")
Back to Journals » International Journal of Nanomedicine » Volume 15
Transferrin Receptor-Targeted PEG-PLA Polymeric Micelles for Chemotherapy Against Glioblastoma Multiforme
Authors Sun P, Xiao Y, Di Q, Ma W, Ma X, Wang Q, Chen W
Received 16 April 2020
Accepted for publication 10 August 2020
Published 11 September 2020 Volume 2020:15 Pages 6673—6687
DOI https://doi.org/10.2147/IJN.S257459
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mian Wang
Ping Sun,1,2 Yue Xiao,1 Qianqian Di,1 Wenjing Ma,1 Xingyu Ma,1 Qingqing Wang,3 Weilin Chen1
1Guangdong Provincial Key Laboratory of Regional Immunity and Diseases, Department of Immunology, Shenzhen University School of Medicine, Shenzhen 518060, People’s Republic of China; 2Key Laboratory for Biomedical Measurements and Ultrasound Imaging, School of Biomedical Engineering, Shenzhen University Health Science Center, Shenzhen 518060, People’s Republic of China; 3Institute of Immunology, Zhejiang University of Medicine School, Hangzhou, Zhejiang 310058, People’s Republic of China
Correspondence: Weilin Chen
Guangdong Provincial Key Laboratory of Regional Immunity and Diseases, Department of Immunology, Shenzhen University School of Medicine, Shenzhen, People’s Republic of China
Email [email protected]
Background: The safe and efficient delivery of chemotherapeutic agents is critical to glioma therapy. However, chemotherapy for glioma is extremely challenging because the blood–brain barrier (BBB) rigorously prevents drugs from reaching the tumor region.
Materials and Methods: TfR-T12 peptide-modified PEG-PLA polymer was synthesized to deliver paclitaxel (PTX) for glioma therapy. TfR was significantly expressed on brain capillary endothelial cells and glioma cells; therefore, TfR-T12 peptide-modified micelles can cross the BBB system and target glioma cells.
Results: TfR-T12-PEG-PLA/PTX polymeric micelles (TfR-T12-PMs) could be absorbed rapidly by tumor cells, and traversed effectively the BBB monolayers. TfR-T12-PMs can effectively inhibit the proliferation of U87MG cells in vitro, and TfR-T12-PMs loaded with paclitaxel presented better antiglioma effect with prolonged median survival of nude mice-bearing glioma than the unmodified PMs.
Conclusion: The TfR-T12-PMs could effectively overcome the BBB barrier and accomplish glioma-targeted drug delivery, thus validating its potential in improving the therapeutic outcome in multiforme.
Keywords: polymeric micelles, transferrin receptor, BBB transcytosis effect, glioblastoma multiforme, targeted delivery
Introduction
Malignant glioma is the second leading cause of death from diseases of the central nervous system, threatening human health for fast development and poor prognosis.1–4 The standard clinical treatments for glioma include surgical resection, radiotherapy, chemotherapy, gene therapy, and immunotherapy.5,6 However, clinical outcome is very limited, the survival rate of patients remains very low after current multimodal treatment-aggressive surgical resection followed by radiation and chemotherapy. In addition, the side effects of radiotherapy and the adverse results of chemotherapy are also the main causes of poor prognosis. In recent decades, an active targeted drug delivery system has attracted wide attention for effective delivery of chemotherapeutics to the tumor site, but the clinical treatment effect is not ideal, because these physiological and pathological barriers such as enzymatic barrier, blood–brain barrier (BBB), and blood–brain tumor barrier (BBTB) prevented drug or drug delivery system from reaching the glioma region.7–10
BBB is mainly formed by capillary endothelial cells and remains a compact structure at the early stage of glioma development and around the invasive tumor edge.11,12 It prevents approximately 98% of small molecule drugs and nearly 100% of large molecule substances from transport into the brain tissue.13 Based on previous reports, receptor-mediated transcytosis (RMT) is exploited as a successful pathway to traverse the BBB. There are many of these receptors that mediate the passage of the BBB such as polymerase δ-interacting protein 2, matrix metalloproteinase-9 and transferrin receptor.14–16 The brain related ligand or monoclonal anti-body was used to modify drug delivery systems to facilitate the transport of chemotherapeutic substances to the brain tissue.17
A variety of glioma-targeted drug delivery systems have been introduced to improve the therapeutic effect in previous reports.18–21 One potential strategy is to couple a drug delivery system with a ligand that can recognize specific receptors on brain tumor cells. Glioblastoma multiforme (GBM) cell surface markers, such as integrins avβ3 and avβ5,22 epidermal growth factor receptor,23 transferrin receptor (TfR),24 can help a drug delivery system to recognize glioma cells specifically.25 Among these target sites, TfR has been identified as one of the most promising target sites since it is overexpressed on both the BBB and GBM cells. Transferrin is a hydrophilic transporter of iron ions in the blood, which can enter cells via TfR-mediated endocytosis. Based on previous reports,26,27 many types of cancerous cells, including GBM, overexpress TfR, whereas TfR is less expressed in noncancerous cells. The overexpression of TfR on the BBB and the GBM cell surfaces but not on the surrounding cells renders TfR a promising target. Thereby, transferrin has been widely explored as a targeting ligand for improving nanocarrier penetration into the BBB. Transferrin receptor-mediated nanodrug delivery system was expected to target deliver chemotherapeutics to glioma cells across the BBB, and circumvent barriers to improve the antiglioma effect in vivo.
Herein, we designed a stable peptide modified PM to overcome BBB/BBTBs and target glioma cells. This glioma-targeting drug delivery was decorated with TfR-T12 peptide, which could mediate the drug system to cross the BBB and specifically recognize glioma cells.28,29 TfR-T12 peptide-modified PEG-PLA has been synthesized for the first time in this study, and has not been reported in the literature. PTX has been selected as a therapeutic drug in this study, and its hydrophobicity can be improved by carrier encapsulation for enhancing the bioavailability in tumor therapy. The objective was to achieve glioma targeting with reduced side effects by overcoming multiple barriers (Figure 1). The efficacy of TfR-T12-PMs in crossing the BBB/BBTB and targeting glioma cells was assessed in vitro and in vivo. The potential antiglioma effect of TfR-T12-PMs was also evaluated in nude mice bearing intracranial U87MG glioma.
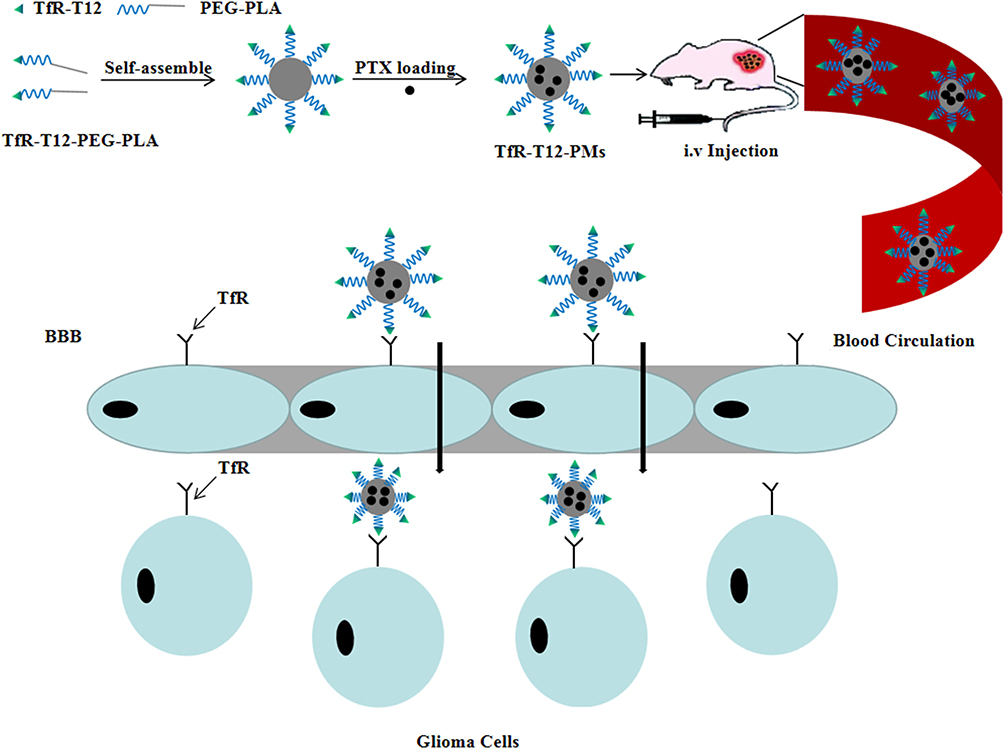 |
Figure 1 Schematic of the intracellular therapeutic mechanism of the TfR-T12-PMs. |
Materials and Methods
Materials, Cells Culture and Animals
The HOOC-PEG5k-PLA5k was purchased from Tanshui Technology Co. Ltd (Guangzhou, China); TfR-T12 peptide (THRPPMWSPVWP) was synthesized by China Peptides Co., Ltd; CCK8 kit was purchased from Yisheng Biotechnology Co., Ltd (Shanghai, China); Annexin V-FITC/PI Apoptosis Detection kit was purchased from Yisheng Biotechnology Co., Ltd; Hoechst 33258 was purchased from Biyuntian Biotechnology Co., Ltd (Shanghai, China); 1,1ʹ-dioctadecyl-3, 3, 3ʹ, 3ʹ-tetramethyl indotricarbocyanine Iodide (DiR) was purchased from Baikang Scientific Instrument Co., Ltd (Shenzhen, China).
