length mutant Htt is expressed at the endogenous level. YAC and BAC HD mice are important models for drug studies as they express full-length mutant human Htt. The toxicity of human mutant Htt may be different from transgenic N-terminal Htt, which carries a large polyQ repeat. Human Htt may have specific post-translational modifications that can also influence its toxicity in transgenic mice.
Since these full-length Htt transgenic mice often show the overweight phenotype, which is not seen in HD patients but can influence rotarod test performance, drugs that can improve rotarod performance should also be considered for their potential effects on metabolism and body weight and need to be verified by examining their protective effects on the HD neuropathology in these mice. HD KI mice express expanded-polyQ Htt at the endogenous level and develop neurological symptoms slowly and to much less of an extent than transgenic HD mice. Thus, they are especially useful for identifying initial pathological changes. For example, one can examine how mutant Htt preferentially accumulates in the striatal neurons in HD KI mice, which mirrors the preferential degeneration of striatal neurons in HD patients. Further, transcriptional and proteomic analysis of HD KI mice during the course of disease development may identify molecular targets that are likely to contribute to disease progression. Although HD is an autosomal dominant neurodegenerative disease caused by polyQ expansion in Htt, environment influences, such as housing and diet, could also influence the age of onset and disease severity in HD animal models. Finally, although HD mice do not show obvious neurodegeneration, use of transgenic large animal models of HD that show similar neuropathology to that in HD patient brains will offer an alternative avenue to identify therapeutics that can reduce or ameliorate neurodegeneration.
Conclusion
A variety of transgenic animal models of HD have been established and provided an important insight into the pathogenesis of HD. Various transgenic mouse models of HD are widely used for identifying therapeutics. Each model possesses unique phenotypes or pathological characteristics of HD, which would allow one to find therapeutics that may have broad protective effects or specific effects in a cell type-dependent manner. Because mutant Htt interacts with a number of proteins and affects multiple cellular functions, the most powerful therapy would be one that selectively suppresses the expression of mutant Htt. Even before we develop such a powerful therapeutic tool, drugs that can selectively target downstream pathways that are impaired in HD can also be beneficial for treating HD. However, combined therapeutics that can synergistically improve the functions of multiple pathways are more likely to achieve the goal of effectively reducing HD pathology and delaying disease progression. Because HD shares many common pathological events with other neurodegenerative diseases that are also caused by accumulation of misfolded proteins, identification of therapeutics using HD animal models could also help find treatment for other neurodegenerative diseases.
Disclosure
The authors report no conflicts of interest in this work.
References
Macdonald ME, Ambrose CM, Duyao MP, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntingtons disease chromosomes. Cell. 1993;72(6):971–983. | ||
Duyao MP, Ambrose CM, Myers RH, et al. Trinucleotide repeat length – instability and age-of-onset in Huntingtons disease. Am J Hum Genet. 1993;53(3):1152. | ||
Stine OC, Pleasant N, Franz ML, Abbott MH, Folstein SE, Ross CA. Correlation between the onset age of Huntingtons-disease and length of the trinucleotide repeat in It-15. Hum Mol Genet. 1993;2(10):1547–1549. | ||
Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet. 1997;60(5):1202–1210. | ||
Harper PS. Huntington’s Disease. 2nd ed: London, UK: WB Saunders; 1996. | ||
Walker FO. Huntington’s disease. Lancet. 2007;369(9557):218–228. | ||
