")
Back to Journals » OncoTargets and Therapy » Volume 7
Transcriptional targeting of sphingosine-1- phosphate receptor S1P2 by epigallocatechin- 3-gallate prevents sphingosine-1-phosphate- mediated signaling in macrophage-differentiated HL-60 promyelomonocytic leukemia cells
Authors Chokor R, Lamy S, Annabi B
Received 19 February 2014
Accepted for publication 19 March 2014
Published 7 May 2014 Volume 2014:7 Pages 667—677
DOI https://doi.org/10.2147/OTT.S62717
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Rima Chokor, Sylvie Lamy, Borhane Annabi
Laboratoire d'Oncologie Moléculaire, Centre de recherche BIOMED, Département de Chimie, Université du Québec à Montréal, Montreal, QC, Canada
Background: Macrophage chemotaxis followed by blood–brain barrier transendothelial migration is believed to be associated with inflammation in the central nervous system. Antineuroinflammatory strategies have identified the dietary-derived epigallocatechin-3-gallate (EGCG) as an efficient agent to prevent neuroinflammation-associated neurodegenerative diseases by targeting proinflammatory mediator signaling.
Methods: Given that high levels of sphingosine kinase and its product, sphingosine-1-phosphate (S1P), are present in brain tumors, we used quantitative reverse-transcription polymerase chain reaction (qRT-PCR) and immunoblotting to test whether EGCG may impact on S1P receptor gene expression and prevent S1P response in undifferentiated and in terminally differentiated macrophages.
Results: Promyelomonocytic human leukemia (HL)-60 cells were differentiated into macrophages, and S1P triggered phosphorylation in extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and P38 mitogen-activated protein kinase (MAPK) intracellular signaling, as shown by Western blot analysis. Pretreatment of cells with EGCG prior to differentiation inhibited the response to S1P in all three pathways, while EGCG abrogated P38 MAPK phosphorylation when present only during differentiation. Terminally-differentiated macrophages were, however, insensitive to EGCG treatment. Using qRT-PCR, gene expression of the S1P receptors S1P1, S1P2, and S1P5 was predominantly induced in terminally-differentiated macrophages, while the S1P2 was decreased by EGCG treatment.
Conclusion: Our data suggest that diet-derived EGCG achieves efficient effects as a preventive agent, targeting signaling pathways prior to cell terminal differentiation. Such properties could impact on cell chemotaxis through the blood–brain barrier and prevent cancer-related neuroinflammation.
Keywords: chemoprevention, green tea EGCG, neuroinflammation, blood–brain barrier, S1P
Introduction
Accumulating evidence suggests that neuroinflammation strongly correlates with and characterizes neurodegenerative processes associated with central nervous system (CNS) disorders. Those inflammatory events occur at the blood–brain barrier (BBB) in diseases such as multiple sclerosis, epilepsy, and Alzheimer’s disease.1 Inflammatory response in the CNS is known to mobilize different types of immune cells, including macrophages, mast cells, T and B lymphocytes, and dendritic cells, as well as CNS-resident cells, such as microglia, astrocytes, and neurons.2 During neuroinflammation, responses to proinflammatory cues and subsequent chemotaxis therefore represent important events in the recruitment of these cells to the CNS.3 As a consequence of proinflammatory growth factor- or cytokine-mediated intracellular signaling, passage through the BBB endothelium allows these cells to reach the site of inflammation. If this process is not controlled, the prolonged inflammation can then be the cause of important cerebral damage.
Inflammation has also been closely linked to various forms of cancer and particularly, to brain tumor development.4,5 However, very little is known about the role of inflammation in glioma, especially at the initiation stage. It has been suggested that bidirectional communications between immune cells and glioma cells create an immunosuppressed microenvironment that promotes tumor survival and growth.6 Such potential tumor-initiating roles of inflammation in glioma prompted researchers to develop several immunotherapy approaches, in order to reverse glioma development and progression and its ability to evade the immune system. In one example of this tumor-initiating role, glioma secrete high levels of the bioactive lipid sphingosine-1-phosphate (S1P),7 and high expression levels of the enzyme that forms S1P, sphingosine kinase 1, correlate with shorter survival time for glioblastoma multiforme patients.8 Furthermore, S1P is a biologically active metabolite of plasma-membrane sphingolipids, which is essential for immune-cell trafficking. Much of the immune function of S1P results from the engagement of a family of five G-protein-coupled S1P receptors (S1P1-5). Recent findings on the role of S1P in immunosurveillance support regulatory mechanisms in S1P-mediated immune-cell trafficking and suggest that any perturbance in the interactions between S1P and its receptors may affect immunity.9
Recently the anti-inflammatory activity of plant and diet-derived compounds, most belonging to the chemical groups of alkaloids, coumarins, flavonoids, polyphenols and terpenoids, was demonstrated.10,11 Although flavonoids have been used in inflammatory pathway–targeting,12,13 it is not known how these may modulate the recruitment and infiltration of macrophages into the tumor, where the microenvironment subsequently activates them to support the malignant progression of cancer cells. Being the most potent secreted matrix metalloproteinase (MMP) to permeate the BBB,14 MMP-9 has been ascribed a crucial role and provided the molecular rationale explaining how the disruption of the BBB15,16 and how subsequent transendothelial migration of terminally differentiated human leukemia (HL-60) macrophages occur.17 Collectively, these processes are thought to lead to cerebral infections and development of brain pathologies. However, while green tea–derived epigallocatechin-gallate (EGCG) was shown to efficiently inhibit the inflammation biomarker cyclooxygenase (COX)-2, it was unable to inhibit MMP-9 secretion in differentiated-macrophages.18
Given the selective biomarker-targeting of EGCG, we questioned whether pro-oncogenic-mediated differentiation of HL-60 promyelomonocytic leukemia cells into macrophages affected the cellular response to EGCG. In the current study, we therefore explored the capacity of diet-derived EGCG to prevent S1P-mediated signaling, which could possibly contribute to macrophage mobilization through the BBB.
