")
Back to Journals » Journal of Inflammation Research » Volume 14
Transcriptional Interactomic Inhibition of RORα Suppresses Th17-Related Inflammation
Authors Ho CC , Kim G , Mun CH, Kim JW, Han J, Park JY, Park YB, Lee SK
Received 15 October 2021
Accepted for publication 8 December 2021
Published 21 December 2021 Volume 2021:14 Pages 7091—7105
DOI https://doi.org/10.2147/JIR.S344031
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Chun-Chang Ho,1 Giha Kim,1 Chin Hee Mun,2 Ju-Won Kim,3 Jieun Han,3 Ji Yoon Park,1 Yong-Beom Park,2– 4 Sang-Kyou Lee1,5
1Department of Biotechnology, College of Life Science and Biotechnology, Yonsei University, Seoul, Republic of Korea; 2Division of Rheumatology, Department of Internal Medicine, Yonsei University College of Medicine, Seoul, Republic of Korea; 3Department of Medical Science, Brain Korea 21 PLUS Project, Yonsei University, Seoul, Republic of Korea; 4Institute for Immunology and Immunological Diseases, Yonsei University College of Medicine, Seoul, Republic of Korea; 5Good T Cells, Inc., Seoul, Republic of Korea
Correspondence: Sang-Kyou Lee
Department of Biotechnology, College of Life Science and Biotechnology, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul, Republic of Korea
Tel +82 2 2123 2889
Email [email protected]
Purpose: Th17 cells and their cytokines are implicated in the pathogenesis of various autoimmune diseases. Retinoic acid-related orphan receptor alpha (RORα) is a transcription factor for the differentiation and the inflammatory functions of Th17 cells. In this study, we generated the nucleus-transducible form of transcription modulation domain of RORα (nt-RORα-TMD) to investigate the functional roles of RORα in vitro and in vivo under normal physiological condition without genetic alteration.
Methods: The functions of nt-RORα-TMD were analyzed in vitro through flow cytometry, luciferase assay, ELISA, and transcriptome sequencing. Finally, the in vivo therapeutic effects of nt-RORα-TMD were verified in dextran sulfate sodium (DSS)-induced colitis mice.
Results: nt-RORα-TMD was effectively delivered into the cell nucleus in a dose- and time-dependent manner without any cellular toxicity. nt-RORα-TMD competitively inhibited the RORα-mediated transcription but not RORγt-mediated transcription. Secretion of IL-17A from the splenocytes was suppressed by nt-RORα-TMD without affecting the secretion of Th1- or Th2-type cytokine and T cell activation events such as induction of CD69 and CD25. The differentiation potential of naïve T cells into Th17 cells, not into Th1, Th2, or Treg cells, was significantly blocked by nt-RORα-TMD. Consistently, mRNA sequencing analysis showed that nt-RORα-TMD treatment down-regulated the expression of the genes related to the differentiation and functions of Th17 cells. Treatment of DSS-induced colitis mice with nt-RORα-TMD improved the overall symptoms of colitis, such as body weight change, colon length, infiltration of inflammatory cells, and the level of inflammatory cytokines in the serum. In the mesenteric lymph node (MLN) of the nt-RORα-TMD-treated mice, the population of CD4+IL-17A+ Th17 cells was reduced, and the population of CD4+Foxp3+ Treg cells increased.
Conclusion: nt-RORα-TMD has a potential to be developed as a novel therapeutic reagent for treating various inflammatory diseases in which Th17 cells are the leading pathological player.
Keywords: retinoic acid-related orphan receptor, T helper 17, transcriptional modulation, DSS-induced colitis, inflammatory disease
Introduction
Th17 cells play an important role in the pathogenesis of various autoimmune diseases such as rheumatoid arthritis (RA), multiple sclerosis (MS), systemic lupus erythematosus (SLE), and inflammatory bowel disease (IBD).1–4 Th17 cells secrete pro-inflammatory cytokines such as IL-17, IL-21, IL-22, and GM-CSF for host defense against extracellular bacterial and fungal infections.5 However, systemic or organ-specific inflammatory disorders can arise when Th17 cells become dysregulated after extracellular pathogenic infection or when the immunological balance among T helper cells is disrupted.6
IBD, such as Crohn’s disease and ulcerative colitis, is a representative autoimmune disease characterized by chronic relapsing inflammation of the gastrointestinal tissue with common symptoms of diarrhea, fecal occult blood, or weight loss.7,8 Although the exact etiology of IBD is still unknown, immune system malfunction is believed to be one of the main causes. Over the last few years, it has been elucidated that Th17 cells and their cytokines are closely implicated in the pathogenesis of IBD and are crucial mediators for inducing and amplifying the inflammatory processes of IBD.9
RORα, one of the nuclear receptors, regulates various biological processes such as cerebellar development, cellular metabolism, cancer progression, or immunological responses.10–13 In previous studies, it has been demonstrated that RORα plays a key role as a lineage-associated transcription factor for Th17 cells together with retinoic acid-related orphan receptor gamma t (RORγt). The overexpression of RORα promoted Th17 cell differentiation, and RORα deficiency reduced the expression of IL-17 both in vitro and in vivo. Double deficiencies of RORγt and RORα led to complete abrogation of Th17 cell development and prevented the mice from developing experimental autoimmune encephalomyelitis (EAE). In contrast, only a single deficiency of RORγt failed to abrogate Th17 cytokine expression completely.14
To date, various studies have suggested therapeutic strategies for targeting Th17 cells to treat various inflammatory diseases including IBD. For example, Ustekinumab, Mirikizumab, or Risankizumab, the monoclonal antibody targeting IL-23, which is a cytokine important for maintaining the functions of Th17 cells, is used in clinical trials for the treatment of IBD (NCT02407236, NCT02589665, and NCT02031276). However, these antibodies still have limitations in that the therapeutic effect was shown only in a limited number of patients.15–17 In other studies, anti-IL-17 antibody (Secukinumab) and anti-IL-17RA antibody (Brodalumab) showed therapeutic efficacy in patients with psoriasis. However, the clinical trials using these antibodies for patients with other autoimmune diseases such as RA or Crohn’s disease failed to show therapeutic efficacy (NCT01009281, NCT00771030). In some cases, several adverse effects were observed.18,19 Therefore, a novel therapeutic strategy targeting the key molecule to regulate the differentiation or the functions of Th17 cells in a multifaceted manner is required to treat autoimmune disorders in which Th17 cells are significantly implicated.
In this study, it was examined whether the inflammatory functions of Th17 cells can be regulated by inhibiting the transcriptional function of RORα. To modulate the functions of RORα in vitro and in vivo without genetic manipulation, novel strategy was used to deliver the TMD of RORα into the nucleus of the cells and competitively inhibit the transcriptional activity of the endogenous RORα. nt-RORα-TMD consists of DBD (DNA Binding Domain) and hinge region of RORα with Hph-1-PTD, a human-origin PTD (Protein Transduction Domain), which could effectively deliver the target protein into the nucleus of the cells in vitro and in vivo. We hypothesized that inhibition of the transcriptional function of RORα by nt-RORα-TMD can regulate the inflammatory functions of Th17 cells and show therapeutic effects in inflammatory disease models in which Th17 cells are pathogenically implicated. To prove this hypothesis, the intracellular delivery kinetics and transcriptional inhibition function of nt-RORα-TMD were examined, and the effects of nt-RORα-TMD on CD4+ T cells, especially on Th17 cells, were analyzed. Finally, the therapeutic efficacy of nt-RORα-TMD in DSS-induced colitis mice was verified.
Materials and Methods
DNA Constructs for the Recombinant Proteins
The cDNA of mouse RORα (NM_001289916.1) was purchased from Korea Human Gene Bank, Medical Genomics Research center, KRIBB, Korea. The base pairs of the DNA binding domain and hinge region encoded amino acid from 10 to 215 of the wildtype RORα were amplified via PCR and inserted into a pET28a(+) vector containing a Hph-1-PTD and a FLAG tag. The form containing point mutations (R42A/R43G) in the DBD was generated through pfu DNA polymerase (Agilent) as a negative control. All the cloned DNA constructs were sequenced to confirm the fidelity of the reading frame.