Pouladi MA, Morton AJ, Hayden MR. Choosing an animal model for the study of Huntington’s disease. Nat Rev Neurosci. 2013;14(10):708–721. | ||
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP. Neuropathological classification of Huntingtons-disease. J Neuropathol Exp Neurol. 1985;44(6):559–577. | ||
Menalled LB, Chesselet MF. Mouse models of Huntington’s disease. Trends Pharmacol Sci. 2002;23(1):32–39. | ||
Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57(5):369–384. | ||
Jackson GR, Salecker I, Dong XZ, et al. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron. 1998;21(3):633–642. | ||
Yang SH, Cheng PH, Banta H, et al. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature. 2008;453(7197):921–924. | ||
Heng MY, Detloff PJ, Albin RL. Rodent genetic models of Huntington disease. Neurobiol Dis. 2008;32(1):1–9. | ||
Kim J, Bordiuk OL, Ferrante RJ. Experimental models of HD and reflection on therapeutic strategies. Int Rev Neurobiol. 2011;98:419–481. | ||
Crook ZR, Housman D. Huntington’s disease: can mice lead the way to treatment? Neuron. 2011;69(3):423–435. | ||
Southwell AL, Patterson PH. Gene therapy in mouse models of Huntington disease. Neuroscientist. 2011;17(2):153–162. | ||
Munoz-Sanjuan I, Bates GP. The importance of integrating basic and clinical research toward the development of new therapies for Huntington disease. J Clin Invest. 2011;121(2):476–483. | ||
Appl T, Kaltenbach L, Lo DC, Terstappen GC. Targeting mutant huntingtin for the development of disease-modifying therapy. Drug Discov Today. 2012;17(21–22):1217–1223. | ||
Lee CY, Cantle JP, Yang XW. Genetic manipulations of mutant huntingtin in mice: new insights into Huntington’s disease pathogenesis. FEBS J. 2013;280(18):4382–4394. | ||
Mangiarini L, Sathasivam K, Seller M, et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87(3):493–506. | ||
Tang B, Seredenina T, Coppola G, et al. Gene expression profiling of R6/2 transgenic mice with different CAG repeat lengths reveals genes associated with disease onset and progression in Huntington’s disease. Neurobiol Dis. 2011;42(3):459–467. | ||
Li SH, Li XJ. Aggregation of N-terminal huntingtin is dependent on the length of its glutamine repeats. Hum Mol Genet. 1998;7(5):777–782. | ||
DiFiglia M, Sapp E, Chase KO, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277(5334):1990–1993. | ||
Landles C, Sathasivam K, Weiss A, et al. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J Biol Chem. 2010;285(12):8808–8823. | ||
Schilling G, Klevytska A, Tebbenkamp AT, et al. Characterization of huntingtin pathologic fragments in human Huntington disease, transgenic mice, and cell models. J Neuropathol Exp Neurol. 2007;66(4):313–320. | ||
Gutekunst CA, Li SH, Yi H, et al. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci. 1999;19(7):2522–2534. | ||
Turmaine M, Raza A, Mahal A, Mangiarini L, Bates GP, Davies SW. Nonapoptotic neurodegeneration in a transgenic mouse model of Hunting-ton’s disease. Proc Natl Acad Sci U S A. 2000;97(14):8093–8097. | ||
Schilling G, Becher MW, Sharp AH, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;8(5):397–407. | ||
Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers – their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91(12):5355–5358. | ||
Davies SW, Turmaine M, Cozens BA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90(3):537–548. | ||
Gray M, Shirasaki DI, Cepeda C, et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28(24):6182–6195. | ||
Slow EJ, van Raamsdonk J, Rogers D, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12(13):1555–1567. | ||
Hodgson JG, Agopyan N, Gutekunst CA, et al. A YAC mouse model for Huntington’s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23(1):181–192. | ||
Pouladi MA, Xie Y, Skotte NH, et al. Full-length huntingtin levels modulate body weight by influencing insulin-like growth factor 1 expression. Hum Mol Genet. 2010;19(8):1528–1538. | ||
Chiang C, Jacobsen JC, Ernst C, et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet. 2012;44(4):390–397. | ||