U87MG, U118, A172 cells and human umbilical vascular endothelial cells (HUVECs) were purchased from ATCC, and grown in DMEM supplemented with 10% FBS at 37°C under a humidified atmosphere containing 5% CO2.
BALB/C nude mouse (four to six weeks old, male) were purchased from Guangdong Medical Laboratory Animal Center, and kept in specific pathogen free (SPF) conditions. All procedures used in this study were conducted in accordance with the Guide for the Care and Use of Laboratory Animals Center of Shenzhen University and approved by the Experimental Animal Ethics Committee of Shenzhen University (Permit No. 20180425).
Synthesis of TfR-T12 Peptide Decorated PEG-PLA Copolymer
Briefly, 50mg/mL HOOC-PEG5k-PLA5k copolymer was activated by EDC/NHS (10mg EDC/5.8mg NHS) for 3h, and TfR-T12 peptide was added into the DMSO solution, the reaction was maintained for another 24h under moderate stirring.30 Then, the reacted mixture was dialyzed (MW cut off 3500Da) against in distilled water for 48h to remove the unconjugated TfR-T12 peptide, the dialysate was freeze-dried for 24h.
Characterization of TfR-T12 Peptide Decorated PEG-PLA Copolymer
The structures of TfR-T12 peptide, PEG-PLA and TfR-T12-PEG-PLA were characterized by 1H-nuclear magnetic resonance (1H-NMR) spectroscopy.
A172 cells, U87MG, and U118 were seeded in 96-well plates at a density of 8000/well to estimate the toxicity of TfR-T12-PEG-PLA copolymer. Thereafter, the medium was replaced with different concentration copolymer samples (1~100mg/mL), and cell viability was measured using CCK8 kit after 48h incubation.
Preparation of TfR-T12-PEG-PLA/PTX Micelles
Briefly, 100mg TfR-T12-PEG-PLA copolymer and 10mg PTX were dissolved in 2mL DMSO, and then added dropwise into 50mL water under moderate stirring, the mixture was stirred for 30min at RT, and dialyzed for 48h in distilled water (MW cut off 3500Da). The unentrapped PTX was removed by filtration using a 0.45µm filter.
Characterization of TfR-T12-PEG-PLA/PTX Micelles
The encapsulation efficiency (EE) and loading efficiency (LE) of PTX in TfR-T12-PMs were determined by Ultraviolet Spectrophotometry (UV) (Shimadzu UV-2550PC spectrophotometer) at 230nm wavelength. The EE% and LE% were calculated as indicated below:
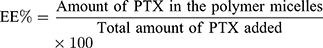

The particle size of TfR-T12-PMs was determined using a Nano laser particle size analyzer (DelsaMax CORE). Zeta potential of PEG-PMs and TfR-T12-PMs was determined using Malvern (Zetasizer Nano-ZS90). The morphology of TfR-T12-PMs was observed by Transmission Electron Microscope (TEM) (JEM-2100 and Aztec Energy TEM SP X-MaxN 80T). The sample solution of TfR-T12-PMs were prepared at the concentration 1mg/mL, measured accurately 20μL and place it on a copper net, and placed the copper net under a lamp for 10minutes to dry, then the morphology of TfR-T12-PMs were observed using TEM.
The in vitro release of PTX from TfR-T12-PMs was assessed up to 48h under physiological condition and the free PTX and PEG-PLA/PTX (PEG-PMs) were as control. Briefly, 1 mL sample containing 0.45mg PTX was added into a dialysis bag. Then, the bags were immersed in 50mL PBS (pH 7.4, containing 0.1% [v/v] Tween 80), incubated in an orbital shaker at 37°C. At predetermined time points, 0.2mL of each sample was withdrawn from the medium and the same volume of fresh medium was added.31 The concentration of PTX was analyzed by UV.
Cellular Uptake Assay
To estimate the uptake efficiency and localization of copolymer micelles, PEG-PMs and TfR-T12-PMs were prepared, DiR was a poorly water-soluble fluorescent substance, which was encapsulated in the carrier instead of PTX. U87MG cells were seeded into a 12-well plate at a density of 6×104 cells/well and incubated overnight at 37°C, then the medium was replaced with free cell culture medium containing various formulation of PMs with 200ng/mL DiR per well for 4h. Cellular uptake of PEG-PMs and TfR-T12-PMs were evaluated by flow cytometry (CYTOFLEX S) and confocal laser scanning microscopy (CLSM) (INVIA REFLEX).
In vitro Cytotoxicity Assay
In briefly, U87MG cells were seeded in 96-well plates and cultured for overnight for confluence. Then the medium was removed and nano-micelle or free PTX with serial different formulations were added. After 24h incubation, 10μL CCK8 working solution was added for 30min, the percent of viability was calculated for each well.
Detection of Apoptosis
Flow cytometry and hoechst staining were used to determine the apoptotic effect. Summarily, the cells were seeded at 5×104 cells per well into 12-well plates and incubated for 24h with the free PTX, PEG-PMs and TfR-T12-PMs, then the cells were stained with annexin V-FITC/PI kit. The cell apoptosis were analyzed by a flow cytometer.
For the hoechst staining assay, cells were fixed with 4% paraformaldehyde for 15min, and then stained with hoechst 33258 reagent at a concentration of 5µg/mL for 10min and washed 3 times with PBS buffer. After that, the nuclear morphology was observed with a fluorescence microscope.
Migration and Invasion Assays
Migration and invasion assays were performed as previously reported.32 Briefly, U87MG cells were used to coat the upper surface of the transwell chamber, and the lower chamber was filled with culture medium containing 20% FBS, after 24h incubation, the medium was replaced by various formulation including PBS, free PTX, PEG-PMs and TfR-T12-NPs, incubated at 37°C for 24h. The cells were fixed in 4% formaldehyde for 15min and then stained with 0.1% crystal violet for 30min. Images of the cell migration were produced using a microscope, and 5 migration area was measured.
Evaluation Transcytosis Effect in HUVEC/U87MG Co-Culture Model
The HUVECs/U87MG co-culture model was established based on previous report.33 Briefly, 5000 HUVECs cells and 2.5×104 U87MG cells were seeded into transwell inserts co-culturing for 72h, HUVECs were seed in upper chamber of the transwell insert, and U87 cells were seeded at lower chamber of the transwell insert. The upper inserts were transferred to another 24-well plate, which was previously seeded with U87MG cells at concentration 5×104 U87MG cells/well, and then treated with free DiR and different DiR formulations. The inserts were removed after 1h of incubation, and HUVECs viability was measured by the CCK8 assay. DiR in U87MG cells was detected by flow cytometer. In addition, in order to further test the penetration effect of TfR-T12-NPs, after HUVECs/U87MG co-culture for 72h, and PTX and different PTX formulations was replaced. After another 24h of incubation, then stained with an apoptosis detection kit and hoechst solution, and the percentage of apoptosis was analyzed using a flow cytometer.
To investigate brain targeted effect of PEG-PMs and TfR-T12-PMs, healthy nude mice were intravenously injected with PEG-PMs or TfR-T12-PMs at the DiR dose of 50μg/kg, all mice were euthanized later, and the heart, liver, spleen, lung, kidney, and brain were excised and tested by three dimensional multimodal imaging system for live small animals (IVIS Spectrum CT).