Materials and methods
Materials
Sodium dodecyl sulfate (SDS) and bovine serum albumin (BSA) were purchased from Sigma-Aldrich Corp (St Louis, MO, USA). Cell culture media was obtained from Life Technologies Corp (Carlsbad, CA, USA). Electrophoresis reagents were purchased from Bio-Rad Laboratories (Hercules, CA, USA). The HyGLO™ Chemiluminescent HRP (horseradish peroxidase) Antibody Detection Reagents were from Denville Scientific Inc. (Metuchen, NJ, USA). Micro bicinchoninic acid (BCA) protein assay reagents were from Pierce (Micro BCA™ Protein Assay Kit; Thermo Fisher Scientific, Waltham, MA, USA). The polyclonal antibodies against extracellular signal-regulated kinase (ERK), phosphorylated (P)-ERK, c-Jun N-terminal kinase (JNK), P-JNK, P38, and P-P38 were all purchased from Cell Signaling Technology, Inc (Danvers, MA, USA). HRP-conjugated donkey anti-rabbit and anti-mouse immunoglobulin (Ig) G secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). EGCG was from MP Biomedicals (Santa Ana, CA, USA). All other reagents were from Sigma-Aldrich Corp.
Cell culture and treatments
The HL-60 promyelomonocytic cell line was purchased from American Type Culture Collection (Manassas, VA, USA) and maintained in Iscove’s Modified Dulbecco’s Medium (Life Technologies Corp) containing 20% (v/v) fetal bovine serum (FBS) (HyClone™; Thermo Fisher Scientific), 2 mM glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin (WISENT Inc., Québec, QC, Canada), and were cultured at 37°C, under a humidified atmosphere containing 5% CO2. The tumor-promoting agent phorbol-12-myristate-13-acetate (PMA) was used to trigger macrophage differentiation. Slides of PMA-treated HL-60 cells were then mounted for light microscopy and air-dried, stained with Diff-Quick Solution (Baxter Healthcare Corp, Deerfield, IL, USA), and examined for morulas, as previously validated.19 Given that numerous protocols can be found in the literature to differentiate resting HL-60 cells into “macrophage-like cells” using PMA (between 2 to 8 days with various PMA concentrations, alone or in combination with other molecules), we wish to emphasize that, throughout the text the “HL-60 macrophage differentiation” condition represents the adherent subpopulation of HL-60 cells immediately harvested upon PMA treatment. We use “terminally differentiated macrophages” to refer to those same adherent cells, which were subsequently maintained in culture for another 24–48 hours. Cell starvation is defined as: treatments of the cells, either with PMA, EGCG, or S1P, in medium from which only FBS was removed.
Gelatin zymography
Gelatin zymography was used to assess the extent of pro-MMP-9 gelatinolytic activity, as previously described.18 Briefly, an aliquot (20 μL) of the culture medium was subjected to SDS–polyacrylamide gel electrophoresis (PAGE) in a gel containing 0.1 mg/mL gelatin, a substrate that is efficiently hydrolyzed by pro-MMP-9. The gels were then incubated in 2.5% Triton™ X-100 and rinsed in nanopure distilled H2O. The gels were further incubated at 37°C for 20 hours in 20 mM NaCl, 5 mM CaCl2, 0.02% Brij®-35, and 50 mM tris(hydroxymethyl)aminomethane (Tris)-HCl buffer, pH 7.6, then stained with 0.1% Coomassie Brilliant blue R-250 and distained in 10% acetic acid and 30% methanol in H2O. Gelatinolytic activity was detected as unstained bands on a blue background.
Immunoblotting procedures
Proteins from control and treated cells were separated by SDS-PAGE. After electrophoresis, proteins were electrotransferred to polyvinylidene difluoride membranes (Thermo Fisher Scientific), which were then blocked for 1 hour, at room temperature, with 5% nonfat dry milk in Tris-buffered saline (150 mM NaCl, 20 mM Tris–HCl, pH 7.5) containing 0.3% Tween™ 20 (TBST). The membranes were further washed in TBST and incubated with the primary antibodies (1/1,000 dilution) in TBST containing 3% BSA and 0.1% sodium azide, followed by a 1-hour incubation with HRP-conjugated anti-rabbit or anti-mouse IgG (1/2,500 dilution) in TBST containing 5% nonfat dry milk. Immunoreactive material was visualized by chemiluminescence.
Total RNA isolation, cDNA synthesis, and qRT-PCR
Total ribonucleic acid (RNA) was extracted from cell suspensions or monolayers using TRIzol® reagent (Life Technologies Corp). For complementary deoxyribonucleic acid (cDNA) synthesis, 2 μg of total RNA were reverse-transcribed using a high capacity cDNA reverse-transcription kit (Life Technologies Corp). cDNA was stored at −80°C prior to quantitative reverse-transcription polymerase chain reaction (qRT-PCR). Gene expression was quantified by qRT-PCR using iQ™ SYBR® Green Supermix (Bio-Rad Laboratories). DNA amplification was carried out using an iCycler iQ™5 (Bio-Rad Laboratories), and product detection was performed by measuring binding of the fluorescent dye SYBR Green I to double-stranded DNA. The following QuantiTect primer sets were obtained from Qiagen (Venlo, the Netherlands): S1P1 (QT00208733), S1P2 (QT00230846), S1P3 (QT00244251), S1P4 (QT01192744), and S1P5 (QT00234178). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primer sets were synthesized by Biocorp (Dollard-des-Ormeaux, QC, Canada), with the following sequences: forward CCATCACCATCTTCCAGGAG and reverse CCTGCTTCACCACCTTCTTG. The relative quantities of target gene messenger (m)RNA, compared against two internal controls, GAPDH and β-actin mRNA, were measured by following a Δ cycle threshold (CT) method employing an amplification plot (fluorescence signal versus [vs] cycle number). The difference (ΔCT) between the mean values in the triplicate samples of target gene and those of GAPDH and β-actin mRNAs were calculated using iQ5 Optical System Software (Version 2.0; Bio-Rad Laboratories), and the relative quantified value was expressed as 2−ΔCT.