Shelbourne PF, Killeen N, Hevner RF, et al. A Huntington’s disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum Mol Genet. 1999;8(5):763–774. | ||
Wheeler VC, White JK, Gutekunst CA, et al. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9(4):503–513. | ||
Lin CH, Tallaksen-Greene S, Chien WM, et al. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum Mol Genet. 2001;10(2):137–144. | ||
Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington’s disease with 140 CAG repeats. J Comp Neurol. 2003;465(1):11–26. | ||
Wheeler VC, Auerbach W, White JK, et al. Length-dependent gametic CAG repeat instability in the Huntington’s disease knock-in mouse. Hum Mol Genet. 1999;8(1):115–122. | ||
Heng MY, Tallaksen-Greene SJ, Detloff PJ, Albin RL. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington’s disease. J Neurosci. 2007;27(34):8989–8998. | ||
Woodman B, Butler R, Landles C, et al. The Hdh(Q150/Q150) knock-in mouse model of HD and the R6/2 exon 1 model develop comparable and widespread molecular phenotypes. Brain Res Bull. 2007;72(2–3):83–97. | ||
Heikkinen T, Lehtimaki K, Vartiainen N, et al. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington’s disease. PLoS One. 2012;7(12):e50717. | ||
Menalled LB, Kudwa AE, Miller S, et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington’s disease: zQ175. PLoS One. 2012;7(12):e49838. | ||
Wang CE, Tydlacka S, Orr AL, et al. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington’s disease. Hum Mol Genet. 2008;17(17):2738–2751. | ||
Leavitt BR, Wellington CL, Hayden MR. Recent insights into the molecular pathogenesis of Huntington disease. Semin Neurol. 1999;19(4):385–395. | ||
Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015;38(1):26–35. | ||
Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012;11(9):709–730. | ||
Kim YJ, Yi Y, Sapp E, et al. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci U S A. 2001;98(22):12784–12789. | ||
Wellington CL, Singaraja R, Ellerby L, et al. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem. 2000;275(26):19831–19838. | ||
O’Brien T, Lee D. Prospects for caspase inhibitors. Mini Rev Med Chem. 2004;4(2):153–165. | ||
Gafni J, Hermel E, Young JE, Wellington CL, Hayden MR, Ellerby LM. Inhibition of calpain cleavage of huntingtin reduces toxicity: accumulation of calpain/caspase fragments in the nucleus. J Biol Chem. 2004;279(19):20211–20220. | ||
Miller JP, Holcomb J, Al-Ramahi I, et al. Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington’s disease. Neuron. 2010;67(2):199–212. | ||
Lunkes A, Lindenberg KS, Ben-Haiem L, et al. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10(2):259–269. | ||
Graham RK, Deng Y, Slow EJ, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125(6):1179–1191. | ||
Gafni J, Papanikolaou T, Degiacomo F, et al. Caspase-6 activity in a BACHD mouse modulates steady-state levels of mutant huntingtin protein but is not necessary for production of a 586 amino acid proteolytic fragment. J Neurosci. 2012;32(22):7454–7465. | ||
Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25(4):385–389. | ||
Tepper JM, Tecuapetla F, Koos T, Ibanez-Sandoval O. Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. 2010;4:150. | ||
Wang N, Gray M, Lu XH, et al. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nat Med. 2014;20(5):536–541. | ||
Rosas HD, Lee SY, Bender AC, et al. Altered white matter microstructure in the corpus callosum in Huntington’s disease: implications for cortical “disconnection”. Neuro Image. 2010;49(4):2995–3004. | ||
Muller HP, Glauche V, Novak MJ, et al. Stability of white matter changes related to Huntington’s disease in the presence of imaging noise: a DTI study. PLoS Curr. 2011;3:RRN1232. | ||
Ciarmiello A, Cannella M, Lastoria S, et al. Brain white-matter volume loss and glucose hypometabolism precede the clinical symptoms of Huntington’s disease. J Nucl Med. 2006;47(2):215–222. | ||
Stoffers D, Sheldon S, Kuperman JM, Goldstein J, Corey-Bloom J, Aron AR.Contrasting gray and white matter changes in preclinical Huntington disease: an MRI study. Neurology. 2010;74(15):1208–1216. | ||