In vivo Assessment of Anti-Glioma Efficacy of PTX Formulations in Subcutaneous Tumor Model
Male nude mice (4–6weeks) were subcutaneously implanted with 1×107 U87MG cells in the right leg.34,35 2 weeks later, when the solid tumor size reached approximately 80mm3, the nude mice were randomly divided into 4 groups (n=5) and received a intravenous injection of saline, PTX, PEG-PMs and TfR-T12-PMs with a dosage of 5mg/kg. Tumor volume and mice weight was measured every 2 days and tumor volume were calculated as the following formula: tumor volume = (length)×(width)2/2. After 5 times administration, the nude mice were euthanized and their hearts, livers, spleens, lungs, kidneys, tumors were collected for tumor immunohistochemistry and H&E histochemical analysis. In addition, three dimensional multimodal imaging system was carried out to detect distribution of PMs in vivo.
Immunohistochemistry Analysis
Tumors or brains from tumor-bearing mice were fixed in 4.0% paraformaldehyde solution, embedded in paraffin and sectioned. TUNEL staining was used to detect apoptotic cells according to a previously reported method,36 CD31 staining was performed to observe the microvessels,37 and Ki67 staining was detected to observe the tumor cell proliferation.38
In vivo Assessment of Anti-Glioma Efficacy of PTX Formulations in Orthotopic Tumor Model
The orthotropic glioma model was established according to a previously reported method.39 In brief, male nude mice (6–8weeks) were anesthetized with chloralhydrate, and 5×105 U87MG cells were implanted into the right brain (2.0mm lateral to the bregma with 3.5 mm depth) with a stereotactic apparatus (Stoelting). A total of 14 days after tumor cell inoculation, tumor-bearing mice were randomly divided into 4 groups (n=5), and received intravenous injection of saline, PTX, PEG-PMs and TfR-T12-PMs with a dosage of 5mg/kg. A total 5 time of administration, the mice in PEG-PMs and TfR-T12-PMs group were randomly selected and euthanized, and collected their brain for three dimensional multimodal imaging. To estimate the safety of PMs on tumor-bearing mice, after administration, the tumor-bearing mice were euthanized, and tissue sections of vital organs were stained with hematoxylin and eosin (H&E) for histochemical analysis.
The orthotropic glioma model was established as the same as above method, 14 days inoculation later, the tumor-bearing mice were randomly divided into 4 groups (n=7), and received a intravenous injection of saline, PTX, PEG-PMs and TfR-T12-PMs with a dosage of 5mg/kg, the survival ratio of tumor-bearing mice was observed after 5 times of administration. For mouse survival study, Kaplan–Meier survival curves were generated and analyzed by Gehan-Breslow-Wilcoxon test.
Statistical Analysis
All results shown represent means ± SEM from triplicate experiments performed in a parallel manner, unless otherwise indicated. Significant differences between groups were analyzed by one-way ANOVA. All comparisons were made relative to control group, and the significance of difference is indicated as *p < 0.05, **p < 0.01.
Results and Discussion
Synthesis and Characterization of TfR-T12-PEG-PLA Copolymer
To develop a more effective and safer delivery system for glioma therapy, di-block copolymer TfR-T12-PEG-PLA was prepared, and the synthetic route of conjugation of TfR-T12-PEG-PLA copolymer was illustrated in Figure S1. The amine groups from TfR-T12 peptide were conjugated with carboxylic acid group from HOOC-PEG5k-PLA5k to produce amide bond using EDC and NHS as carboxyl activating agent. 1H-NMR spectrum of the TfR-T12-PEG-PLA copolymer was shown in Figure 2A. The methylene group of the PEG segment in the PEG-PLA was detected in the signal of 3.65 ppm, and the signals of CH and CH3 of the lactide from the PLA segment are shown at 5.3 and 1.6 ppm, respectively (Figure S2). The chemical shift at 7.0–8.0 ppm is assigned to the H protons in TfR-T12 peptide blocks. The result indicated that the TfR-T12 peptide is successfully conjugated to PEG-PLA polymer.
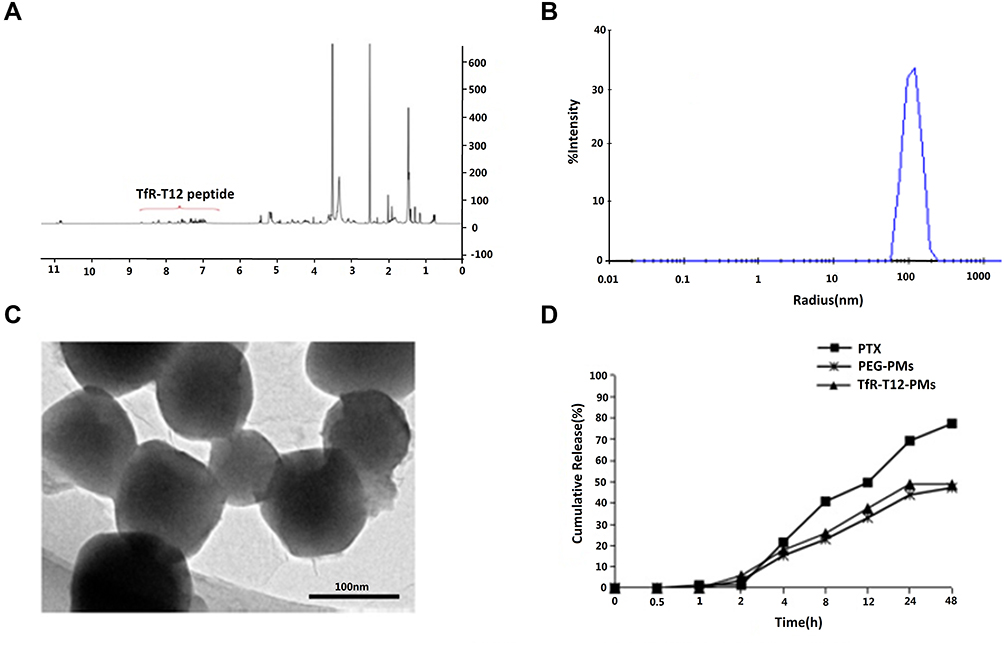 |
Figure 2 The synthesis of TfR-T12-PEG-PLA polymer and characterization of TfR-T12-PMs. (A) 1H-NMR of TfR-T12-PEG-PLA polymer; (B) Particle size of TfR-T12-PMs; (C) The TEM of TfR-T12-PMs, bar=100 nm; (D) Cumulative release property of TfR-T12-PMs. |
The cell toxicity of TfR-T12-PEG-PLA copolymer with different concentration (0–100 mg/mL) was investigated, and the result indicated that the cell viability of the TfR-T12-PEG-PLA copolymer found no obvious difference as concentration increased, the cell viability reached above 90% in all groups (Figure S3). Therefore, it came to the conclusion that TfR-T12-PEG-PLA copolymer was safe for drug delivery.
Preparation and Characterization of TfR-T12-PMs
PEG-PMs and TfR-T12-PMs were similar in size as well as encapsulation efficiency and loading efficiency. The average size of TfR-T12-PMs was 110 nm (Polydispersity Index (PI): 0.217) (Figure 2B), and the zeta potential of PEG-PMs and TfR-T12-PMs was −7.91 and −4.77, respectively (Figure S4), because of the carboxyl group at the end of PEG, PEG-PMs is negatively charged, while the zeta potential of TfR-T12-PMs increased slightly with the modification of TfR-T12. From the TEM morphological images (Figure 2C), the TfR-T12-PMs are spherical and well distributed. EE and LE of PTX in the TfR-T12-PMs were 90% and 2.5%, respectively. PTX loading in TfR-T12-PMs and PEG-PMs were released slowly compared to free PTX in pH 7.4 PBS at 37 °C, about 50% of PTX was released from TfR-T12-PMs and unmodified PEG-PMs at 24 h, and 70% free PTX was released at the same time point (Figure 2D). Thus, the peptide had no significant effect on release of PTX from PMs.