Statistical data analysis
Unless otherwise stated, data are representative of three or more independent experiments. Statistical significance was assessed using Student’s unpaired t-test. Probability values of less than 0.05 were considered significant, and an asterisk identifies such significance in the figures. Error bars in all figures represent standard error of the mean (SEM) values.
Results
PMA triggers promyelocytic HL-60 cell differentiation into adherent macrophages
Among the agents shown to induce differentiation of HL-60 cells, the tumor-promoting agent PMA triggers a terminal differentiated monocytic/macrophage phenotype.20 Here, we treated HL-60 cells with increasing concentrations of PMA for 24 hours, then discarded any cells that remained in suspension and took phase contrast pictures (Figure 1A). We validated that the optimal dose of PMA that triggered optimal cell adhesion was 3 μM (Figure 1B). Finally, we demonstrated, by zymography, increased secretion of MMP-9, characteristic of the adhesive phenotype of terminally differentiated macrophages, in agreement with previous reports.18,19
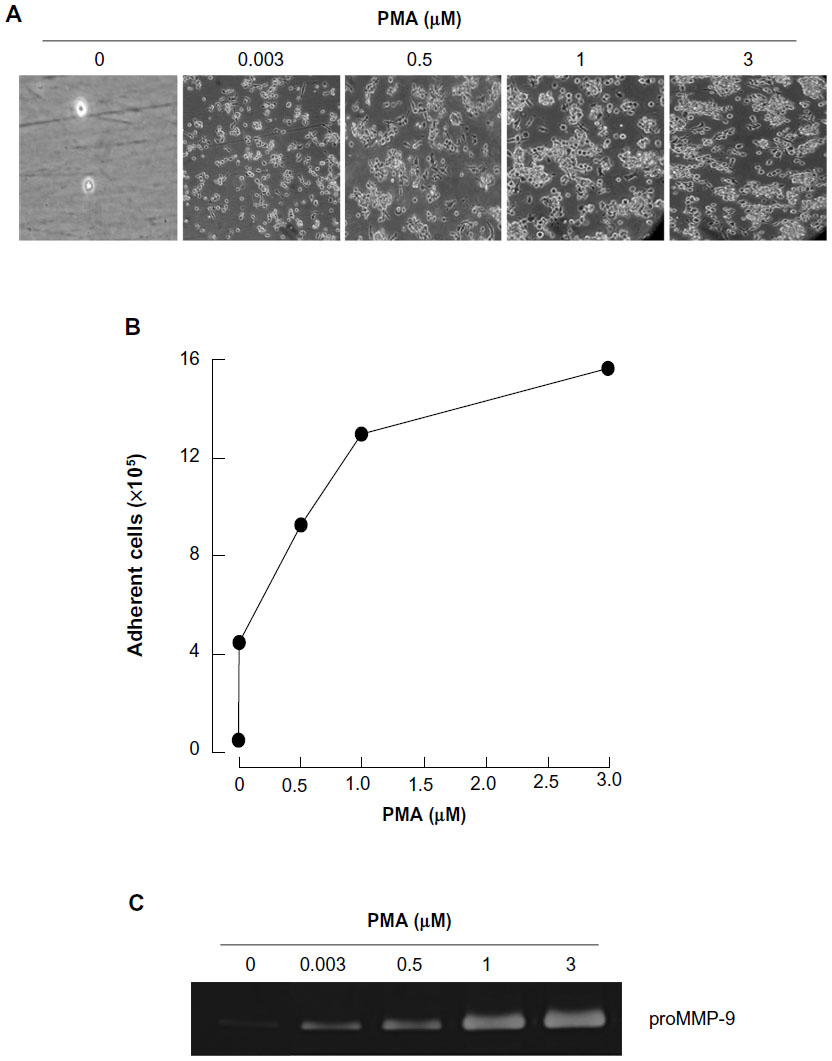 | Figure 1 PMA triggers promyelocytic HL-60 cell differentiation into adherent macrophages. |
Macrophage differentiation correlates with significant increases in S1P1, S1P2, and S1P5 gene expression
We next evaluated the gene expression profiles of the S1P receptor family, in macrophage/monocyte terminally differentiated HL-60 cells. Promyelocytic HL-60 cells were treated with increased PMA concentrations, total RNA extracted from adherent cells, and cDNA synthesis performed, as described in the “Methods” section. We first validated that each of our specific primer sets amplified a single amplicon corresponding to the S1P1, S1P2, S1P3, S1P4, and S1P5 genes, as seen on an ethidium bromide-stained agarose gel (Figure 2A). qRT-PCR then allowed us to assess the extent of changes in gene expression; we concluded that S1P1, S1P2, and S1P5 were significantly increased in adherent cells, while only moderate increases were observed for S1P3 and S1P4 (Figure 2B). Collectively, these results suggest that macrophage-like cells could be responsive to S1P.