Bohanna I, Georgiou-Karistianis N, Sritharan A, et al. Diffusion tensor imaging in Huntington’s disease reveals distinct patterns of white matter degeneration associated with motor and cognitive deficits. Brain Imaging Behav. 2011;5(3):171–180. | ||
Dumas EM, van den Bogaard SJ, Hart EP, et al. Reduced functional brain connectivity prior to and after disease onset in Huntington’s disease. Neuroimage Clin. 2013;2:377–384. | ||
Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci U S A. 2009;106(52):22480–22485. | ||
Sapp E, Kegel KB, Aronin N, et al. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol. 2001;60(2):161–172. | ||
Crotti A, Benner C, Kerman BE, et al. Mutant huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014;17(4):513–521. | ||
Xu Q, Huang S, Song M, et al. Synaptic mutant huntingtin inhibits synapsin-1 phosphorylation and causes neurological symptoms. J Cell Biol. 2013;202(7):1123–1138. | ||
Kitazawa M, Medeiros R, Laferla FM. Transgenic mouse models of Alzheimer disease: developing a better model as a tool for therapeutic interventions. Curr Pharm Des. 2012;18(8):1131–1147. | ||
Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron. 2010;66(5):646–661. | ||
Li XJ, Li S. Influence of species differences on the neuropathology of transgenic Huntington’s disease animal models. J Genet Genomics. 2012;39(6):239–245. | ||
Niu Y, Guo X, Chen Y, et al. Early Parkinson’s disease symptoms in alpha-synuclein transgenic monkeys. Hum Mol Genet. 2015;24(8):2308–2317. | ||
Cheng PH, Li CL, Her LS, et al. Significantly differential diffusion of neuropathological aggregates in the brain of transgenic mice carrying N-terminal mutant huntingtin fused with green fluorescent protein. Brain Struct Funct. 2013;218(1):283–294. | ||
Yang DS, Wang CE, Zhao BT, et al. Expression of Huntington’s disease protein results in apoptotic neurons in the brains of cloned transgenic pigs. Hum Mol Genet. 2010;19(20):3983–3994. | ||
Baxa M, Hruska-Plochan M, Juhas S, et al. A transgenic minipig model of Huntington’s disease. J Huntingtons Dis. 2013;2(1):47–68. | ||
Jacobsen JC, Bawden CS, Rudiger SR, et al. An ovine transgenic Huntington’s disease model. Hum Mol Genet. 2010;19(10):1873–1882. | ||
Kordasiewicz HB, Stanek LM, Wancewicz EV, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74(6):1031–1044. | ||
Stanek LM, Sardi SP, Mastis B, et al. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum Gene Ther. 2014;25(5):461–474. | ||
Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell. 2000;101(1):57–66. | ||
Harper SQ, Staber PD, He X, et al. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A. 2005;102(16):5820–5825. | ||
Carroll JB, Warby SC, Southwell AL, et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19(12):2178–2185. | ||
Sah DW, Aronin N. Oligonucleotide therapeutic approaches for Huntington disease. J Clin Invest. 2011;121(2):500–507. | ||
Duyao MP, Auerbach AB, Ryan A, et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science. 1995;269(5222):407–410. | ||
Nasir J, Floresco SB, O’Kusky JR, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81(5):811–823. | ||
Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. 1995;11(2):155–163. | ||
van Bilsen PH, Jaspers L, Lombardi MS, Odekerken JC, Burright EN, Kaemmerer WF. Identification and allele-specific silencing of the mutant huntingtin allele in Huntington’s disease patient-derived fibroblasts. Hum Gene Ther. 2008;19(7):710–719. | ||
Pfister EL, Kennington L, Straubhaar J, et al. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr Biol. 2009;19(9):774–778. | ||
Boudreau RL, McBride JL, Martins I, et al. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther. 2009;17(6):1053–1063. | ||
Drouet V, Perrin V, Hassig R, et al. Sustained effects of nonallele-specific huntingtin silencing. Ann Neurol. 2009;65(3):276–285. | ||
Rodriguez-Lebron E, Denovan-Wright EM, Nash K, Lewin AS, Mandel RJ. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol Ther. 2005;12(4):618–633. | ||