Cellular Uptake Study
Cellular uptake of PEG-PMs and TfR-T12-PMs by U87MG cells was studied with flow cytometry and confocal microscope, and DiR was used as probe for evaluation of the cellular uptake of formulations. After six hours of incubation at 37°C with different formulations of PMs, the uptake of TfR-T12-PMs was significantly greater than that of PEG-PMs and free DiR, and the percentage of fluorescein-positive cells after treatment with TfR-T12-PMs was increased significantly in comparison to PEG-PMs and free DiR (Figure 3A). It suggested that the presence of TfR-T12 effectively increased PMs uptake by U87MG cells. These results indicated the presence of TfR-T12 on the surface of PMs executed obvious influence on the uptake ability of PMs.
 |
Figure 3 Cellular uptake study on U87MG cells. (A) Cellular uptake was detected by flow cytometer. (B) Intracellular fluorescence was observed by CLSM, bar=10 μm. |
The intracellular accumulation and distribution of PMs in U87MG cells were also studied by confocal microscopy (Figure 3B). The U87MG cells were treated with free DiR for four hours at 37°C, a weak red intracellular fluorescence was observed because most of the DiR had not entered into the cells. While the red intracellular fluorescence in unmodified PEG-PMs and TfR-T12-PMs group was shown, and the fluorescence intensity in the TfR-T12-PMs group was much stronger than the unmodified PEG-PMs, which might be due to the interaction of TfR-T12 with TfR receptor on U87MG cell surface. The data indicated that the enhanced uptake and absorption of DiR by cells were due to the surface modification of PMs with the TfR-T12 peptide.
TfR-T12-PMs Inhibits Glioma Cell Proliferation
The antiproliferative effect of PTX on U87MG cells was dose-dependent. The cells viability deceased significantly with the concentration of PTX increasing (Figure 4A). All of the formulations including free PTX, PEG-PMs and TfR-T12-PMs showed an increased antiproliferative effect on U87MG cells compared to the blank group (Figure 4B). The increased cytotoxicity in these cancer cells is considered a promising improvement in the therapeutic efficacy of PTX when PTX is entrapped in the TfR-T12-modified PEG-PLA copolymer, such a feature may be ascribed to a combination of higher PTX delivery to cancer cells and stable release of PTX from PMs within the cells. Accordingly, the increased PTX toxicity of TfR-T12-PMs could be attributed to the higher uptake of PTX in the U87MG cancer cells. Furthermore, there is obvious significant difference between the uptake profiles of TfR-T12-PMs and PEG-PMs. In conclusion, the antiproliferative effect of the formulations on U87MG cells followed the order as TfR-T12-PMs> PEG-PMs>PTX.
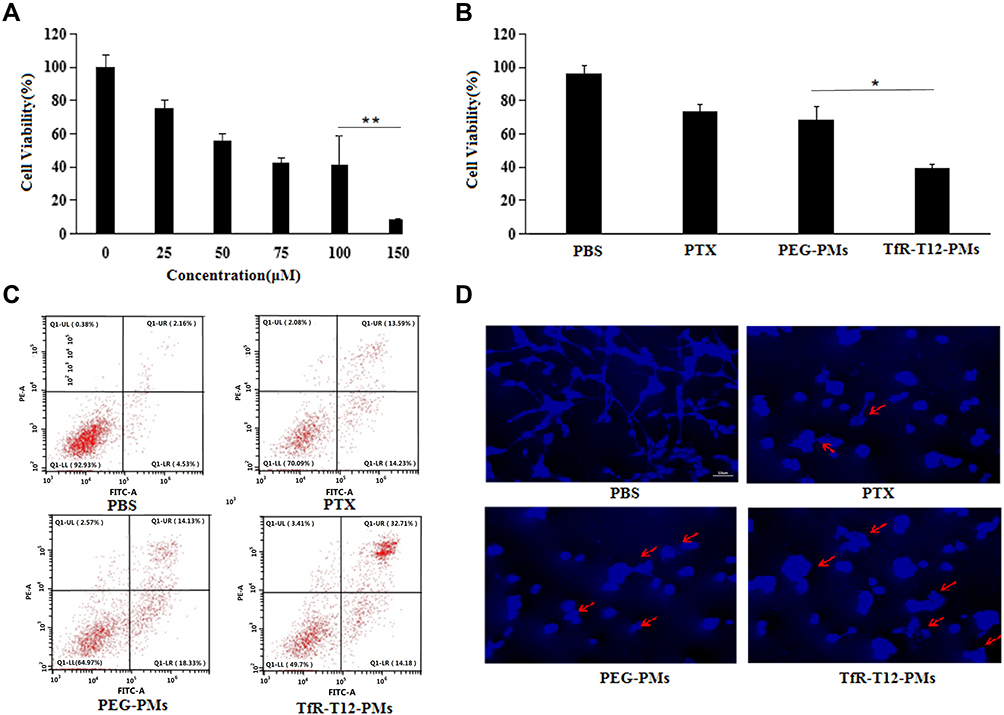 |
Figure 4 Antiproliferative and apoptotic effect of different PTX formulation. Cells viability of U87MG cells treated with different concentration PTX (A) or different PTX formulation; (B) was measured by CCK8 assay. *p<0.05, **p<0.01. (C) Apoptotic analysis of U87MG cells was determined using FITC/PI staining; Necrotic cells were found in the upper left quadrant, normal cells in the lower left quadrant, early apoptotic cells in the upper right quadrant, and late apoptotic cells in the lower right quadrant. (D) Hoechst stained for U87MG cells treated different PTX formulation, and the red arrow represents U87MG apoptosis bodies. |
TfR-T12-PMs Induces Glioma Cell Apoptosis
To determine whether the inhibition of U87MG cell proliferation by these PMs was a consequence of apoptosis, we conducted an annexin V-FITC and PI double-staining assay on U87MG cells. Indeed, a high level of apoptosis after 24 h of treatment was induced by TfR-T12-PMs (Figure 4C), which was comparable to the level induced by PEG-PMs and free PTX. Meanwhile, similar results also appear in Hoechst staining experiments, the U87MG cells treated by different PTX formulations were stained by Hoechst solution, and cell apoptosis was increased sharply in TfR-T12-PMs group (Figure 4D). Data above indicated that TfR-T12-PMs could promote cell apoptosis in U87MG cells effectively.
TfR-T12-PMs Inhibits Glioma Cell Migration and Invasion
We checked the effect of TfR-T12-PMs on U87MG cell migration by performing the transwell assay. U87MG cells treated with the PTX, PEG-PMs and TfR-T12-PMs exhibited a significantly lower migration rate than the blank group. The number of migratory cells was notably lower in U87MG cells after treatment by TfR-T12-PMs in contrast to PTX and PEG-PMs group (Figure 5A), and TfR-T12-PMs reduced the migration area of U87MG cells compared to PEG-PMs (Figure 5B). From the invasion assay result, the cell invasion rate was inhibited by free PTX and PEG-PMs slightly, while TfR-T12-PMs showed obvious inhibition effect (Figure 5C and D). Taken together, TfR-T12-PMs reduced glioma cells migration and invasion significantly.
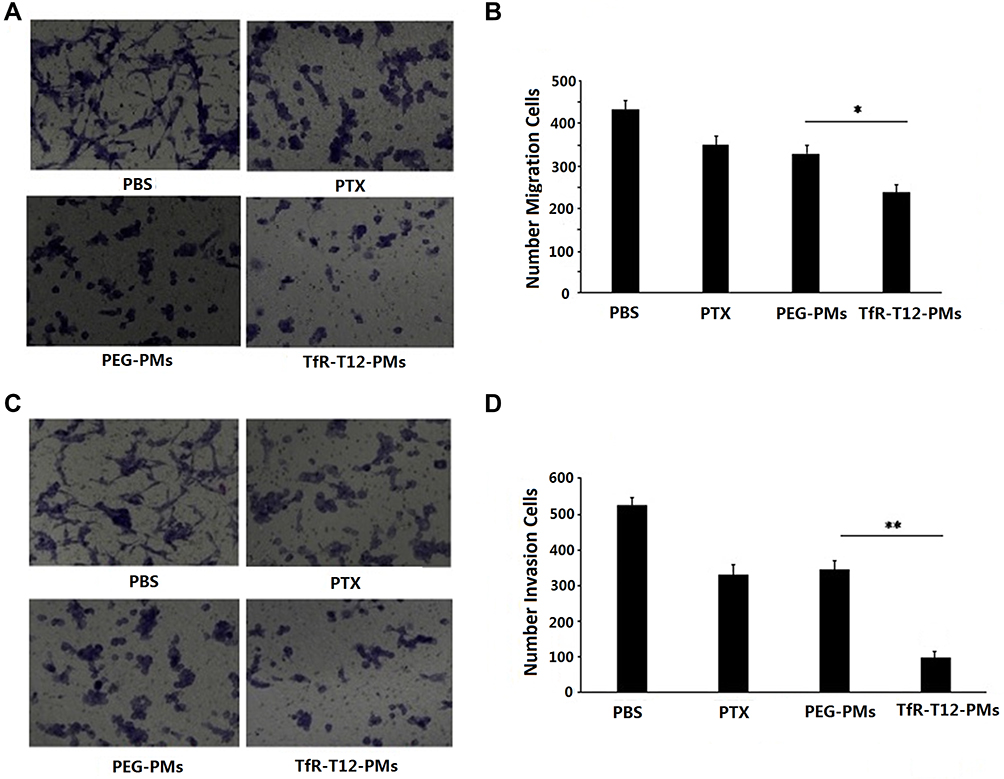 |
Figure 5 Effect of different PTX formulations on migration and invasion of U87MG cells. (A) Migration of U87MG cells treated with different PTX formulation was studied using a microscope. (B) Five areas of the migrated cells per field was measured. (C) Representative images of the invaded U87MG cells were observed using a microscope. (D) Five areas of the invaded cells per field were measured. Data are depicted as mean ±SD. *p<0.05 **p<0 0.01. |
In vitro and in vivo of Transcytosis Effect
It was reported that HUVECs were stimulated by tumor cell-endowed endothelial cells with some characteristics of angiogenesis, so HUVECs exhibited higher proliferative rate after being co-cultured with U87MG cells for three days. HUVECs/U87MG co-culture model was established as a BBTB model to assess the BBTB transcytosis efficiency of the PMs (Figure S5). The result showed that the cell viability of the HUVECs cell treated with TfR-T12-PMs was decreased slightly (Figure 6A). After 24 h incubation, the uptake and apoptosis on U87MG cell was significantly elevated in TfR-T12-PMs group compared to PEG-PMs group (Figure 6B and C), and the apoptotic phenomenon were also observed in TfR-T12-PMs group obviously (Figure S6), indicated that TfR-T12 modification PMs boosted transcytosis across the BBTB and targeted tumor cells through the interaction between TfR-T12 peptide and TfR.