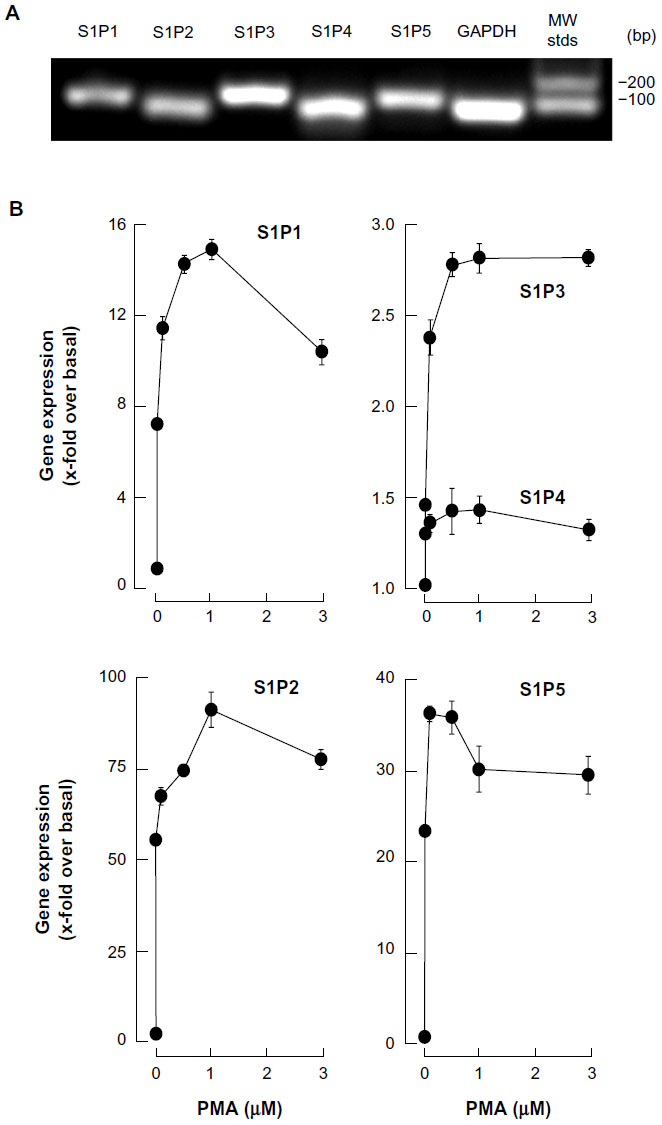 | Figure 2 Macrophage differentiation correlates with significant increases in S1P1, S1P2, and S1P5 gene expression. |
S1P triggers rapid intracellular signaling through multiple pathways in macrophage-differentiated HL-60 cells
In order to assess whether differentiated HL-60 cells were capable of responding to S1P, we measured the extent of phosphorylation of ERK, P38 MAPK, and JNK. We observed that S1P rapidly triggered the phosphorylation of all the aforementioned signaling intermediates (Figure 3A, C, and E), typically reaching an optimal effect between 30 seconds and 1 minute of stimulation (Figure 3B, D, and F). A prior assessment of dose–response ERK phosphorylation, by increasing concentrations of S1P, found that 1 μM S1P triggered an optimal signal (not shown).
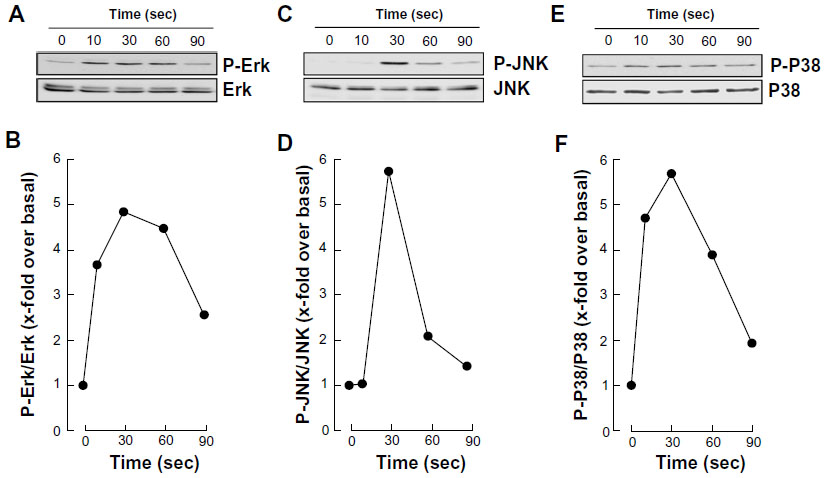 | Figure 3 S1P triggers rapid intracellular signaling through multiple pathways in macrophage-differentiated HL-60 cells. |
EGCG pretreatment of promyelocytic HL-60 inhibits S1P response in macrophage-differentiated HL-60 cells
We next assessed the potential ability of EGCG to affect the S1P cellular response. To this end, we evaluated EGCG effects either prior (predifferentiation) to inducing HL-60 cells differentiation with PMA, simultaneously (codifferentiation) with HL-60 differentiation, or on cells that were already differentiated (postdifferentiation). Cell lysates were isolated following stimulation with S1P, and the extents of ERK, P38 MAPK, and JNK phosphorylation were evaluated by Western blotting (not shown). Scanning densitometry of the autoradiograms showed that PMA-differentiated cells that had been pretreated with EGCG had an abrogated global S1P response as ERK, P38 MAPK, and JNK phosphorylation were inhibited (Figure 4A–C). Where EGCG had been added to cells simultaneously with PMA, only the S1P-mediated phosphorylation of P38 MAPK was inhibited (Figure 4D–F). Finally, where EGCG was added to terminally differentiated cells, none of the S1P-induced phosphorylated intermediates were diminished (Figure 4G–I). Altogether, these observations suggest that EGCG efficiently prevents subsequent responses to S1P in macrophages, while it selectively abrogates the cell response during differentiation and is completely inefficient in terminally differentiated cells.