Machida Y, Okada T, Kurosawa M, Oyama F, Ozawa K, Nukina N. rAAV-mediated shRNA ameliorated neuropathology in Huntington disease model mouse. Biochem Biophys Res Commun. 2006;343(1):190–197. | ||
DiFiglia M, Sena-Esteves M, Chase K, et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci U S A. 2007;104(43):17204–17209. | ||
Mantha N, Das SK, Das NG. RNAi-based therapies for Huntington’s disease: delivery challenges and opportunities. Ther Deliv. 2012;3(9):1061–1076. | ||
Kapphahn RJ, Bigelow EJ, Ferrington DA. Age-dependent inhibition of proteasome chymotrypsin-like activity in the retina. Exp Eye Res. 2007;84(4):646–654. | ||
Keller JN, Hanni KB, Markesbery WR. Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev. 2000;113(1):61–70. | ||
Keller JN, Huang FF, Markesbery WR. Decreased levels of proteasome activity and proteasome expression in aging spinal cord. Neuroscience. 2000;98(1):149–156. | ||
Tydlacka S, Wang CE, Wang XJ, Li SH, Li XJ. Differential activities of the ubiquitin-proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci. 2008;28(49):13285–13295. | ||
Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292(5521):1552–1555. | ||
Bhat KP, Yan S, Wang CE, Li S, Li XJ. Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc Natl Acad Sci U S A. 2014;111(15):5706–5711. | ||
Yin P, Tu Z, Yin A, et al. Aged monkey brains reveal the role of ubiquitin-conjugating enzyme UBE2N in the synaptosomal accumulation of mutant huntingtin. Hum Mol Genet. 2015;24(5):1350–1362. | ||
Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36(6):585–595. | ||
Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. Biol Chem. 1998;273(7):3963–3966. | ||
Li XJ, Li SH, Sharp AH, et al. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378(6555):398–402. | ||
Li SH, Cheng AL, Zhou H, et al. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol. 2002;22(5):1277–1287. | ||
Dunah AW, Jeong H, Griffin A, et al. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 2002;296(5576):2238–2243. | ||
Wang CE, Zhou H, McGuire JR, et al. Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant huntingtin. J Cell Biol. 2008;181(5):803–816. | ||
Southwell AL, Ko J, Patterson PH. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington’s disease. J Neurosci. 2009;29(43):13589–13602. | ||
Messer A, Joshi SN. Intrabodies as neuroprotective therapeutics. Neurotherapeutics. 2013;10(3):447–458. | ||
Ripaud L, Chumakova V, Antonin M, et al. Overexpression of Q-rich prion-like proteins suppresses polyQ cytotoxicity and alters the polyQ interactome. Proc Natl Acad Sci U S A. 2014;111(51):18219–18224. | ||
Jia HQ, Pallos J, Jacques V, et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol Dis. 2012;46(2):351–361. | ||
Thomas EA, Coppola G, Desplats PA, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci U S A. 2008;105(40):15564–15569. | ||
Steffan JS, Bodai L, Pallos J, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413(6857):739–743. | ||
Gauthier LR, Charrin BC, Borrell-Pages M, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118(1):127–138. | ||
Chen LW, Horng LY, Wu CL, et al. NGF has a role in the potentiation of neuroregeneration in a mouse model of Huntington’s disease. Mov Disord. 2011;26:S57–S58. | ||
Jin K, LaFevre-Bernt M, Sun Y, et al. FGF-2 promotes neurogenesis and neuroprotection and prolongs survival in a transgenic mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. 2005;102(50):18189–18194. | ||
Zuccato C, Liber D, Ramos C, et al. Progressive loss of BDNF in a mouse model of Huntington’s disease and rescue by BDNF delivery. Pharmacol Res. 2005;52(2):133–139. | ||
Schilling G, Coonfield ML, Ross CA, Borchelt DR. Coenzyme Q10 and remacemide hydrochloride ameliorate motor deficits in a Huntington’s disease transgenic mouse model. Neurosci Lett. 2001;315(3):149–153. | ||
Killoran A, Biglan K, Julian-Baros E, Yoritomo N, Ross C, PREQUEL HSG. Update on PREQUEL: a study of coenzyme Q10 in premanifest Huntington’s disease. Neurotherapeutics. 2013;10(1):181–182. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.