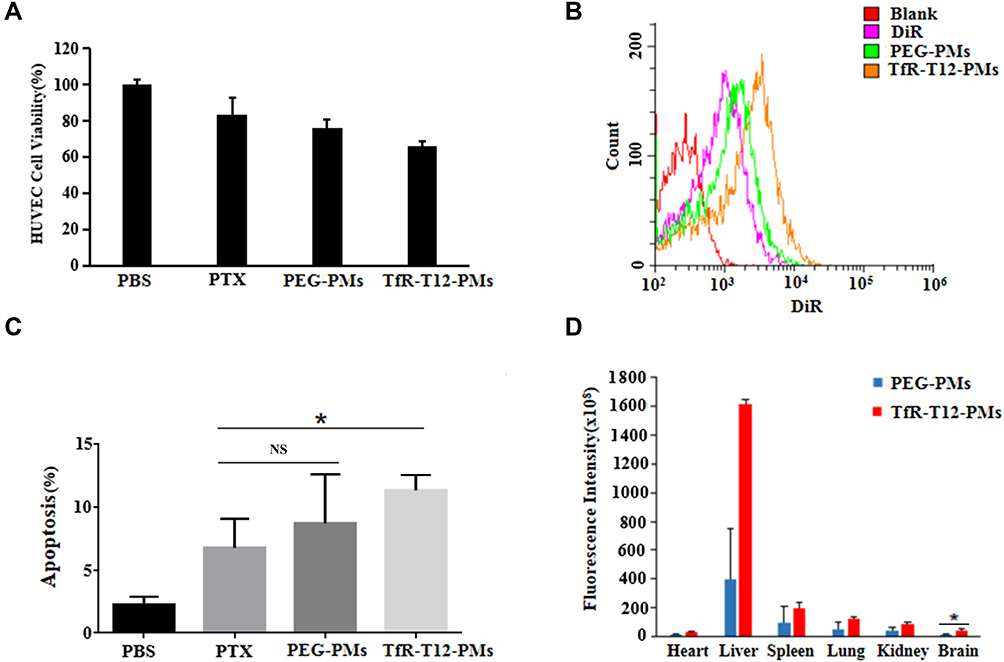 |
Figure 6 The BBTB/BBB transcytosis effect of different PTX formulations in vitro and in vivo. (A) HUVEC viability was measured by the CCK8 assay. (B) The intake efficiency of U87MG treated with different PTX formulations was measured by flow cytometer. (C) Apoptosis ratio of U87MG cell induced by different PTX formulations was measured by flow cytometer. (D) BBB traversal effect in healthy mice was detected by 3D multimodal imaging system, data represent mean ±SD; n=3. *p<0.05; NS, not significant. |
To investigate in vivo distribution of PEG-PMs and TfR-T12-PMs in brain tissues. DiR loaded in PEG-PMs and TfR-T12-PMs were injected by caudal vein in healthy mice, and then mice were euthanized and their hearts, livers, spleens, lungs, kidneys, brains were collected. The result showed that TfR-T12-PMs was significantly higher brain accumulation than PEG-PMs (Figures 6D and S7), likely due to effective active targeting from ready access to the BBB vasculature. Therefore, TfR-T12-PMs displayed an even higher accumulation than PEG-PMs due to its active targeting capability. It should be noted that TfR-T12 peptide-modified micelles accumulated more in liver tissue than unmodified PEG-PMs group, which may be due to more distribution of TfR in liver tissue.
TfR-T12-PMs Suppresses Tumor Growth in vivo on a Subcutaneous Tumor Model
The antitumor efficacy of PMs is illustrated using xenograft model in Figure 7. The tumor growth was remarkably inhibited after the successive intravenous injection of PTX formulations compared to the control, in particular, the group treated with TfR-T12-PMs presented a more notable decrease in the tumor growth. It demonstrated the superiority of combining the TfR-T12 peptide strategy with nanoparticle-based delivery platforms. By comparison, untreated mice from the saline group showed rapid tumor growth, with tumor volume reaching approximately 140 mm3 by day 33 (Figure 7A), and body weight of mice was decreased (Figure 7B), tumor weight was quadrupled (Figure 7C and D). The distribution and antitumor effect of drugs in vivo depend on the size, surface characteristics and shape of PMs to a great extent. It is worth noting that the efficacy of PMs in vivo is closely related to its cytotoxicity in vitro, and the value of this parameter in the design of more effective chemotherapy drugs delivery carrier in the future is emphasized. The uptake efficiency of PMs by tumor cells was greatly enhanced by TfR-T12 peptide modification (Figure S8), which is also the reason for the increase of antitumor effect in vivo. At the end of administration, the tumor-bearing mice were killed and tumor tissues were separated, the tumor sections from each group were respectively stained with TUNEL, Ki67 and CD31 for evaluation of apoptosis, antiproliferation and the observation of angiogenesis. TUNEL and Ki67 was the marker of apoptosis and proliferation inhibition of tumor cells, respectively. The results showed that the apoptosis of tumor cells increased and the proliferation of tumor cells was significantly inhibited after TfR-T12-PMs treatment compared to PEG-PMs group. Compared to the saline group, PTX and PEG-PMs, TfR-T12-PMs reduced angiogenesis evidently. So the conclusion is drawn that tumors treated with TfR-T12-PMs displayed higher apoptosis, lower proliferation effect, and less angiogenesis due to the enhanced drug localization at the tumor target (Figure 7E).