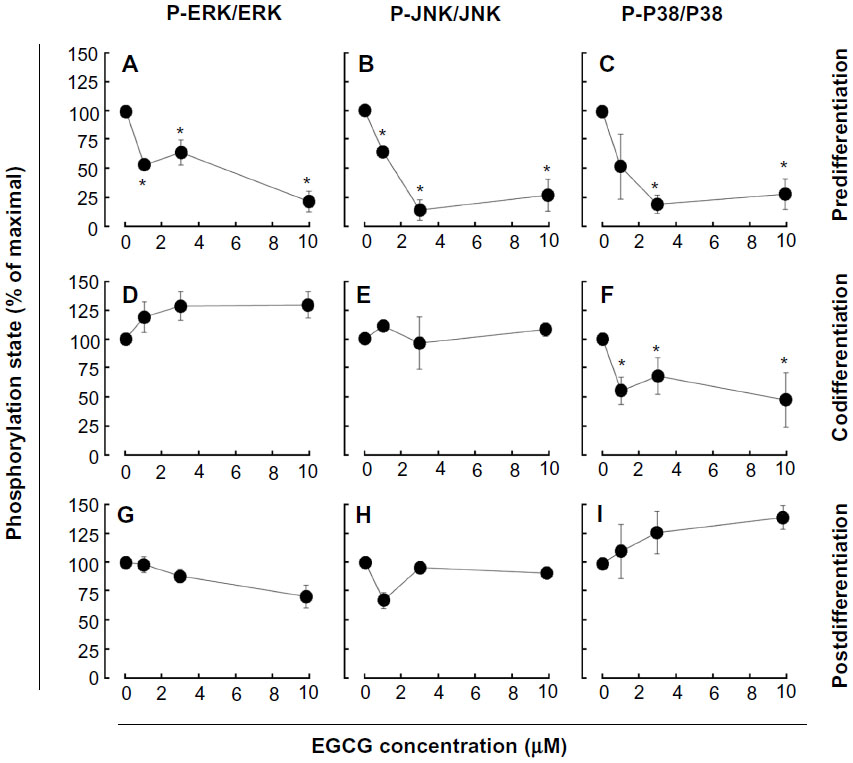 | Figure 4 EGCG pretreatment of promyelocytic HL-60 inhibits S1P response in macrophage-differentiated HL-60 cells. |
EGCG selectively alters S1P receptor gene expression in macrophage- differentiated HL-60 cells
We next examined whether expression of the induced S1P1, S1P2, or S1P5 genes were affected by EGCG. Total RNA was extracted from cells treated as described above and gene expression assessed by qRT-PCR. EGCG was found to only significantly downregulate S1P2 gene expression in pre- and codifferentiation conditions, while it did not alter the increased expression of all three receptors in the postdifferentiation condition (Figure 5).
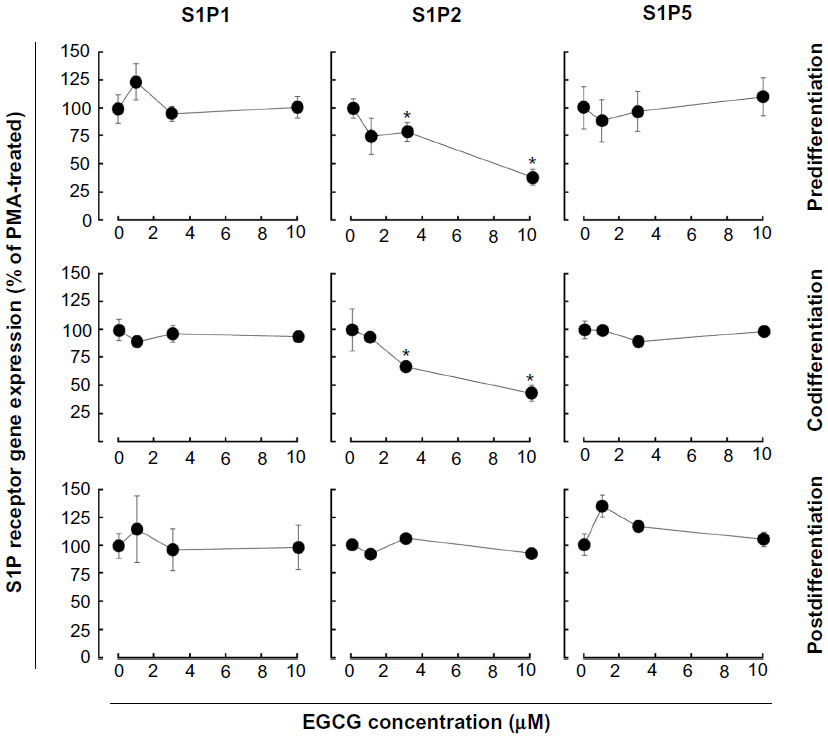 | Figure 5 EGCG selectively alters S1P receptor gene expression in macrophage-differentiated HL-60 cells. Three different conditions (predifferentiation, codifferentiation, and postdifferentiation) were used to evaluate EGCG impact on S1P1, S1P2, and S1P5 gene expression. In all three conditions, total RNA was extracted from adherent cells, and qRT-PCR performed. |
Discussion
The BBB concept, originally suggested to limit access of blood-borne immune cells into the healthy CNS,21,22 has now been challenged by demonstration of leukocyte penetration and by increased recruitment of microglia, which resemble immature antigen-presenting cells.23 Less documented are secreted factors that may contribute to such recruitment and enable modulation of the immunosuppressive environment of glioma. While factors such as transforming growth factor (TGF)-β, COX-2 and interleukin (IL)-10 have been suggested to contribute to the recruitment and expansion of regulatory T cells,24 we now provide new evidence that S1P, which can originate from brain tumor cells where sphingosine kinase 1 is highly expressed, can also be a major actor in the regulation of undifferentiated and terminally differentiated macrophage functions. Given that recruitment of macrophages plays a critical role in cancer and tumor angiogenesis,25,26 we hypothesized that diet-derived molecules, such as the green tea–derived polyphenol EGCG, could sustain a pharmacological intervention targeting cell transformation, affect S1P-mediated signaling through various cell differentiation processes, and ultimately prevent the brain tumor proinflammatory phenotype occurrence.