Expression and Purification of the Recombinant Proteins
The cloned DNA plasmids were transformed into a BL21-Codon Plus (DE3)-RIPL strain (Novagen) of Escherichia coli for expression. For protein induction, 1 mM of IPTG (Duchefa Biochemie) was added. The cells were harvested and resuspended with native lysis buffer (10 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, pH 8.0) and then sonicated. Lysates were centrifuged, and the supernatants were mixed with Ni-NTA resin (Qiagen). The recombinant proteins were purified using native wash buffer (30 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, pH 8.0) and native elution buffer (250 mM imidazole, 50 mM NaH2PO4, 300 mM NaCl, pH 8.0). The eluted recombinant proteins were desalted with PD-10 Sephadex G-25 (GE Healthcare) using 10% glycerol PBS. The proteins were then mixed with SP Sepharose Fast Flow (GE Healthcare) in binding buffer (50 mM NaH2PO4, 200 mM NaCl, pH 6.0). After washing with the binding buffer, bound proteins were eluted with elution buffer (50 mM NaH2PO4, 2 M NaCl, pH 6.0) and desalted using PD-10 Sephadex G-25. The endotoxin level in the purified recombinant protein was within the safe range, approximately 6 EU/mL. There were no symptoms of the endotoxin-induced immune response in the animal experiment using recombinant proteins, such as anaphylactic shock.
Western Blot
The purified recombinant proteins were loaded on the gel through SDS-PAGE and transferred to a PVDF membrane (Bio-Rad). The membrane was washed with TBST (Tris-buffered saline with 0.1% Tween 20) and blocked with blocking buffer (4% Bovine Serum Albumin in TBST). The anti-FLAG antibody (Cell signaling) and anti-mouse IgG-HRP antibody (Abcam) were used to detect the recombinant proteins. ECL reagent (Bio-Rad) was added onto the membrane, and the chemiluminescence signal was detected using a ChemiDoc (Bio-Rad).
Cell Culture
Jurkat cells (Clone E6-1, ATCC TIB-152) and HEK293 cells (ATCC CRL-1573) were purchased from ATCC (USA). Jurkat cells were cultured in RPMI 1640 (Lonza) with 10% heat-inactivated FBS (Hyclone), 2 mM L-glutamine (Lonza), and 100 μg/mL penicillin–streptomycin (Lonza). HEK293 cells were cultured in DMEM (Lonza) with 10% heat-inactivated FBS, 2 mM L-glutamine, and 100 μg/mL penicillin-streptomycin, 1 mM sodium pyruvate, and NEAA (Lonza). Mouse primary lymphocytes were cultured in RPMI 1640 with 7.5% heat-activated FBS, 2 mM L-glutamine, 100 μg/mL penicillin-streptomycin, and 50 μM β-Mercaptoethanol (Sigma Aldrich). All cells were cultured at 37°C in a humidified atmosphere of 5% CO2.
Protein Transduction
2x105 Jurkat cells were treated and incubated with nt-RORα-TMD in a dose-dependent manner or in a time-dependent manner. After transduction, cells were harvested and washed with PBS. Cells were then fixed and permeabilized with Fixation/Permeabilization Buffer (eBioscience), stained with anti-FLAG antibody-PE (BioLegend), and analyzed through flow cytometry (SA3800 Spectral Cell Analyzer) (Sony Biotechnology).
Immunocytochemistry
1x106 splenocytes were treated with PBS, 2 μM nt-RORα-TMD, or 2 μM RORα-TMD in 96 well-plate and incubated. The splenocytes were harvested and seeded on top of a round microscope cover glass 18-mm in diameter placed on the bottom of a 12-well culture plate. Cells were fixed with 10% formalin (Sigma-Aldrich) and permeabilized with 0.5% Triton X-100 (Sigma-Aldrich). After blocking with 1% BSA in PBS, anti-FLAG M2-FITC antibody (Sigma-Aldrich) was used to stain the recombinant proteins. 4ʹ,6-diamino-2-phenylindole (DAPI) was used to counterstain nuclei, and cells were analyzed with a confocal microscope (Carl Zeiss).
Cell Viability Assay
nt-RORα-TMD and nt-RORα-TMD (R42A/R43G) were tested for cytotoxicity using Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies). 2×105 Jurkat cells or mouse splenocytes were incubated with nt-RORα-TMD or nt-RORα-TMD (R42A/R43G) for 1 h, and CCK-8 reagent was added and incubated for an additional 4 h. Cell viability was measured using a microplate reader (Bio-Rad) at an absorbance of 450 nm.
Transient Transfections and Luciferase Reporter Gene Assays
1 μg of SPORT6 vector containing wildtype RORα or 1 μg of pEGFP-N1 vector containing wildtype RORγt were co-transfected along with IL-17A promoter-luciferase vector into 5×105 HEK293 cells using Lipofectamine Reagents (Invitrogen) diluted with Opti-MEM (Gibco). After 4 h incubation, cells were treated with nt-RORα-TMD or nt-RORα-TMD (R42A/R43G). After overnight incubation, cells were washed with PBS and lysed using Cell Culture Lysis 5X Reagent (Promega). Lysates were mixed with Luciferase Assay Substrate (Promega), and luciferase activity was measured by a luminometer (Promega).
Cytokine Measurement Through ELISA
Primary mouse splenocytes were treated with nt-RORα-TMD or nt-RORα-TMD (R42A/R43G) and incubated along with plate-bound 1 μg/mL of anti-CD3 (BD Bioscience) and anti-CD28 (BD Bioscience) antibodies for stimulation. After 72 h incubation, the supernatant from the culture medium was collected. The levels of IFN-γ, IL-4, and IL-17A were measured by ELISA according to the manufacturer’s instructions (Invitrogen). Blood was obtained from DSS-induced colitis mice for the mice serum samples, and the serum was separated by centrifuge. The levels of TNF-α, IL-1β, and IL-6 were measured by ELISA according to the manufacturer’s instructions (Invitrogen).
T Cell Isolation, Activation, and Differentiation
CD4+CD62L+T Cell Isolation Kit (Miltenyi Biotec) was used to MACS-purify naïve T cells from spleens of seven-week-old mice. Naïve T cells were activated in 96 well plates coated with 1 μg/mL of anti-CD3 (BD Bioscience) and anti-CD28 (BD Bioscience) antibodies for 3 days. To differentiate naïve T cells into Th1, Th2, Th17, or iTreg cells, different cytokines were added in addition to the plate-bound anti-CD3 and anti-CD28 antibodies: for Th1-skewing condition, recombinant mouse IL-12 (25 ng/mL; PeproTech) and anti-IL-4 antibody (2 μg/mL; BioLegend); for Th2-skewing condition, recombinant mouse IL-4 (25 ng/mL; PeproTech), and anti-IFN-γ antibody (2 μg/mL; BioLegend); for Th17-skewing condition, recombinant mouse TGF-β1 (1 ng/mL; R&D Systems), recombinant mouse IL-6 (25 ng/mL; PeproTech), anti-IFN-γ antibody (2 μg/mL), and anti-IL-4 antibody (2 μg/mL); for iTreg-skewing condition, recombinant mouse TGF-β1 (5 ng/mL) and recombinant mouse IL-2 (20 ng/mL; PeproTech). Along with the cytokines, nt-RORα-TMD or nt-RORα-TMD (R42A/R43G) were added to each well and incubated for 72 h.
T Cell Analysis Using Flow Cytometry
To analyze activated T cells using FACS, cells were stained with either anti-CD69-FITC antibody (BD Bioscience) or anti-CD25-PE antibody (BD Bioscience) and then analyzed through flow cytometry. For Th1, Th2, Th17, and iTreg cells, after 72 h incubation with the recombinant proteins, cells were restimulated with Cell Stimulation Cocktail (500X) (eBioscience). After 4 h restimulation, cells were fixed and permeabilized using Fixation/Permeabilization Buffer (eBioscience), and intracellular proteins were stained with appropriate antibodies for each cell type: Th1 cells were stained with anti-T-bet antibody-APC (eBioscience) and anti-IFN-gamma antibody-PE (eBioscience); Th2 cells were stained with anti-Gata-3 antibody-APC (eBioscience) and anti-IL-4 antibody-PE (eBioscience); Th17 cells were stained with anti-ROR gamma (t) antibody-PE (eBioscience) and anti-IL-17A antibody-APC (eBioscience); Treg cells were stained with anti-Foxp3 antibody-APC (eBioscience) and anti-IL-10 antibody-PE (eBioscience). Stained cells were analyzed through flow cytometry. To analyze mesenteric lymph node (MLN) cells from DSS-induced mice, MLN cells were isolated and restimulated for 4 h using Cell Stimulation Cocktail (500X). After restimulation, cells were stained with anti-CD4 antibody-AF700 (eBioscience) and were fixed and permeabilized using Fixation/Permeabilization Buffer, and then cells were stained with anti-IFN-gamma antibody-PE, anti-IL-17A antibody-APC, anti-ROR gamma (t) antibody-PE, or anti-Foxp3 antibody-PE.