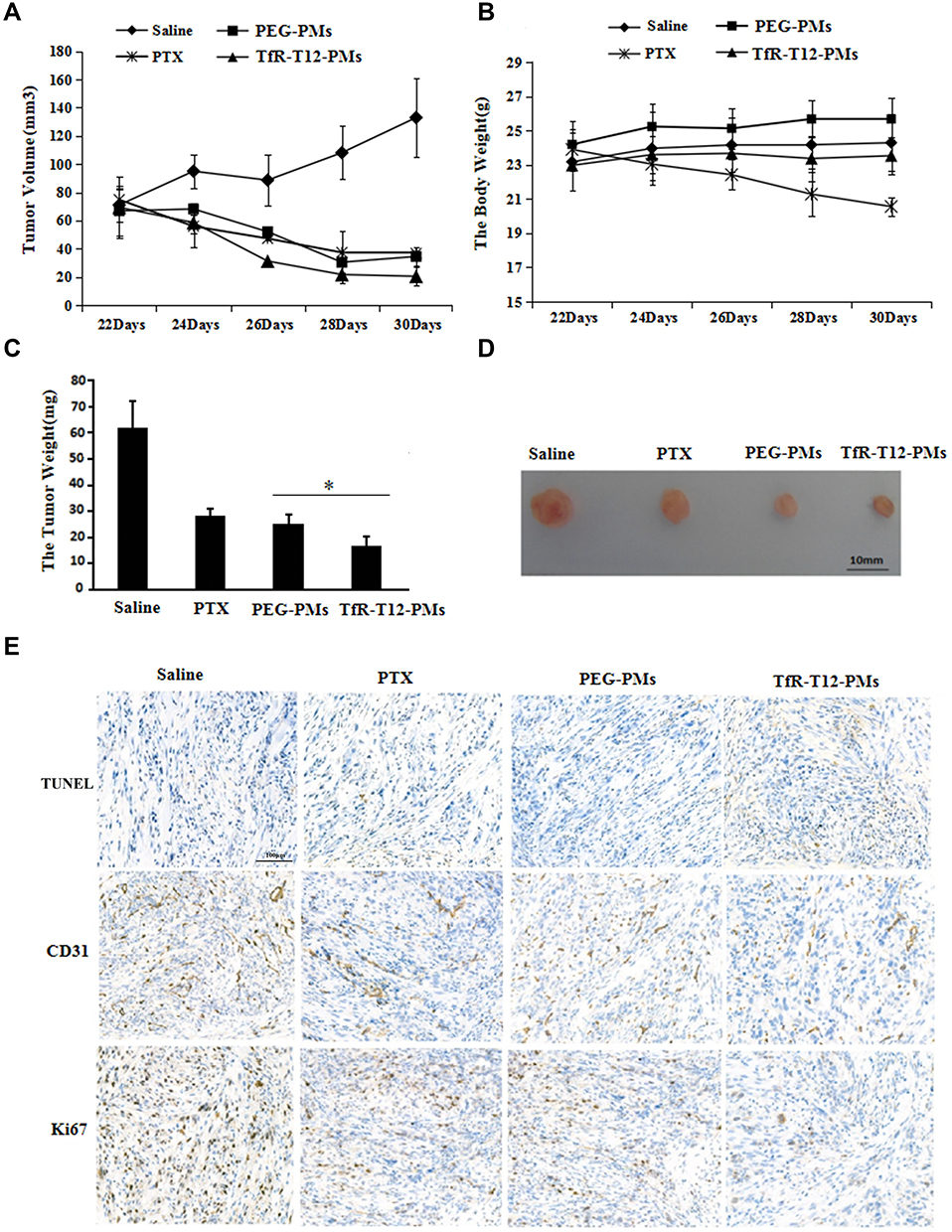 |
Figure 7 Antitumor efficacy in subcutaneous glioma tumor model in vivo. (A) Tumor volumes were measured every two days after administration (n=5). (B) The body weight of mice were measured (n=5). (C) Tumor weight was measured at the end of administration (n=5). *p<0.05. (D) Tumor size was observed, bar=10 mm. (E) TUNEL, CD31 and Ki67 staining of tumor sections were observed by fluorescence microscopy, the scale bar=100 μm. |
The Safety of TfR-T12-PMs on Subcutaneous Tumor-bearing Mice
The body weight changes of tumor-bearing mice can directly reflect the side effects of the preparation. We found that the body weight of the mice receiving treatment with PEG-PMs and TfR-T12-PMs remained stable (Figure 7B). There is no obvious trend of decrease, suggesting a low systemic toxicity in this two groups. The body weight of the mice in the PTX group decreased significantly, it even decreased more than in normal saline mice, this may be due to the poor solubility of PTX in PBS, so cosolvent T-80 must be added, indicating that free PTX is showing obvious side effects in glioma therapy. The H&E staining results of heart, liver, spleen, lung, and kidney further confirmed this conclusion (Figure S9), and liver injury was observed only in PTX group, compared with other groups, PTX group had liver enlargement and necrosis.
Biodistribution and Antitumor Efficacy on an Orthotropic Glioma Model
To investigate the in vivo distribution and antiglioma efficacy of TfR-T12-PMs, mice-bearing orthotropic U87MG gliomas were established in Figure 8A. The DiR loading PMs was used to estimate brain distribution of PMs. After administration for three hours, brain tissue is separated and measured by three dimensional multimodal imaging. Small amount fluorescence of PEG-PMs could be detected in the brains due to the existence of the BBB/BBTB. TfR-T12-modified PMs increased drug accumulation to a greater extent at time points because TfR-T12-PMs could traverse the BBB/BBTB by TfR-mediated transcytosis (Figure 8B). Similarly, the in vivo antiglioma results showed that the survival ratio in PTX and PEG-PMs groups slightly improved compared to the saline group. The median survival in TfR-T12-PMs group was longer than PEG-PMs group, registering a median survival of 63 days vs 46 days for the PEG-PMs group (Figures 8C and S10). The rapid weight loss of tumor-bearing mice in PTX group suggests that there are serious side effects when treated with PTX, and the body weight of tumor-bearing mice did not change significantly before and after administration in TfR-T12-PMs group (Figure 8D). These results indicated that TfR-T12-PMs exhibited a significant improvement in antitumor activities and had no obvious side effects. The H&E staining of heart, liver, spleen, lung, and kidney were also visualized for safe estimation. Liver necrosis and lung injury was observed in PTX group, and heart injury was found in PEG-PMs group, and no organ injury was found in TfR-T12-PMs groups (Figure 8E). When PTX was coated with carrier, its side effect was reduced. Thus, TfR-T12 PMs were expected to develop brain tumor targeting preparation to lay a theoretical foundation for clinical application in the future.
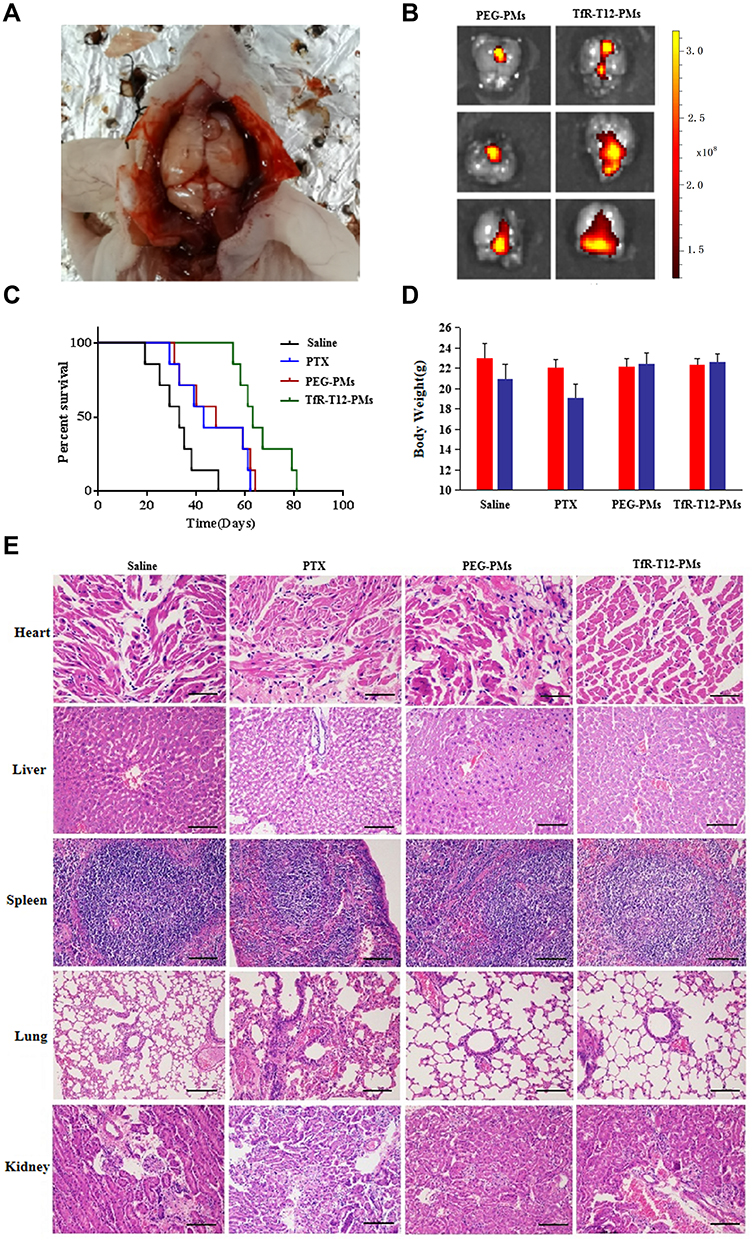 |
Figure 8 Antitumor efficacy in orthotopic glioma tumor model in vivo. (A) The orthotopic tumor model was established with a stereotactic apparatus. (B) Biodistribution of DiR loaded in PEG-PMs and TfR-T12-PMs in brain was quantified by 3D multimodal imaging system (n=3). (C) Cumulative survival of orthotopic tumor-bearing mice was detected (n=7). (D) The body weights were measured at the beginning and then at the end of the study. (E) H&E stained for heart, liver, spleen, lung, and kidney sections of every group, scale bar=100 µm. |
Glioma is a serious malignant tumor, and its treatment has always been an open question.39 Gliomas can grow evenly in the contralateral hemisphere, hide in the skull and be protected by the BBB.40 In addition, surgery or radiotherapy cannot completely eliminate glioma cells, recurrence is inevitable. Therefore, chemotherapy continues to play an important role in the treatment of glioma.41,42 BBB/BBTBs are the main obstacles of chemotherapy, it is urgent to explore new chemotherapy strategies. In this study, PEG-PLA micelles modified with TfR-T12 peptide were used to prepare a multifunctional drug sustained-release vesicle. One advantage of this study is that the TfR-T12-PEG-PLA as carrier was synthesized firstly, and preparation of TfR-T12-PMs are simple and easy.29,43 TfR-T12-PMs can be transported by RMT through the BBB/BBTB, and specifically enter the glioma through receptor mediated endocytosis (RME). To eliminate the glioma by triggering the necrosis and apoptosis of glioma, the transport effect of TfR-T12-PMs was evaluated by BBB model. In the co-cultured BBTB model, HUVEC proliferated and formed a close connection, forming BBTB in vitro. The uptake and inhibition rates of U87MG glioma cells in the BBTB model reflect the transport across the BBTB. According to the results, TfR-T12-PMs has transport capacity across the BBTB and has obvious killing effect on U87MG glioma cells. It is most likely that these mechanisms are related to RMT-mediated cell transfer, TfR-T12 peptide and TfR interact specifically on HUVECs cells in the BBTB model. To a large extent, the distribution of drugs in mice determines how the drug takes effect. The distribution of TfR-T12-PMs in the organs of healthy mice was observed by 3D imaging. The results showed that the fluorescent probe of