Chemopreventive assessment of EGCG has, in fact, established this class of diet-derived molecule as a signal transduction inhibitor. In support of this, a screening of 89 nuclear factor (NF)-κB-regulated genes was performed in carcinogen-induced macrophage differentiation of HL-60 cells, where several gene expression profiles were found to be induced during the cell differentiation, immune response, and inflammation functions, and to be significantly reversed by EGCG.18 However, although leukemic cell-derived MMP-9 increased the BBB permeability,16 EGCG was found inefficient to decrease MMP-9 secretion in terminally differentiated macrophages from HL-60 promyelomonocytic cells,18 suggesting alternate transendothelial migration and cell response events could be preferentially targeted. In the current study, we confirm that S1P effectively triggers multiple signaling cascades, in agreement with previous reporting,27 in both undifferentiated and terminally differentiated macrophages. We show that the S1P receptor expression profile was significantly inducible upon macrophage differentiation for S1P1, S1P2, and S1P5, with very marginal inductions for S1P3 and S1P4. Finally, this expression profile was found to be altered by EGCG only for S1P2, under both predifferentiation and codifferentiation treatment conditions. More importantly, the inhibition by EGCG of ERK, JNK, and P38 MAPK signaling pathways in the predifferentiation condition could be explained by either direct downstream targeting of each S1P receptor, or by the reduced S1P2 receptor expression that we report (Figure 6A) and which perfectly matches those functions reported for S1P2.27 Interestingly, although S1P2 expression remained reduced in the codifferentiation condition, EGCG was unable to inhibit S1P-mediated ERK and JNK phosphorylation (Figure 6B), suggesting that those two pathways may be relayed through a S1P receptor signaling function27 and their gene expression is not affected by EGCG, as in the case for S1P1 and S1P5 in the current study. S1P2 downstream functions remain controversial as they relay both a repressive signal in macrophage recruitment,28 while they also potentiate human lung fibroblast chemotaxis.29 In fact, the HL-60 promyelomonocytic cell model was chosen in the current study to address the impact of EGCG treatments on the cellular response of a given cell differentiation status. This model is well recognized to reflect the molecular signature of both the macrophage-differentiated and of the undifferentiated circulation leukemia cells. Given that terminally differentiated macrophages (postdifferentiation condition) became unresponsive to EGCG inhibition (Figure 6C), it becomes tempting to suggest that efficient EGCG impact can only be achieved in a chemopreventive context, where tumor-derived growth factor-mediated chemotaxis, such as can be triggered by S1P, could still be abrogated. Furthermore, given the inability of EGCG to also inhibit MMP-9 expression/function in terminally differentiated HL-60 macrophages, our current data further confirm that such a pharmacological targeting strategy may only affect predifferentiated cells.
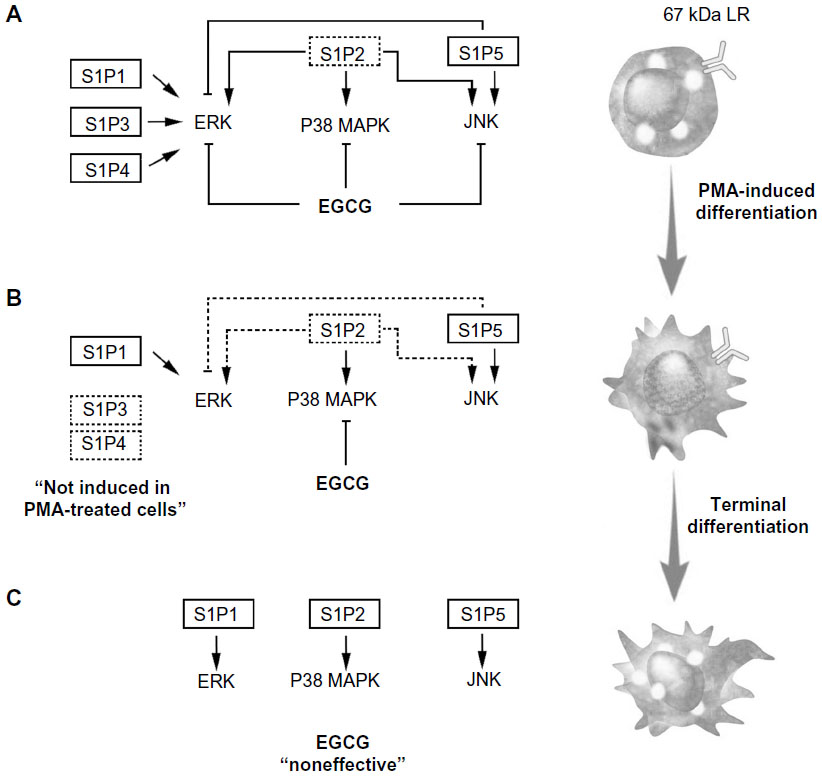 | Figure 6 Schematic representation of pretreatment, cotreatment, or postdifferentiation cell treatments with EGCG, on S1P-mediated signaling pathways. |
S1P fulfills tasks in the immune system through auto- and/or paracrine mechanisms, ultimately regulating macrophage trafficking.30,31 Accordingly, our current assessment of S1P responsiveness in all pre-, co-, and postdifferentiation conditions will shed light and impact on events in macrophage biology, such as apoptosis, migration and cell proliferation,32 but will require further investigation. In our cellular model, S1P treatment was reported to not induce macrophage cell death.32 Therefore, the impact of S1P signaling and of its inhibition by EGCG can be anticipated on mechanisms involved in cell proliferation and/or chemotaxis. Interestingly, modern approaches in cancer therapy are currently aimed at neutralizing cancer stem cell responsiveness to circulating growth factors.33 To this end, a cluster of differentiation (CD)133(+) cell population isolated from U87 glioblastoma cells was recently shown to be highly responsive to S1P signaling and to require the cooperation of membrane-associated, type-I (MT1)-MMP,34 a cell surface MMP shown to be efficiently targeted by EGCG.35,36 Whether such cooperative MT1-MMP to S1P signaling is physiologically relevant was further explored in alternate circulating cell models, such as T cells37 and bone marrow-derived mesenchymal stromal cells,38 where MT1-MMP functions were targeted by EGCG. Evidence possibly supporting such a cooperative MT1-MMP to S1P signaling axis was recently based on the fact that S1P induced the association of MT1-MMP with p130Cas in endothelial cells39 and that this interaction represents a novel mechanism in the macrophage cell fusion machinery.40