Transcriptome Sequencing and Gene Set Enrichment Analysis (GSEA)
Cells differentiated in Th17-skewing condition along with or without nt-RORα-TMD were harvested on day 3, and RNA isolation and sequencing were performed by Macrogen (Republic of Korea). After isolation of total RNA from cells, mRNA was isolated using an mRNA purification kit, and sequencing was performed using cDNA fragments generated by mRNA. Aligned reads were generated by mapping the reads to the reference genome using the HISAT2 program. Using the reference-based aligned reads information, the transcript assembly was performed through the StringTie program. The expression levels obtained through transcript quantification of each sample were extracted as a normalization value. GSEA was analyzed using GSEA v4.1.0 software. ImmuneSigDB.v7.4. symbols (Godec et al 2016) were used as a gene sets database, and the number of permutations and permutation type was set to 1000 and “gene set”. The chip platform was set to “Mouse_Gene_Symbol_Remapping_Human_Orthologs_MSigDB.v7.4. chip”. Transcripts per kilobase million (TPM) values for 11551 genes from 2 samples of nt-RORα-TMD-treated and non-treated Th17 cells were used for the analysis.
Animals
Male and female C57BL/6N inbred mice 7–8 weeks were purchased from Orient Bio (Republic of Korea) and maintained under semi-specific-pathogen-free (SPF) conditions. All experimental procedures with animals were approved by the Institutional Animal Care and Use Committee (IACUC) of Yonsei Laboratory Animal Research Center (YLARC) and performed under the YLARC-IACUC guidelines for the ethical use of animals (IACUC-A-202012-1186-03, 2019-0279).
DSS-Induced Colitis Mice and Disease Activity Index (DAI) Scoring
Seven-week-old male C57BL/6N mice were purchased from Orient Bio (Republic of Korea) and maintained under semi-SPF conditions. Mice were acclimatized for the first 1 week and were given water containing 2% DSS ad libitum from day 0 to day 7, and the water containing DSS was changed every other day. Mice were randomly divided into Normal, DSS, Anti-IL-17A antibody treatment, nt-RORα-TMD treatment, and nt-RORα-TMD (R42A/R43G) treatment groups. The normal group was given sterile water, and the other groups were given water containing 2% DSS. nt-RORα-TMD (20 or 100 μg/mouse), nt-RORα-TMD (R42A/R43G) (100 μg/mouse) or anti-IL-17A antibody (BioXCell) (100 μg/mouse) were injected intraperitoneally once daily from day 1 to day 7, and the body weight and DAI score were measured daily from day 0 to day 7. All groups of mice were given sterile water from day 7 and were sacrificed for the analysis on day 8. The spleen and colon (from the cecum to the end of the rectum) were obtained from each mouse to measure the size and length, and mesenteric lymph nodes were obtained to analyze the T cell population. The length of the colon was measured from the beginning of the proximal colon to the end of the distal colon. The DAI score was measured by C-C Ho, G Kim, CH Mun, J-W Kim, J Han, and JY Park in a blinded fashion according to the following index. Body weight change: 0, no change; 1, 1–5%; 2, 5–10%; 3, 10–20%; 4, >20%. Stool: 0, normal; 1, some soft but still formed; 2, very soft; 3, diarrhea. Fecal occult blood: 0, negative hemoccult; 1, positive hemoccult; 2, blood traces in stool; 3, rectal bleeding.
Histopathological Examination
The middle part of the colon isolated from the mice of each group was cut and fixed with 4% phosphate-buffered paraformaldehyde. Each colon sample was then embedded in paraffin, and paraffin sections were stained with hematoxylin and eosin (H&E) (Dako) and periodic acid-Schiff (PAS). The histological images were obtained through an optical microscope (Olympus BX51) (Olympus Corporation). The degree of inflammatory cell infiltration, goblet cell depletion, and crypt damage was measured by CH Mun, J-W Kim, and J Han in a blinded fashion according to the following index. Inflammatory cell infiltration: 0, no infiltrate; 1, mild infiltrate <25%; 2, moderated infiltrate <50%; 3, marked infiltrate >50%. Goblet cell depletion: 0, none; 1, mild depletion <25%; 2, moderate depletion <50%; 3, marked depletion >50%. Crypt damage: 0, none; 1, some crypt damage; 2, larger spaces between crypts; 3, large areas without crypts.
Statistical Analysis
The results are expressed in terms of a mean ± SEM (n≥3). Statistical analysis of group differences was performed using ANOVA analysis followed by Dunnett’s multiple comparison test. The number of asterisks indicated the following significance: ns (not significant), *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001. All data were analyzed using GraphPad Prism 8.
Results
The Efficiency and Kinetics of the Intra-Nuclear Delivery of nt-RORα-TMD
RORα is known as a transcription factor essential for the development and functions of Th17 cells.20 However, RORα is also considered functionally redundant to RORγt. To investigate the functions of RORα in vitro and in vivo under normal physiological condition without genetic alteration such as gene knockout, knock-down, or overexpression of RORα, nt-RORα-TMD was generated by fusing Hph-1-PTD with RORα-TMD composed of DBD and hinge region of RORα (Figure 1A). In addition to nt-RORα-TMD, RORα-TMD without Hph-1-PTD (RORα-TMD) was used as a negative control. The mutant form of nt-RORα-TMD (nt-RORα-TMD(R42A/R43G)) was also generated, which might have a decreased binding capacity to the RORα-binding sites due to the mutations in two arginines located at the base of the DNA-binding zinc finger motif of RORα. The proteins were expressed in an Escherichia coli expression system and purified under native conditions. The identity of the proteins was confirmed by SDS-PAGE and Western blot analysis using anti-FLAG antibody (Figure 1B). The transduction kinetics of nt-RORα-TMD was examined using Jurkat T cells or primary T cells treated with different concentrations of nt-RORα-TMD or for different periods. nt-RORα-TMD can be delivered into the cells in a concentration- and time-dependent manner (Figure 1C and D). The delivered nt-RORα-TMD remained stable inside the cells and gradually degraded 24 h after transduction (Figure 1D). The intranuclear localization of the delivered nt-RORα-TMD was visually confirmed by confocal microscopy. nt-RORα-TMD was detected in nucleus of the cells, but not in the cells treated with PBS or RORα-TMD (Figure 1E). To determine whether the proteins have any cellular toxicity, Jurkat T cells or mouse splenocytes were treated with different concentrations of nt-RORα-TMD or nt-RORα-TMD (R42A/R43G). As shown in Figure 1F, the treatment of the cells with these proteins did not affect the cell viability. Therefore, nt-RORα-TMD can be delivered into the nucleus of the cells effectively and stably and in a concentration- and time-dependent manner without cellular cytotoxicity.