labeled PMs mainly accumulated in the brain. These phenomena indicated that TfR-T12-PMs was easier to enter the brain tissue through the BBB than PEG-PMs. An uptake test showed that TfR-T12 peptide played an important role in the cell uptake of PMs. Due to the high expression of TfR in glioma cells, its mechanism is also related to RME. In the cytotoxic test, the in vitro effect of TfR-T12-PMs was confirmed in glioma cells. Although free PTX and PEG-PMs have a good therapeutic effect on glioma, TfR-T12-PMs has a strong killing ability on glioma cells. The improvement of therapeutic effect is mainly due to the increased endocytosis of RME mediated by TfR-T12 peptide. The role of TfR-T12-PMs in subcutaneous glioma of nude mice has been further confirmed. In vivo imaging results showed that the fluorescence probe labeled TfR-T12-PMs accumulated more in the glioma site, which could significantly inhibit the growth of glioma compared with the control group. Immunofluorescence analysis also showed that TfR-T12-PMs could induce apoptosis of tumor-bearing mice and inhibit the proliferation and angiogenesis. In order to study the anti-glioma effect of TfR-T12-PMs, a mouse with orthotropic U87MG glioma was established. After intravenous injection of TfR-T12-PMs, the overall effect of mouse glioma treatment further showed the increase of survival time. The side effects of TfR-T12-PMs were also observed, liver necrosis and lung injury were observed in PTX group. TfR-T12-PMs had no obvious organ damage to tumor-bearing mice, suggesting that TfR-T12-PEG-PLA coating PTX could reduce the side effects. Although TfR-T12-PMs has many advantages in the treatment of glioma, there are still some shortcomings in this project, such as the in vitro stability of TfR-T12-PMs and the study of long-term toxic and side effects, which need to be further investigated.
Conclusion
In summary, we have successfully established TfR-T12-PMs with high drug loading and small particle size. TfR-T12-PMs is used in the treatment of glioma, which demonstrated the advantage of crossing the BBTB/BBB capacity and controlled intracellular release of chemotherapeutics. It was also confirmed that TfR-T12 peptide enabled the PMs to target GBM cells and enhance the internalization efficiency. This research result showed that TfR-T12-PMs internalized into brain microvascular cells might be beneficial for the drug delivery through the BBTB/BBB. These favorable biological properties resulted in higher toxicity to glioma cells. Further studies to illustrate the potential of TfR-T12-PMs will be conducted in an orthotopic glioma model with an intact BBB. To sum up, our findings suggest that TfR-T12-PM is a promising new candidate for targeted glioma treatment, and the systematic in vitro and in vivo evaluations established here shall be a solid foundation for brain tumor therapy.
Abbreviations
PEG-PLA, Poly(ethylene glycol)-b-poly(L-lactide-co-glycolide); TfR-T12-PMs, TfR-T12-PEG-PLA/PTX polymeric micelles; PEG-PMs, PEG-PLA/PTX polymeric micelles; PMs, polymeric micelles; EDC, 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide-2- hydrochloride; EE, encapsulation efficiency; LE, loading efficiency; NHS, N-hydroxysuccinimide; PTX, paclitaxel; CLSM, confocal laser scanning microscopy; 1H-NMR, 1H-nuclear magnetic resonance; TEM, transmission electron microscope; TfR, transferrin receptor; UV, ultraviolet spectrophotometry; BBB, blood−brain barrier; BBTB, blood–brain tumor barrier; RMT, receptor-mediated transcytosis; GBM, glioblastoma multiforme; TfR-T12, transferrin receptor-T12 peptide; SPF, specific pathogen free; DiR, 1,1ʹ-dioctadecyl-3, 3, 3ʹ, 3ʹ-tetramethyl indotricarbocyanine Iodide, PI, polydispersity index.
Acknowledgments
This work was supported by grants from the Shenzhen Science and Technology Innovation Commission grant no. JCYJ20180507182253653; the National Natural Science Foundation of China (No. U1801283, 31870908, 31670914); China Post Doctoral Science Foundation (No. 2020M672820); and Guangdong Provincial Science and Technology Program (No. 2019B030301009). Thanks to Instrumental Analysis Center of Shenzhen University for providing instrument support.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Barua NU, Gill SS, Love S. Convection-enhanced drug delivery to the brain: therapeutic potential and neuropathological considerations. Brain Pathol. 2014;24:117–127. doi:10.1111/bpa.12082
2. Strauss SB, Meng A, Ebani EJ, et al. Imaging glioblastoma posttreatment: progression, pseudoprogression, pseudoresponse, radiation necrosis. Radiol Clin North Am. 2019;57:1199. doi:10.1016/j.rcl.2019.07.003
3. Yang K, Niu L, Bai Y, et al. Glioblastoma: targeting the autophagy in tumorigenesis. Brain Res Bull. 2019;153:334–340. doi:10.1016/j.brainresbull.2019.09.012
4. Liu HW, Lee PM, Bamodu OA, et al. Enhanced hsa-miR-181d/p-STAT3 and hsa-miR-181d/p-STAT5A ratios mediate the anticancer effect of garcinol in STAT3/5A-addicted glioblastoma. Cancers (Basel). 2019;11:1888–1912. doi:10.3390/cancers11121888
5. Ozyerli-Goknar E, Sur-Erdem I, Seker F, et al. The fungal metabolite chaetocin is a sensitizer for pro-apoptotic therapies in glioblastoma. Cell Death Dis. 2019;10:894–914. doi:10.1038/s41419-019-2107-y
6. Zhang C, Liu CF, Chen AB, et al. Prognostic and clinic pathological value of cx43 expression in glioma: a meta-analysis. Front Oncol. 2019;9:1209–1220. doi:10.3389/fonc.2019.01209
7. Tinkle CL, Simone B, Chiang JCH, et al. Defining optimal target volumes of conformal radiation therapy for diffuse intrinsic pontine glioma. Int J Radiat Oncol Biol Phys. 2019;S0360–3016:34056–34058.