Conclusion
The fact that only 5%–10% of all cancer cases are due to genetic defects and that the remaining 90%–95% are due to environment and lifestyle provides opportunities for preventing cancer.10 Accordingly, among the various phytochemicals identified in fruits, vegetables, spices, and grains that exhibit chemopreventive potential, our current study on the impact of EGCG clearly demonstrates its safety and ability to act in preventive rather than therapeutic settings. Accordingly, its lack of cytotoxicity in undifferentiated HL-60 cells is in good agreement with previous studies.41 Tea polyphenols have been ascribed the ability to cross the BBB,42–44 but given the lack of EGCG action on S1P-mediated signaling in terminally differentiated macrophages (Figure 6C) in our study, one possible explanation may be provided by the downregulation of the putative 67-kDa laminin receptor (67LR) EGCG receptor.18 Such adaptive processes may, in part, explain the molecular basis for the cancer-preventive activity of EGCG prior to oncogenic cell transformation in response to carcinogens, and will help in the promotion of new diet strategies to prevent brain tumor-associated neuroinflammation consequent, in part, to macrophage infiltration of the CNS.
Acknowledgments
This study was supported by the “Chaire de Recherche en Prévention et Traitement du Cancer”, held by BA at the Université du Québec à Montréal.
Disclosure
The authors report no conflicts of interest in this work.
References
Trippier PC, Jansen Labby K, Hawker DD, Mataka JJ, Silverman RB. Target- and mechanism-based therapeutics for neurodegenerative diseases: strength in numbers. J Med Chem. 2013;56(8):3121–3147. | |
Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122(4):1164–1171. | |
Chavarria A, Alcocer-Varela J. Is damage in central nervous system due to inflammation? Autoimmun Rev. 2004;3(4):251–260. | |
King JG, Khalili K. Inhibition of human brain tumor cell growth by the anti-inflammatory drug, flurbiprofen. Oncogene. 2001;20(47):6864–6870. | |
Tafani M, Di Vito M, Frati A, et al. Pro-inflammatory gene expression in solid glioblastoma microenvironment and in hypoxic stem cells from human glioblastoma. J Neuroinflammation. 2011;8:32. | |
Galvão RP, Zong H. Inflammation and gliomagenesis: bi-directional communication at early and late stages of tumor progression. Curr Pathobiol Rep. 2013;1(1):19–28. | |
Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst. 2007;99(21):1583–1593. | |
Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J Neuropathol Exp Neurol. 2005;64(8):695–705. | |
Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8(10):753–763. | |
Anand P, Kunnumakkara AB, Kunnumakara AB, et al. Cancer is a preventable disease that requires major lifestyle changes. Pharm Res. 2008;25(9):2097–2116. | |
Chen W, Li Z, Bai L, Lin Y. NF-kappaB in lung cancer, a carcinogenesis mediator and a prevention and therapy target. Front Biosci (Landmark Ed). 2011;16:1172–1185. | |
Luqman S, Pezzuto JM. NFkappaB: a promising target for natural products in cancer chemoprevention. Phytother Res. 2010;24(7):949–963. | |
Prasad S, Phromnoi K, Yadav VR, Chaturvedi MM, Aggarwal BB. Targeting inflammatory pathways by flavonoids for prevention and treatment of cancer. Planta Med. 2010;76(11):1044–1063. | |
Lakhan SE, Kirchgessner A, Tepper D, Leonard A. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol. 2013;4:32. | |
Feng SR, Chen ZX, Cen JN, Shen HJ, Wang YY, Yao L. [Disruption of blood brain-barrier by leukemic cells in central nervous system leukemia]. Zhonghua Xue Ye Xue Za Zhi. 2011;32(5):289–293. Chinese. | |
Feng S, Cen J, Huang Y, et al. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS One. 2011;6(8):e20599. | |
Seidel G, Böcker K, Schulte J, et al. Pertussis toxin permeabilization enhances the traversal of Escherichia coli K1, macrophages, and monocytes in a cerebral endothelial barrier model in vitro. Int J Med Microbiol. 2011;301(3):204–212. | |
Vézina A, Chokor R, Annabi B. EGCG targeting efficacy of NF-κB downstream gene products is dictated by the monocytic/macrophagic differentiation status of promyelocytic leukemia cells. Cancer Immunol Immunother. 2012;61(12):2321–2331. | |