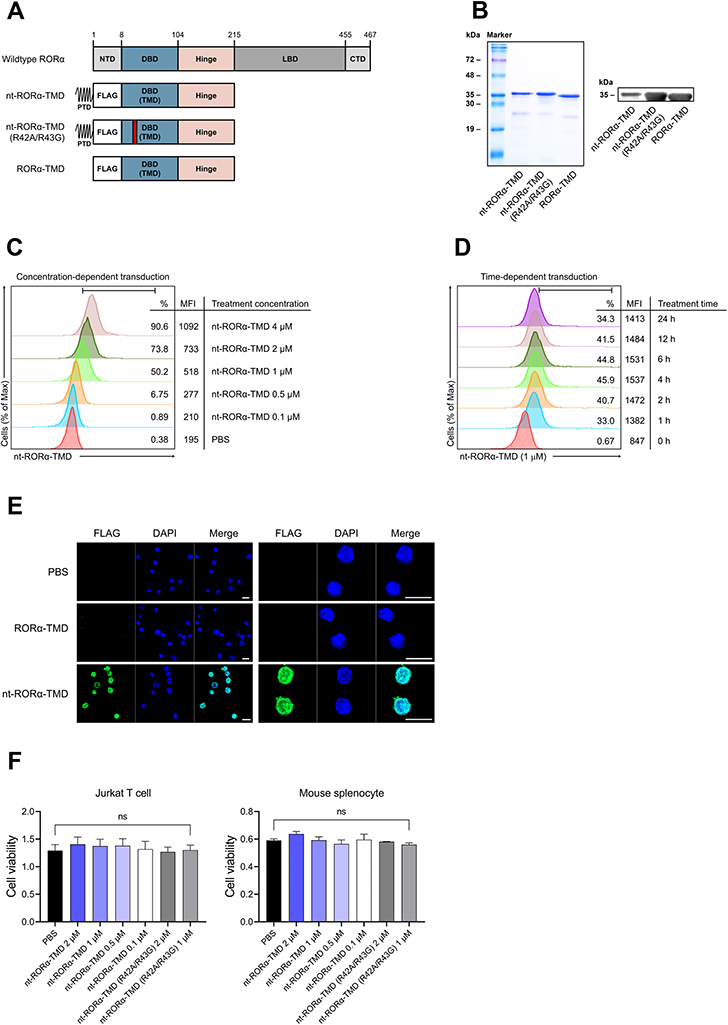 |
Figure 1 Generation of nt-RORα-TMD and verification of its intranuclear delivery kinetics. (A) Structure of nt-RORα-TMD, nt-RORα-TMD (R42A/R43G) with mutated arginine 42 and 43, and RORα-TMD without Hph-1-PTD. (B) The identity of nt-RORα-TMD, nt-RORα-TMD (R42A/R43G), and RORα-TMD were confirmed by SDS-PAGE and Western blot after protein purification. (C and D) Jurkat T cells were treated with different concentrations of nt-RORα-TMD for 1 h (C) or with 1 μM of nt-RORα-TMD for the different periods (D), and the amount of the delivered protein inside the cells was analyzed by flow cytometry using anti-FLAG antibody. (E) The intracellular localization of nt-RORα-TMD was confirmed by confocal microscope using DAPI and anti-FLAG antibody. Scale bar = 10 μm (F) Jurkat T cells and mouse splenocytes were treated with nt-RORα-TMD or nt-RORα-TMD (R42A/R43G), and the cellular cytotoxicity was examined by CCK-8 analysis. The graphs are represented as mean ± SEM (n=3). Abbreviation: ns, not significant. |
Suppression of IL-17A Expression by nt-RORα-TMD Through the Specific Inhibition of RORα-Mediated Transcription Without Affecting T Cell Activation
It has been known that RORα binds to the promoter region and induces the expression of the IL-17A gene.14,20 To confirm the inhibitory function of nt-RORα-TMD on RORα-mediated IL-17A expression, wild-type RORα-expressing vector and IL-17A promoter-luciferase vector were co-transfected into HEK293 cells. Then, the transfected cells were incubated with either nt-RORα-TMD or nt-RORα-TMD (R42A/R43G). nt-RORα-TMD was able to reduce the luciferase activity in a dose-dependent manner, and the level of inhibition by nt-RORα-TMD (R42A/R43G) was minimal (Figure 2A). To examine the functional specificity of nt-RORα-TMD toward RORα, wildtype RORγt-expressing vector was co-transfected instead of wildtype RORα-expressing vector. As shown in Figure 2B, nt-RORα-TMD did not exert the inhibitory effect on RORγt-mediated IL-17A transcription activity. Next, the inhibitory function of nt-RORα-TMD toward the secretion of IL-17A was investigated using the mouse splenocytes stimulated with the plate-bound anti-CD3 and anti-CD28 antibodies in the presence of either nt-RORα-TMD or nt-RORα-TMD (R42A/R43G). nt-RORα-TMD was able to decrease the amount of IL-17A secreted in the media dose-dependently, while the secretion of IFN-γ or IL-4 remained unaffected (Figure 2C). Next, we examined whether nt-RORα-TMD influences the signaling events leading to T cell activation. Mouse naïve CD4+ T cells were activated with plate-bound anti-CD3 and anti-CD28 antibodies in the presence of nt-RORα-TMD or nt-RORα-TMD (R42A/R43G) and the induced expression of CD69 or CD25 on the surface was analyzed, which serves as the early and late markers for T cell activation, respectively. Neither nt-RORα-TMD nor nt-RORα-TMD (R42A/R43G) treatment affected the level of CD69 or CD25 on the surface (Figure 2D). These results indicate that nt-RORα-TMD can specifically reduce the expression of IL-17A at the transcriptional level without affecting the expression of other cytokines specific to Th1 or Th2 subset, or the T cell activation.
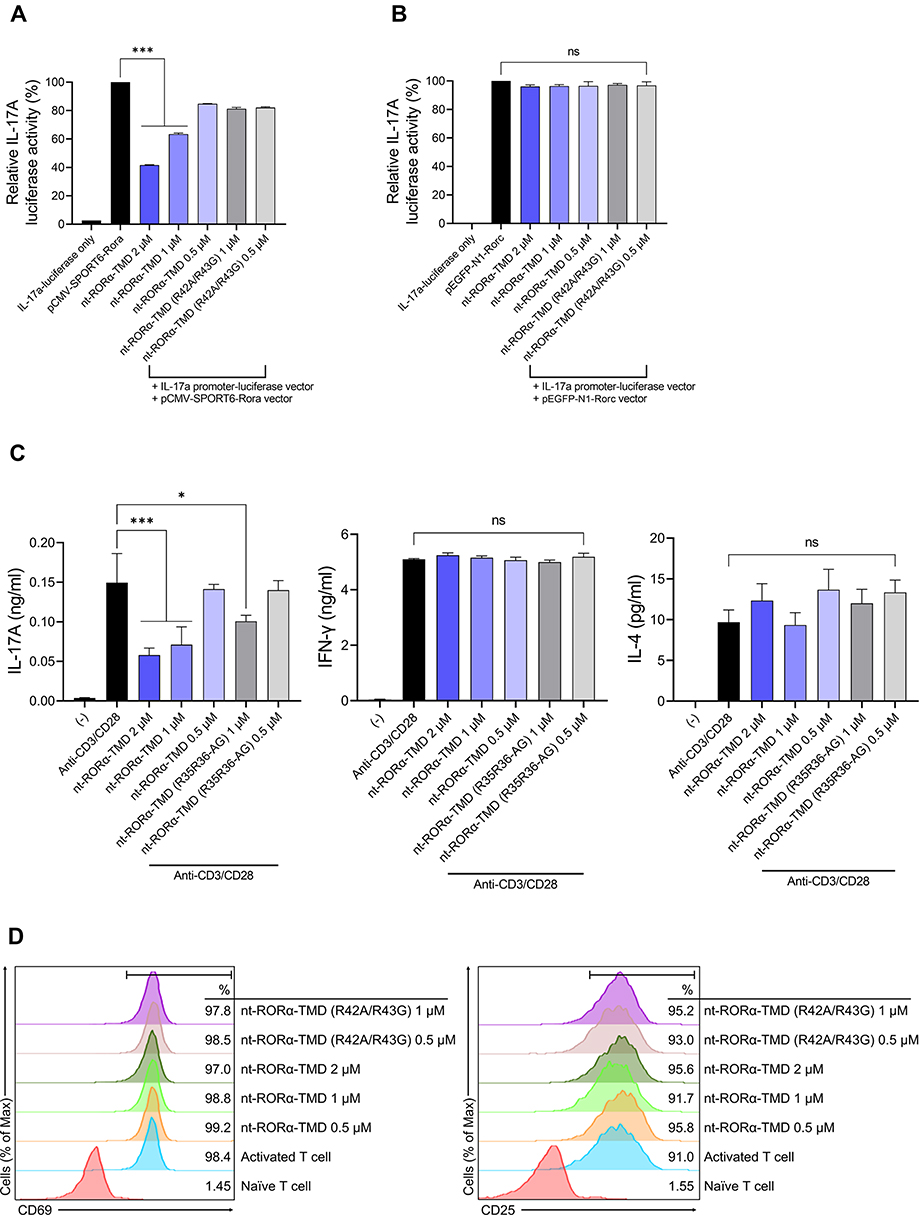 |
Figure 2 nt-RORα-TMD specifically suppressed IL-17A secretion through the inhibition of RORα-mediated transcription. (A and B) The competitive inhibition of nt-RORα-TMD over the endogenous RORα was examined by luciferase assay using HEK293 cells co-transfected with IL-17A promoter-luciferase vector and a vector expressing wild-type RORα (A) or a vector expressing wild-type RORγt (B). At 24 hours after the treatment of the transfected cells with nt-RORα-TMD or nt-RORα-TMD (R42A/R43G), the luciferase activity was measured by a luminometer. (C) Mouse splenocytes were activated with anti-CD3 and anti-CD28 antibodies in the presence of nt-RORα-TMD or nt-RORα-TMD (R42A/R43G), and the amount of IL-17A, IFN-γ, and IL-4 in the medium was measured by ELISA. (D) Mouse naïve CD4+CD62L+ T cells were isolated through MACS and activated with anti-CD3 and anti-CD28 antibodies treated in the presence of nt-RORα-TMD or nt-RORα-TMD (R42A/R43G). The level of CD69 or CD25 on the surface was analyzed by flow cytometry after 1 and 3 days of activation, respectively. The graphs are represented as mean ± SEM (n=3), *P<0.05, ***P<0.001. Abbreviation: ns, not significant. |
nt-RORα-TMD Specifically Inhibited Th17 Cell Differentiation and Down-Regulated the Expression of Th17-Related Genes
To investigate whether nt-RORα-TMD could affect the differentiation potential of naïve T cells into various T cell subsets (Th1, Th2, Th17, or Treg), naive T cells were isolated from mice splenocytes. Differentiation of each T cell subset was induced under each T cell subset-skewing condition in the presence of nt-RORα-TMD or nt-RORα-TMD (R42A/R43G). The level of differentiation for each T cell subset was analyzed by intracellular staining of the lineage-specific transcription factor for each T cell subset such as T-bet, GATA-3, RORγt, or Foxp3. Treatment of nt-RORα-TMD substantially reduced the population of RORγt+ T cells under Th17 skewing condition in a dose-dependent manner and also inhibited the expression of IL-17A among RORγt+ T cells (Figure 3A). The level of Th17 cell differentiation inhibition by nt-RORα-TMD (R42A/R43G) was less than that by nt-RORα-TMD. However, nt-RORα-TMD neither affects the differentiation into Th1, Th2, or Treg cells (Figure 3B), nor the expression of IFN-γ, IL-4, or IL-10, the major cytokines of each T cell subset (Figure 3C). To find out the genes in Th17 cells, of which expression is affected by nt-RORα-TMD, we compared the gene expression profile between Th17 cells and Th17 cells treated with nt-RORα-TMD by mRNA sequencing. The genes of Th17-related transcription factors, cytokines, or chemokines such as Rorc, Stat3, Il17a, Il21, Ccl3, and Ccl20 were down-regulated in nt-RORα-TMD-treated Th17 cells and the expression of the genes known to suppress the development or activation of Th17 cells such as Osm, Socs3, Irf7, and Eomes was induced by nt-RORα-TMD treatment.21,22 In addition, it was found that the naïve T cell-related genes were more enriched in nt-RORα-TMD-treated Th17 cells (Figure 3D). Consistently, Gene set enrichment analysis (GSEA) suggested that the nt-RORα-TMD-treated Th17 cells were highly enriched for the signature genes associated with naive CD4+ T cells (Figure 3E). These results demonstrated that nt-RORα-TMD explicitly inhibits Th17 cell differentiation from naïve T cells through the modulation of RORα-mediated transcription to express Th17-related genes.