8. Gupta SK, Kizilbash SH, Daniels DJ, et al. Editorial: targeted therapies for glioblastoma: a critical appraisal. Front Oncol. 2019;9:1216–1220. doi:10.3389/fonc.2019.01216
9. Goebel AM, Gnekow AK, Kandels D, et al. Natural history of pediatric low-grade glioma disease-first multi-state model analysis. J Cancer. 2019;10:6314–6326. doi:10.7150/jca.33463
10. Silva VAO, Rosa MN, Tansini A, et al. Semi-synthetic ingenol derivative from euphorbia tirucalli inhibits protein kinase C isotypes and promotes autophagy and S-phase arrest on glioma cell lines. Molecules. 2019;24:4265–4283. doi:10.3390/molecules24234265
11. Zhu Y, Liu C, Pang Z. Dendrimer-based drug delivery systems for brain targeting. Biomolecules. 2019;9:790–819. doi:10.3390/biom9120790
12. Lovejoy DA, Hogg DW, Dodsworth TL, et al. Synthetic peptides as therapeutic agents: lessons learned from evolutionary ancient peptides and their transit across blood-brain barriers. Front Endocrinol (Lausanne). 2019;10:730–742. doi:10.3389/fendo.2019.00730
13. Shan Y, Tan S, Lin Y, et al. The glucagon-like peptide-1 receptor agonist reduces inflammation and blood-brain barrier breakdown in an astrocyte-dependent manner in experimental stroke. J Neuroinflammation. 2019;16:242–262. doi:10.1186/s12974-019-1638-6
14. Kikuchi DS, Campos ACP, Qu H, et al. Poldip2 mediates blood-brain barrier disruption in a model of sepsis-associated encephalopathy. J Neuroinflammation. 2019;16:241–252. doi:10.1186/s12974-019-1575-4
15. Wu MY, Gao F, Yang XM, et al. Matrix metalloproteinase-9 regulates the blood brain barrier via the hedgehog pathway in a rat model of traumatic brain injury. Brain Res. 2019.
16. Johnsen KB, Burkhart A, Thomsen LB, et al. Targeting the transferrin receptor for brain drug delivery. Prog Neurobiol. 2019;181:101665–101695. doi:10.1016/j.pneurobio.2019.101665
17. Teles CM, Lammoglia LC, Juliano MA, et al. Novel anticancer PdII complexes: the effect of the conjugation of transferrin binding peptide and the nature of halogen coordinated on antitumor activity. J Inorg Biochem. 2019;199:110754–110764. doi:10.1016/j.jinorgbio.2019.110754
18. Shi D, Mi G, Shen Y, et al. Glioma-targeted dual functionalized thermosensitive ferri-liposomes for drug delivery through an in vitro blood-brain barrier. Nanoscale. 2019;11:15057–15071. doi:10.1039/C9NR03931G
19. Shirvalilou S, Khoei S, Khoee S, et al. Development of a magnetic nano-graphene oxide carrier for improved glioma- targeted drug delivery and imaging: in vitro and in vivo evaluations. Chem Biol Interact. 2018;295:97–108. doi:10.1016/j.cbi.2018.08.027
20. Wang H, Wang X, Xie C, et al. Nanodisk-based glioma-targeted drug delivery enabled by a stable glycopeptide. J Control Release. 2018;284:26–38. doi:10.1016/j.jconrel.2018.06.006
21. An S, He D, Wagner E, et al. Peptide-like polymers exerting effective glioma- targeted siRNA delivery and release for therapeutic application. Small. 2015;11:5142–5150. doi:10.1002/smll.201501167
22. Zhao YZ, Lin Q, Wong HL, et al. Glioma-targeted therapy using cilengitide nanoparticles combined with UTMD enhanced delivery. J Control Release. 2016;224:112–125. doi:10.1016/j.jconrel.2016.01.015
23. Chou ST, Patil R, Galstyan A, et al. Simultaneous blockade of interacting CK2 and EGFR pathways by tumor-targeting nanobioconjugates increases therapeutic efficacy against glioblastoma multiforme. J Control Release. 2016;244:14–23. doi:10.1016/j.jconrel.2016.11.001
24. Choudhury H, Pandey M, Chin PX, et al. Transferrin receptors-targeting nanocarriers for efficient targeted delivery and transcytosis of drugs into the brain tumors: a review of recent advancements and emerging trends. Drug Deliv Transl Res. 2018;8:1545–1563. doi:10.1007/s13346-018-0552-2
25. Wu LP, Ahmadvand D, Su J, et al. Crossing the blood-brain-barrier with nanoligand drug carriers self-assembled from a phage display peptide. Nat Commun. 2019;10:4635–4651. doi:10.1038/s41467-019-12554-2
26. Ramalho MJ, Sevin E, Gosselet F, et al. Receptor- mediated PLGA nanoparticles for glioblastoma multiforme treatment. Int J Pharm. 2018;545(1–2):84–92. doi:10.1016/j.ijpharm.2018.04.062
27. Jhaveri A, Deshpande P, Pattni B, et al. Transferrin-targeted, resveratrol-loaded liposomes for the treatment of glioblastoma. J Control Release. 2018;277:89–101. doi:10.1016/j.jconrel.2018.03.006
28. Mu LM, Bu YZ, Liu L, et al. Lipid vesicles containing transferrin receptor binding peptide TfR-T 12 and octa-arginine conjugate stearyl-R 8 efficiently treat brain glioma along with glioma stem cells. Sci Rep-UK. 2017;7(1):3487–3499. doi:10.1038/s41598-017-03805-7
29. Sonali S, Agrawal P, Singh RP, et al. Transferrin receptor-targeted vitamin E TPGS micelles for brain cancer therapy: preparation, characterization and brain distribution in rats. Drug Deliv. 2016;23(5):1788–1798. doi:10.3109/10717544.2015.1094681
30. Sun P, Huang W, Kang L, et al. siRNA loaded in poly(histidine-arginine)6 modified chitosan nanoparticle for suppressing metastasis of breast tumor with enhancing cell penetration and endosome escape. Int J Nanomed. 2017;12:3221–3234. doi:10.2147/IJN.S129436
31. Jin MJ, Jin GM, Kang L, et al. Smart polymeric nanoparticles with pH-responsive and PEG-detachable properties for co-delivering paclitaxel and survivin siRNA to enhance antitumor outcomes. Int J Nanomed. 2018;13:2405–2426. doi:10.2147/IJN.S161426
32. Zhang L, Wang J, Fu Z, et al. Sevoflurane suppresses migration and invasion of glioma cells by regulating miR-146b-5p and MMP16. Artif Cells Nanomed Biotechnol. 2019;47:3306–3314. doi:10.1080/21691401.2019.1648282
33. Chai ZL, Ran DN, Lu LW, et al. Ligand-modified cell membrane enables the targeted delivery of drug nanocrystals to glioma. ACS Nano. 2019;13:5591–5601. doi:10.1021/acsnano.9b00661
34. Pineda B, Sánchez García FJ, Olascoaga NK, et al. Malignant glioma therapy by vaccination with irradiated C6 cell-derived microvesicles promotes an antitumoral immune response. Mol Ther. 2019;27:1612–1620. doi:10.1016/j.ymthe.2019.05.016
35. Qu F, Wang P, Zhang K, et al. Manipulation of Mitophagy by “All-in-One” nanosensitizer augments sonodynamic glioma therapy. Autophagy. 2019;1–23.
36. Luo H, Xu R, Chen B, et al. MicroRNA-940 inhibits glioma cells proliferation and cell cycle progression by targeting CKS1. Am J Transl Res. 2019;11:4851–4865.
37. Wang C, Chen Y, Wang Y, et al. Inhibition of COX-2, mPGES-1 and CYP4A by isoliquiritigenin blocks the angiogenic Akt signaling in glioma through ceRNA effect of miR-194-5p and lncRNA NEAT1. J Exp Clin Cancer Res. 2019;38:371–385. doi:10.1186/s13046-019-1361-2
38. Tang X, Zhao S, Zhang Y, et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol Ther Oncolytics. 2019;14:279–287. doi:10.1016/j.omto.2019.07.002
39. Song SS, Cheng Y, Ma J, et al. Simultaneous FET-PET and contrast-enhanced MRI based on hybrid PET/MR improves delineation of tumor spatial biodistribution in gliomas: a biopsy validation study. Eur J Nucl Med Mol Imaging. 2020;1–10.
40. Liu F, Huang J, Liu X, et al. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int. 2020;20:7–17. doi:10.1186/s12935-019-1085-6
41. Bonm AV, Ritterbusch R, Throckmorton P, et al. Clinical imaging for diagnostic challenges in the management of gliomas: a review. J Neuroimaging. 2020;1–7.
42. Xie Y, Tan Y, Yang C, et al. Omics-based integrated analysis identified ATRX as a biomarker associated with glioma diagnosis and prognosis. Cancer Biol Med. 2019;16:784–796. doi:10.20892/j.issn.2095-3941.2019.0143
43. Zhang PC, Hu LJ, Yin Q, et al. Transferrin-modified c[RGDfK]-paclitaxel loaded hybrid micelle for sequential blood-brain barrier penetration and glioma targeting therapy. Mol Pharm. 2012;9:1590–1598. doi:10.1021/mp200600t
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.