Annabi B, Currie JC, Moghrabi A, Béliveau R. Inhibition of HuR and MMP-9 expression in macrophage-differentiated HL-60 myeloid leukemia cells by green tea polyphenol EGCg. Leuk Res. 2007;31(9):1277–1284. | |
Huberman E, Callaham MF. Induction of terminal differentiation in human promyelocytic leukemia cells by tumor-promoting agents. Proc Natl Acad Sci U S A. 1979;76(3):1293–1297. | |
Perry VH, Anthony DC, Bolton SJ, Brown HC. The blood-brain barrier and the inflammatory response. Mol Med Today. 1997;3(8):335–341. | |
Miller DW. Immunobiology of the blood-brain barrier. J Neurovirol. 1999;5(6):570–578. | |
Persidsky Y. Model systems for studies of leukocyte migration across the blood – brain barrier. J Neurovirol. 1999;5(6):579–590. | |
Rolle CE, Sengupta S, Lesniak MS. Mechanisms of immune evasion by gliomas. Adv Exp Med Biol. 2012;746:53–76. | |
Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–631. | |
Mantovani A, Savino B, Locati M, Zammataro L, Allavena P, Bonecchi R. The chemokine system in cancer biology and therapy. Cytokine Growth Factor Rev. 2010;21(1):27–39. | |
Taha TA, Argraves KM, Obeid LM. Sphingosine-1-phosphate receptors: receptor specificity versus functional redundancy. Biochim Biophys Acta. 2004;1682(1–3):48–55. | |
Michaud J, Im DS, Hla T. Inhibitory role of sphingosine 1-phosphate receptor 2 in macrophage recruitment during inflammation. J Immunol. 2010;184(3):1475–1483. | |
Hashimoto M, Wang X, Mao L, et al. Sphingosine 1-phosphate potentiates human lung fibroblast chemotaxis through the S1P2 receptor. Am J Respir Cell Mol Biol. 2008;39(3):356–363. | |
Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8(12):1295–1301. | |
Gude DR, Alvarez SE, Paugh SW, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008;22(8):2629–2638. | |
Weigert A, Weis N, Brüne B. Regulation of macrophage function by sphingosine-1-phosphate. Immunobiology. 2009;214(9-10):748–760. | |
Wu HC, Huang CT, Chang DK. Anti-angiogenic therapeutic drugs for treatment of human cancer. J Cancer Mol. 2008;4(2):37–45. | |
Annabi B, Lachambre MP, Plouffe K, Sartelet H, Béliveau R. Modulation of invasive properties of CD133+ glioblastoma stem cells: a role for MT1-MMP in bioactive lysophospholipid signaling. Mol Carcinog. 2009;48(10):910–919. | |
Annabi B, Lachambre MP, Bousquet-Gagnon N, Page M, Gingras D, Beliveau R. Green tea polyphenol (-)-epigallocatechin 3-gallate inhibits MMP-2 secretion and MT1-MMP-driven migration in glioblastoma cells. Biochim Biophys Acta. 2002;1542(1-3):209–220. | |
Yamakawa S, Asai T, Uchida T, Matsukawa M, Akizawa T, Oku N. (-)-Epigallocatechin gallate inhibits membrane-type 1 matrix metalloproteinase, MT1-MMP, and tumor angiogenesis. Cancer Lett. 2004;210(1):47–55. | |
Savinov AY, Strongin AY. Targeting the T-cell membrane type-1 matrix metalloproteinase-CD44 axis in a transferred type 1 diabetes model in NOD mice. Exp Ther Med. 2013;5(2):438–442. | |
Zgheib A, Lamy S, Annabi B. Epigallocatechin gallate targeting of membrane type 1 matrix metalloproteinase-mediated Src and Janus kinase/signal transducers and activators of transcription 3 signaling inhibits transcription of colony-stimulating factors 2 and 3 in mesenchymal stromal cells. J Biol Chem. 2013;288(19):13378–13386. | |
Gingras D, Michaud M, Di Tomasso G, Béliveau E, Nyalendo C, Béliveau R. Sphingosine-1-phosphate induces the association of membrane-type 1 matrix metalloproteinase with p130Cas in endothelial cells. FEBS Lett. 2008;582(3):399–404. | |
Gonzalo P, Arroyo AG. MT1-MMP: A novel component of the macrophage cell fusion machinery. Commun Integr Biol. 2010;3(3):256–259. | |
Xu F, Zhen YS. (-)-Epigallocatechin-3-gallate enhances anti-tumor effect of cytosine arabinoside on HL-60 cells. Acta Pharmacol Sin. 2003;24(2):163–168. | |
Mandel S, Amit T, Reznichenko L, Weinreb O, Youdim MB. Green tea catechins as brain-permeable, natural iron chelators-antioxidants for the treatment of neurodegenerative disorders. Mol Nutr Food Res. 2006;50(2):229–234. | |
Lin LC, Wang MN, Tseng TY, Sung JS, Tsai TH. Pharmacokinetics of (-)-epigallocatechin-3-gallate in conscious and freely moving rats and its brain regional distribution. J Agric Food Chem. 2007;55(4):1517–1524. | |
Nath S, Bachani M, Harshavardhana D, Steiner JP. Catechins protect neurons against mitochondrial toxins and HIV proteins via activation of the BDNF pathway. J Neurovirol. 2012;18(6):445–455. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.