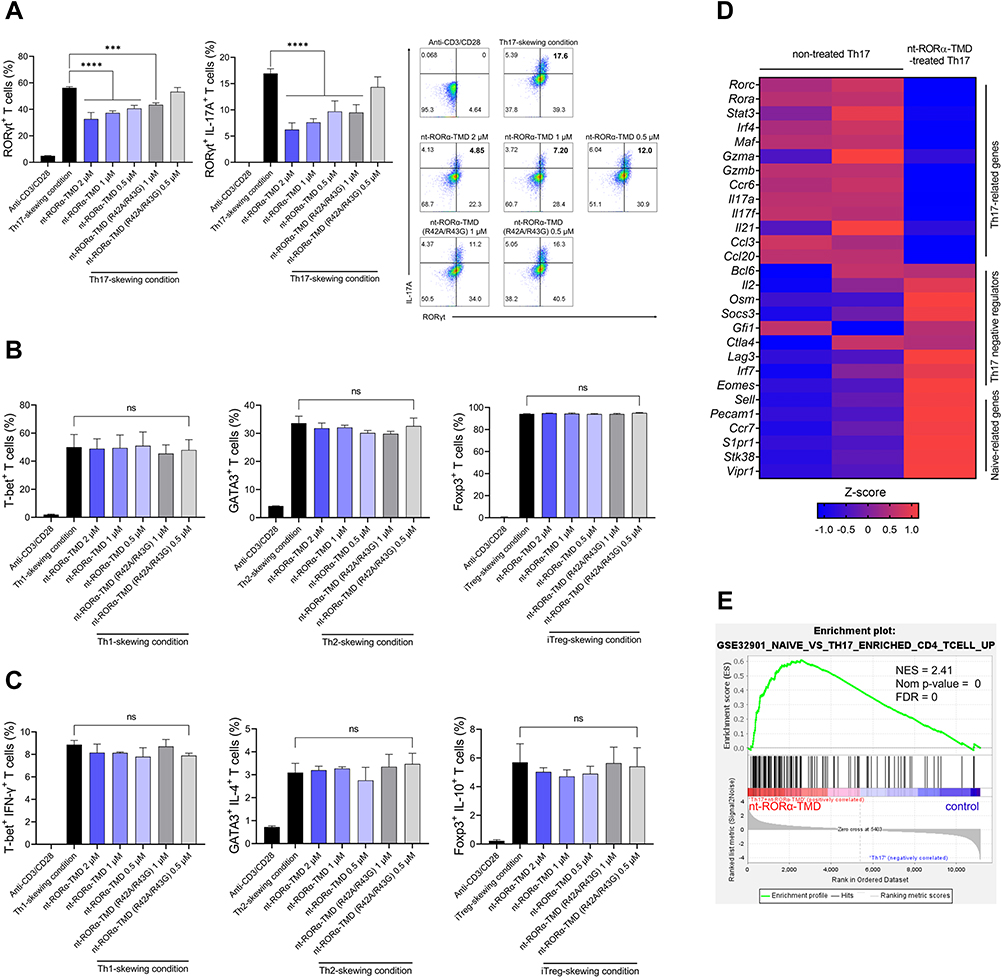 |
Figure 3 Specific inhibition of Th17 differentiation and the expression of Th17-related genes by nt-RORα-TMD. (A) Mouse naïve CD4+CD62L+ T cells were isolated and cultured in Th17-skewing condition in the presence of nt-RORα-TMD or nt-RORα-TMD (R42A/R43G). After 3 days of culture, T cells expressing RORγt and IL-17A were analyzed by flow cytometry using the corresponding mAb. (B and C) Naïve T cells were induced to differentiate into Th1, Th2 or iTreg cells under Th1-, Th2-, or iTreg-skewing condition in the presence of nt-RORα-TMD or nt-RORα-TMD (R42A/R43G), and T cells expressing the transcription factor specific to each T cell subset (B) and its representative cytokines (C) were analyzed by flow cytometry. (D and E) The gene expression profiles of Th17 cells treated with or without nt-RORα-TMD were analyzed by transcriptome sequencing. (D) The TPM counts of the signature genes were normalized and represented as a heatmap. (E) Enrichment plots based on immunologic signature gene sets were analyzed through gene set enrichment analysis (GSEA). The graphs are represented as mean ± SEM (n=3), ***P<0.001, ****P<0.0001. Abbreviation: ns, not significant. |
In vivo Therapeutic Effects of nt-RORα-TMD in DSS-Induced Colitis Animal Model
To verify the in vivo therapeutic efficacy of nt-RORα-TMD, DSS-induced colitis mice were treated with nt-RORα-TMD as the treatment protocol as shown in Figure 4A, and anti-IL-17A antibody was used as a positive control. Body weight loss and DAI, such as manifestation of fecal occult blood or diarrhea, were significantly alleviated by nt-RORα-TMD treatment in a dose-dependent manner. The improvement level of colitis symptoms was comparable to that of anti-IL-17A antibody treatment (Figure 4B and C). The size of the spleen or colon was well maintained by 100 μg of nt-RORα-TMD treatment, whose therapeutic effect is similar to that of IL-17A antibody treatment (Figure 4D and E). These results demonstrate that nt-RORα-TMD has the therapeutic potential to alleviate inflammatory disease.
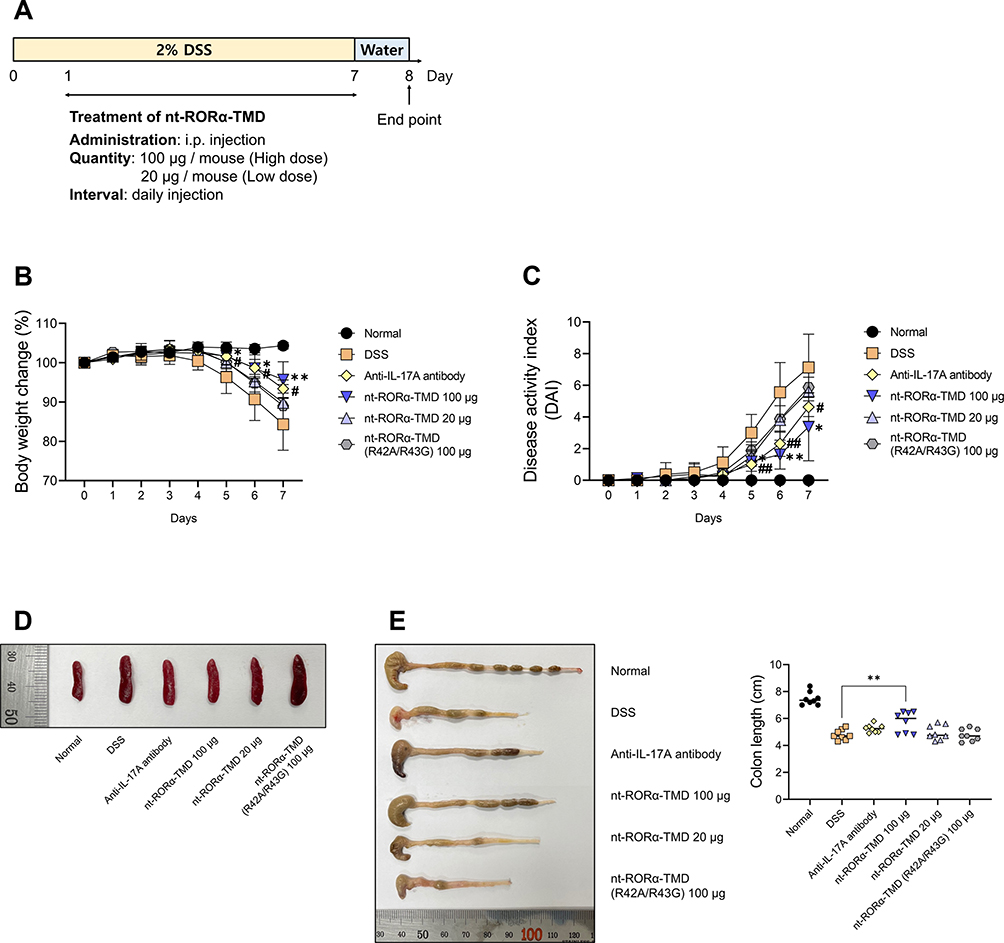 |
Figure 4 Therapeutic efficacy of nt-RORα-TMD in DSS-induced colitis mice. (A) Treatment scheme of DSS-induced colitis mice with PBS (for DSS group), anti-IL-17A antibody (100 μg/mouse), nt-RORα-TMD (100 or 20 μg/mouse) or nt-RORα-TMD (R42A/R43G) (100 μg/mouse). (B) The body weight of the mice was measured once daily. (C) The DAI score was calculated by measuring the clinical score of body weight change, stool, and fecal occult blood. (D) The spleens were harvested from the mice, and each group’s representative spleens were compared. (E) The colons were harvested from the mice and their length was measured. The graphs are represented as mean ± SEM (n=8). *,#P<0.05, **,##P<0.01. *DSS and nt-RORα-TMD 100 μg. #DSS and anti-IL-17A antibody. |
In vivo Therapeutic Efficacy of nt-RORα-TMD is Contributed by Reduction of Inflammatory Cytokines and Pathogenic T Cells
To determine the therapeutic mechanism of action by nt-RORα-TMD in the DSS-induced colitis animal model, the level of major pro-inflammatory cytokines such as TNF-α, IL-1β, or IL-6 in the serum was analyzed by ELISA. As shown in Figure 5A, the concentration of all three pro-inflammatory cytokines substantially decreased in the serum of the mice injected with nt-RORα-TMD dose-dependently. The level of inhibition by nt-RORα-TMD is comparable to that by anti-IL-17A antibody. Next, the sections of the colon were stained with H&E or PAS for analyzing the histopathological status of the colon, such as the level of inflammatory cell infiltration, goblet cell depletion, or crypt damage. In the colons of the mice treated with 100 μg of nt-RORα-TMD, the extent of the infiltrated inflammatory cells or the depleted goblet cells were significantly reduced, and the crypt damage was also less severe compared to the colon of the DSS-induced colitis animals (Figure 5B). To examine the change of the pathogenic T cell population in the DSS-induced colitis animal model, the level of Th17 cells, Th1 cells, or Treg cells in the MLN of the mice treated with the different reagents was analyzed (Figure 5C and D). Among CD4+ T cells, the population of IL-17A-expressing Th17 cells was significantly reduced in the MLN of the mice injected with 100 μg of nt-RORα-TMD. In addition, the number of IFN-γ+ T cells, which have been known to play a pathogenic role in colitis together with Th17 cells, also decreased by nt-RORα-TMD treatment (Figure 5C). Consistent with these results, nt-RORα-TMD treatment markedly reduced the number of CD4+RORγt+ Th17 cells in MLN to the level in the normal healthy animal. The decrease of CD4+RORγt+ Th17 cells was accompanied by the increase of CD4+Foxp3+ Treg cells in MLN of the nt-RORα-TMD-treated mice (Figure 5D). Taking these results together, it is suggested that nt-RORα-TMD exerts the in vivo therapeutic efficacy by reducing pathogenic CD4+IL-17A+RORγt+ Th17 cells and CD4+IFN-γ+ Th1 cells accompanying the increase of Treg cells in DSS-induced colitis animal model, leading to a decrease of pro-inflammatory cytokines in the serum.
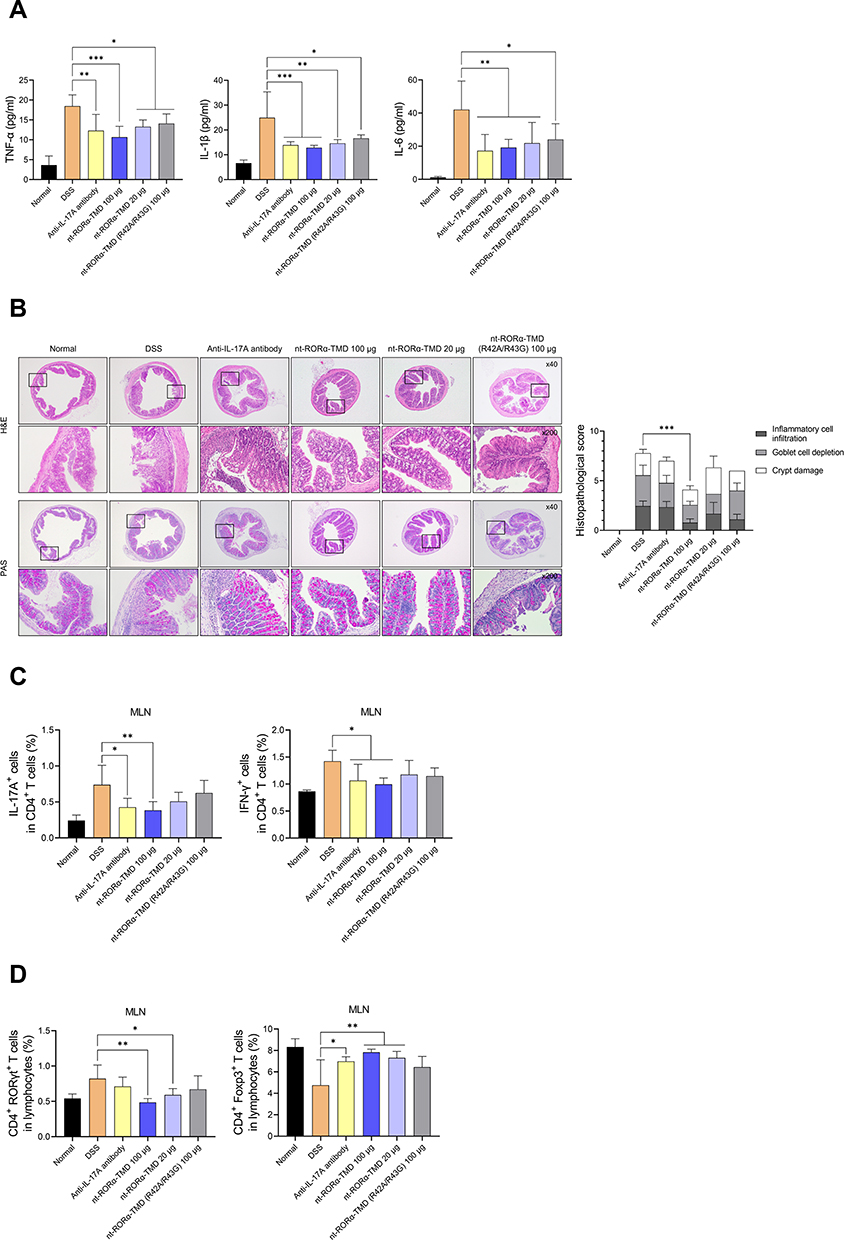 |
Figure 5 nt-RORα-TMD inhibits the secretion of pro-inflammatory cytokines and the level of pathogenic T cells in DSS-induced colitis. (A) The level of pro-inflammatory cytokines such as TNF-α, IL-1β, or IL-6 from the serum of the mice was measured by ELISA. (B) The sections of the colons were stained with H&E or PAS, and the histological images were obtained through an optical microscope. Histopathological scores of inflammatory cell infiltration, goblet cell depletion, and crypt damage were measured. (C and D) MLNs were isolated from the mice, and the cells from MLN were restimulated using the Cell Stimulation Cocktail for 4 hours. After restimulation, the population of IL-17A+ or IFN-γ+ cells among CD4+ T cells (C), and the population of CD4+RORγt+ or CD4+Foxp3+ T cells among whole lymphocytes (D) were analyzed by flow cytometry. The graphs are represented as mean ± SEM (n≥3). *P<0.05, **P<0.01, and ***P<0.001. |
Discussion
Retinoic acid-related orphan receptors (RORs), as a subfamily of nuclear receptors, are mainly composed of a DNA binding domain (DBD), a ligand-binding domain (LBD), and a hinge region connecting DBD and LBD. RORα is a member of the ROR family along with RORβ and RORγ.23 To date, RORα is known to be implicated in various biological processes such as cerebellum development, lipid homeostasis, cancer progression, and immune responses.24 Concerning the immune system, it has been reported that RORα is an essential factor for the activation of macrophages and the development of type 2 innate lymphocyte cells (ILC2) in the thymus.25,26 In addition, RORα has been shown to play a role as an essential regulator of Th2 cell responses in allergic asthma and of the skin-resident Treg cells for repressing skin inflammation in atopic dermatitis.27,28 In Th17 cells, which are significantly implicated in various inflammatory disorders, RORα has been reported as a transcription factor for Th17 differentiation and the expression of Th17-related genes, including its cytokines.14,20,29,30 Although the function of RORα has been revealed in Th17 cells, the studies on regulating the function of Th17 cells targeting RORα under the normal physiological condition without using genetic alteration have not been sufficiently reported.
IBD, such as Crohn’s disease and ulcerative colitis, is known as one of the most prevalent digestive diseases with a high incidence.31 As a representative autoimmune disease, IBD is reported that Th17 cells are strongly implicated in disease pathogenesis. In IBD patients, Th17 cells and their related-cytokines such as IL-17, IL-22, and IL-23 were more enriched in the intestinal mucosal than healthy controls.32,33 For the treatment of IBD, anti-inflammatory or immunosuppressive chemical drugs have been developed and are currently being used. However, many patients with IBD show refractory to these classical drugs. To overcome this limitation, various novel cytokine inhibitors and transcription factor inhibitors targeting Th17 cells have been developed, and their therapeutic effects are currently being evaluated in clinical trials.34
In this study, nt-RORα-TMD was generated to inhibit the function of RORα and thereby regulate the function of Th17 cells. Finally, the therapeutic effect of nt-RORα-TMD was verified in colitis mice. Intranuclear delivery of RORα-TMD was attempted using nt-RORα-TMD, a fusion protein linking the Hph-1-PTD to the DBD domain and hinge region of RORα. nt-RORα-TMD was effectively delivered into the nucleus of the cells in a dose- and time-dependent manner and stably present inside the cells for up to 24 h after delivery without any cellular toxicity. nt-RORα-TMD competitively and specifically inhibited the transcriptional function of endogenous RORα, leading to a significant reduction of IL-17A promoter activity. Importantly, nt-RORα-TMD did not block the IL-17A promoter activity through RORγt, another critical transcription factor for IL-17A expression. These results are consistent with the previous report that the function of RORα is independent of RORγt in developing the pathogenicity of Th17 cells.20 In several studies, small-molecule inhibitors or siRNA have been used to investigate and inhibit the function of RORα.20,35 Here, we generated a biologics recombinant protein that is more target-specific and has a lower risk of side effects than small-molecule inhibitors.
nt-RORα-TMD also inhibited the secretion of IL-17A from the splenocytes upon TCR stimulation. However, the nt-RORα-TMD treatment did not affect the secretion of IFN-γ and IL-4 from the splenocytes and the signaling events for T cell activation. Consistent with these results, nt-RORα-TMD treatment specifically blocked the differentiation of naïve T cells into Th17 cells, not into Th1, Th2, or Treg cells. However, the functions of RORα in Th2 cells in allergic asthma or skin-resident Treg cells have been elucidated.27,28 Therefore, our results may indicate that RORα does not affect the differentiation of Th2 or Treg cells from naïve T cells but plays a role in the functional regulation of Th2 cells or Treg cells in the local microenvironment or the disease-setting. Finally, through mRNA sequencing analysis, it is confirmed that the inhibitory effect of Th17 cell differentiation by nt-RORα-TMD was mediated by the inhibition of the expression of Th17-related genes. In recent studies, the functions of RORα in CD4+ T cells under inflammatory conditions have been studied using RORα-knockout mice.20,36,37 In this study, the role of RORα on the differentiation of each CD4+ T cell subset was newly investigated using nt-RORα-TMD without genetic modification, and it was confirmed that RORα only plays a role in the development of Th17 cells.
Finally, the in vivo therapeutic efficacy of nt-RORα-TMD on the inflammatory disease was verified using a DSS-induced colitis animal model. In colitis mice treated with nt-RORα-TMD, various symptoms of colitis such as body weight loss, DAI score, and maintenance of colon length and spleen size were substantially improved by nt-RORα-TMD treatment. Furthermore, nt-RORα-TMD treatment substantially reduced the level of inflammatory cytokines in the serum, known to be elevated in IBD patients.38 nt-RORα-TMD also improved the histopathological status of colons, such as decreased level of inflammatory cell infiltration, goblet cell depletion, and crypt damage. Consistently, alleviation of the various colitis symptoms by nt-RORα-TMD treatment was mediated by the reduction of CD4+IL-17A+ and CD4+IFN-γ+ T cells in MLN, which are known to be the leading pathogenic populations in IBD.39 Interestingly, the decrease of these pathogenic T cell populations was accompanied by the increase of CD4+Foxp3+ regulatory T cells. Currently, anti-TNF agents are used for the treatment of IBD.34 However, 20–40% of patients were refractory to anti-TNF therapy.40 Recent studies have reported that IL-17A+ and IFN-γ+ T cells expanded in anti-TNF-resistant Crohn’s disease patients more than responding patients.41,42 Based on our results, nt-RORα-TMD decreased pro-inflammatory cytokines and pathogenic T cells by inhibiting Th17-mediated pathogenesis of IBD accompanied by the increase of Treg cells. Therefore, nt-RORα-TMD has the potential to overcome the current barriers in the treatment of IBD.
In this study, nt-RORα-TMD (R42A/R43G) was used as a negative control in which two amino acids were mutated for blocking the bind to the corresponding promoter region. In most in vitro and in vivo experiments, the functional effects of nt-RORα-TMD (R42A/R43G) were partial with different degrees of functional contribution. It is because mutation of two amino acids in TMD may not completely eliminate the DNA binding capacity of TMD to the promoter region. Alternatively, the remaining subdomain other than the DNA-binding motif in TMD may be a crucial interaction domain for the transcriptional function of RORα. For the in vivo studies, the DSS-induced colitis mice model was used to verify the therapeutic effect of nt-RORα-TMD. Since Th17 cells and Th17 producing cytokines were known as essential mediators in the pathogenesis of IBD, in vitro inhibition of Th17 differentiation and its cytokine expression by nt-RORα-TMD led us to choose the DSS-induced colitis mice model for verifying the therapeutic effect of nt-RORα-TMD. Given that nt-RORα-TMD inhibited the differentiation of naive T cells into Th17 cells in our in vitro study, it was considered appropriate to use the acute IBD model in which naïve T cells are expected to differentiate into pathogenic Th17 cells actively. DSS-induced colitis is a commonly used model to study IBD and exhibits various autoimmune disease features. However, for developing nt-RORα-TMD as a novel therapeutic agent for various inflammatory diseases, it is considered to be further verified the therapeutic effects in chronic disease or other Th17-implicated disease models such as EAE.
Conclusion
In conclusion, we demonstrated that nt-RORα-TMD, the competitive inhibitor of endogenous RORα, can specifically and effectively inhibit the functions of Th17 cells in vitro and in vivo without influencing the differentiation and functions of other T cell subsets. Recent studies reported that the severity of colitis or EAE was attenuated in CD4+ T cell-specific RORα CKO mice.20,36 Therefore, nt-RORα-TMD can be a novel therapeutic agent for treating IBD and has the potential to be developed as a reagent for treating inflammatory diseases in which Th17 cells are dominant pathogenic population.
Acknowledgments
We would like to thank Dr. Jae-Seung Moon from Stanford University School of Medicine for his experimental advice.
Funding
This work was supported by a Global Research Laboratory (GRL) Program of the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (NRF-2016K1A1A2912755) and a Basic Science Research Program of the NRF grant funded by the Ministry of Education (2019R1I1A1A01064332), and the Brain Korea 21 (BK21) PLUS Program, Republic of Korea.
Disclosure
The authors declare that there are no competing financial interests and conflicts of interest in this work.
References
1. Azizi G, Jadidi‐Niaragh F, Mirshafiey A. Th17 cells in immunopathogenesis and treatment of rheumatoid arthritis. Int J Rheum Dis. 2013;16(3):243–253. doi:10.1111/1756-185X.12132
2. Moser T, Akgün K, Proschmann U, Sellner J, Ziemssen T. The role of TH17 cells in multiple sclerosis: therapeutic implications. Autoimmun Rev. 2020;19:102647. doi:10.1016/j.autrev.2020.102647
3. Martin JC, Baeten DL, Josien R. Emerging role of IL-17 and Th17 cells in systemic lupus erythematosus. Clin Immunol. 2014;154(1):1–12. doi:10.1016/j.clim.2014.05.004
4. Zhao J, Lu Q, Liu Y, et al. Th17 Cells in inflammatory bowel disease: cytokines, plasticity, and therapies. J Immunol Res. 2021;2021:1–14. doi:10.1155/2021/8816041
5. Aujla SJ, Dubin PJ, Kolls JK. Th17 cells and mucosal host defense.
6. Singh RP, Hasan S, Sharma S, et al. Th17 cells in inflammation and autoimmunity. Autoimmun Rev. 2014;13(12):1174–1181. doi:10.1016/j.autrev.2014.08.019
7. Zhang Y-Z, Li -Y-Y. Inflammatory bowel disease: pathogenesis. World J Gastroenterol. 2014;20(1):91. doi:10.3748/wjg.v20.i1.91
8. Neurath MF. Cytokines in inflammatory bowel disease. Nat Rev Immunol. 2014;14(5):329–342. doi:10.1038/nri3661
9. Gálvez J. Role of Th17 cells in the pathogenesis of human IBD. Int Scholar Res Notices. 2014;2014:928461.
10. Serra HG, Duvick L, Zu T, et al. RORα-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell. 2006;127(4):697–708. doi:10.1016/j.cell.2006.09.036
11. Kim K, Boo K, Yu YS, et al. RORα controls hepatic lipid homeostasis via negative regulation of PPARγ transcriptional network. Nat Commun. 2017;8(1):1–15. doi:10.1038/s41467-016-0009-6
12. Sun X, Dongol S, Qiu C, et al. miR-652 promotes tumor proliferation and metastasis by targeting RORA in endometrial cancer. Mol Cancer Res. 2018;16(12):1927–1939. doi:10.1158/1541-7786.MCR-18-0267
13. Oh SK, Kim D, Kim K, et al. RORα is crucial for attenuated inflammatory response to maintain intestinal homeostasis. Proc Natl Acad Sci. 2019;116(42):21140–21149. doi:10.1073/pnas.1907595116
14. Yang XO, Pappu BP, Nurieva R, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity. 2008;28(1):29–39. doi:10.1016/j.immuni.2007.11.016
15. Sands BE, Sandborn WJ, Panaccione R, et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2019;381(13):1201–1214. doi:10.1056/NEJMoa1900750
16. Sandborn WJ, Ferrante M, Bhandari BR, et al. Efficacy and safety of mirikizumab in a randomized Phase 2 study of patients with ulcerative colitis. Gastroenterology. 2020;158(3):537–549. e510. doi:10.1053/j.gastro.2019.08.043
17. Feagan BG, Sandborn WJ, D’Haens G, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet. 2017;389(10080):1699–1709. doi:10.1016/S0140-6736(17)30570-6
18. Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61(12):1693–1700. doi:10.1136/gutjnl-2011-301668
19. Martin DA, Churchill M, Flores-Suarez LF, et al. A phase Ib multiple ascending dose study evaluating safety, pharmacokinetics, and early clinical response of brodalumab, a human anti-IL-17R antibody, in methotrexate-resistant rheumatoid arthritis. Arthritis Res Ther. 2013;15(5):R164. doi:10.1186/ar4347
20. Wang R, Campbell S, Amir M, et al. Genetic and pharmacological inhibition of the nuclear receptor RORα regulates TH 17 driven inflammatory disorders. Nat Commun. 2021;12(1):1–18. doi:10.1038/s41467-020-20314-w
21. Son H-J, Lee SH, Lee S-Y, et al. Oncostatin M suppresses activation of IL-17/Th17 via SOCS3 regulation in CD4+ T cells. J Immunol. 2017;198(4):1484–1491. doi:10.4049/jimmunol.1502314
22. Tao Y, Zhang X, Chopra M, et al. The role of endogenous IFN-β in the regulation of Th17 responses in patients with relapsing-remitting multiple sclerosis. J Immunol. 2014;192(12):5610–5617. doi:10.4049/jimmunol.1302580
23. Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal. 2009;7(1):
24. Cook DN, Kang HS, Jetten AM. Retinoic acid-related orphan receptors (RORs): regulatory functions in immunity, development, circadian rhythm, and metabolism. Nuclear Recept Res. 2015;2. doi:10.11131/2015/101185
25. Hams E, Roberts J, Bermingham R, Fallon PG. Functions for retinoic acid-related orphan receptor alpha (RORα) in the activation of macrophages during lipopolysaccharide-induced septic shock. Front Immunol. 2021;12:612. doi:10.3389/fimmu.2021.647329
26. Ferreira AC, Szeto AC, Heycock MW, et al. RORα is a critical checkpoint for T cell and ILC2 commitment in the embryonic thymus. Nat Immunol. 2021;22(2):166–178. doi:10.1038/s41590-020-00833-w
27. Lee J-E, Choi G, Cho M, Kim D, Lee M-O, Chung Y. A critical regulation of Th2 cell responses by RORα in allergic asthma. Sci China Life Sci. 2020;64:1–10.
28. Malhotra N, Leyva-Castillo JM, Jadhav U, et al. RORα-expressing T regulatory cells restrain allergic skin inflammation. Sci Immunol. 2018;3(21). doi:10.1126/sciimmunol.aao6923
29. Capone A, Volpe E. Transcriptional regulators of T helper 17 cell differentiation in health and autoimmune diseases. Front Immunol. 2020;11:348. doi:10.3389/fimmu.2020.00348
30. Castro G, Liu X, Ngo K, et al. RORγt and RORα signature genes in human Th17 cells. PLoS One. 2017;12(8):e0181868. doi:10.1371/journal.pone.0181868
31. Guan Q. A comprehensive review and update on the pathogenesis of inflammatory bowel disease. J Immunol Res. 2019;2019:1–16. doi:10.1155/2019/7247238
32. Rismo R, Olsen T, Cui G, Christiansen I, Florholmen J, Goll R. Mucosal cytokine gene expression profiles as biomarkers of response to infliximab in ulcerative colitis. Scand J Gastroenterol. 2012;47(5):538–547. doi:10.3109/00365521.2012.667146
33. Kobayashi T, Okamoto S, Hisamatsu T, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut. 2008;57(12):1682–1689. doi:10.1136/gut.2007.135053
34. Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol. 2017;14(5):269–278. doi:10.1038/nrgastro.2016.208
35. Liang T, Chen T, Qiu J, et al. Inhibition of nuclear receptor RORα attenuates cartilage damage in osteoarthritis by modulating IL-6/STAT3 pathway. Cell Death Dis. 2021;12(10):1–13. doi:10.1038/s41419-021-04170-0
36. Chi X, Jin W, Bai X, et al. RORα is critical for mTORC1 activity in T cell-mediated colitis. Cell Rep. 2021;36(11):109682. doi:10.1016/j.celrep.2021.109682
37. Haim-Vilmovsky L, Henriksson J, Walker JA, et al. Mapping Rora expression in resting and activated CD4+ T cells. PLoS One. 2021;16(5):e0251233. doi:10.1371/journal.pone.0251233
38. Strober W, Fuss IJ. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140(6):1756–1767. e1751. doi:10.1053/j.gastro.2011.02.016
39. Imam T, Park S, Kaplan MH, Olson MR. Effector T helper cell subsets in inflammatory bowel diseases. Front Immunol. 2018;9:1212. doi:10.3389/fimmu.2018.01212
40. Ben-Horin S, Chowers Y. Tailoring anti-TNF therapy in IBD: drug levels and disease activity. Nat Rev Gastroenterol Hepatol. 2014;11(4):243–255. doi:10.1038/nrgastro.2013.253
41. Martin JC, Chang C, Boschetti G, et al. Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell. 2019;178(6):1493–1508. e1420. doi:10.1016/j.cell.2019.08.008
42. Schmitt H, Billmeier U, Dieterich W, et al. Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease. Gut. 2019;68(5):814–828. doi:10.1136/gutjnl-2017-315671
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.