")
Back to Journals » Cancer Management and Research » Volume 11
Transcription factor Nrf2 induces the up-regulation of lncRNA TUG1 to promote progression and adriamycin resistance in urothelial carcinoma of the bladder
Authors Sun Z, Huang G, Cheng H
Received 9 January 2019
Accepted for publication 27 May 2019
Published 4 July 2019 Volume 2019:11 Pages 6079—6090
DOI https://doi.org/10.2147/CMAR.S200998
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lu-Zhe Sun
Zhulei Sun,1 Gui Huang,1 Hepeng Cheng2
1Department of Pathology, Huaihe Hospital of Henan University, Kaifeng, People’s Republic of China; 2Department of Urology, Huaihe Hospital of Henan University, Kaifeng, People’s Republic of China
Background: Taurine-upregulated gene 1 (TUG1) has been documented to be implicated in carcinogenesis and chemoresistance in solid tumors. Here, we explored the biological role and regulatory mechanism of TUG1 in progression and chemoresistance of urothelial carcinoma of the bladder (UCB).
Methods: Nuclear factor-erythroid 2 (NF-E2)-related factor 2 (Nrf2) mRNA and TUG1 expression was determined by quantitative reverse transcription polymerase chain reaction. Western blot was performed to determine the protein levels of Nrf2, p-glycoprotein (p-gp), Ki-67 (Ki67), matrix metalloproteinase (MMP)-2 and MMP-9 and cleaved caspase-3. The effects of either Nrf2 or TUG1 knockdown on the proliferation, invasion, apoptosis and adriamycin (ADM) resistance of UCB cells were evaluated by CCK-8 assay, transwell invasion assay and flow cytometry analysis. Xenograft tumor assay was carried out to confirm the role of Nrf2 and TUG1 in ADM resistance of UCB cells in vivo.
Results: Nrf2 and TUG1 were upregulated in UCB tissues and cell lines. A positive correlation between Nrf2 and TUG1 expression was discovered in UCB tissues. Moreover, Nrf2 and TUG1 expression levels were higher in ADM-resistant cells compared with those in parental cells. Furthermore, Nrf2 positively regulated the expression of TUG1 in UCB cells. Knockdown of either Nrf2 or TUG1 led to the inhibition of cell proliferation and invasion and promotion of cell apoptosis, accompanying with down-regulation of Ki67, MMP-2 and MMP-9 and up-regulation of cleaved caspase-3. Knockdown of either Nrf2 or TUG1 enhanced the sensitivity of BIU-87/ADM and T24/ADM cells to ADM, as indicated by decreased expression of p-gp. Besides, knockdown of either Nrf2 or TUG1 inhibited tumor growth in the absence or presence of ADM in vivo.
Conclusions: Nrf2 induces the up-regulation of TUG1 to promote progression and ADM resistance in UCB.
Keywords: urothelial carcinoma of the bladder, nuclear factor-erythroid 2 (NF-E2)-related factor 2, taurine-upregulated gene 1, adriamycin
Introduction
Urothelial carcinoma of the bladder (UCB) is a malignancy arising from the tissues of the urinary bladder. Metastasis and recurrence are regarded as the main obstacles in the treatment of bladder cancer. Clinically, adriamycin (ADM)-based chemotherapy is an important accessory treatment for bladder cancer. ADM is capable of inhibiting the synthesis of cellular DNA and RNA, which results in cancer cell death.1 Although most patients achieve initial induction remission with this treatment, the 5-year survival rate of bladder cancer patients is still disappointing due to the occurrence of therapeutic drug resistance.2 Hence, understanding the molecular mechanism that underlies chemoresistance can help to develop an effective therapy for UCB.
Nuclear factor-erythroid 2 (NF-E2)-related factor 2 (Nrf2) is a basic leucine zipper transcription factor that plays a vital role in the cellular responses to oxidative stress.3 Nrf2 is capable of regulating cellular redox homeostasis by binding to the antioxidant response elements (AREs) in its target gene promoter. Under normal conditions, Kelch-like ECH-associated protein 1 (keap1), as a major inhibitor of Nrf2, interacts with Nrf2 and anchors Nrf2 in the cytoplasm. Keap1 forms a complex with cullin3, which facilitates the ubiquitination and subsequent proteolysis of Nrf2. Under oxidative stress, Nrf2 dissociates from keap1 and travels into the nucleus where it activates the transcription of ARE-driven genes.4 Nrf2 is a transcription factor master regulator of many diverse cellular mediators and it can reduce sensitivity of cancer cells to chemotherapeutic drugs.5 There is now growing evidence to suggest that Nrf2 hyperactivation contributes to the development of tumors and chemoresistance by regulating its targets, such as ATP-binding cassette, subfamily G, member 2 and ATP-binding cassette sub-family F member 2.6,7 Down-regulation of Nrf2 reduced clonogenicity of acute myeloid leukemia cells and enhanced their chemotherapeutic responsiveness.8 Nrf2 modulated the sensitivity of cancer cells towards platinum, including cervical cancer cell line ME180R, ovarian cancer cell line SKOV3 and lung adenocarcinoma cell line A549 cells.9 These recent data suggest that Nrf2 is an vital mediator in the mechanism of chemotherapeutic drug resistance in cancer cells. However, whether Nrf2 is involved in the ADM resistance in UCB and its underlying mechanism remain elusive.
Long non-coding RNAs (lncRNAs) are actively being investigated for their potential roles in human cancers. Emerging evidence suggests that lncRNAs serve as major regulators in tumorigenesis. Aberrant expression of lncRNAs has been reported to confer tumor growth, cancer cell metastasis, apoptosis and chemoresistance.10–12 As an example, lnc-LBCS functioned as a tumor suppressor in bladder cancer stem cells (BCSCs), which was tightly correlated with tumor grade, chemotherapy response and prognosis. Furthermore, lnc-LBCS inhibited tumorigenesis and enhanced chemosensitivity through inhibiting enhancer of zeste homolog 2/SRY (sex determining region Y)-box 2 axis in BCSCs.13 Taurine-upregulated gene 1 (TUG1), located at chromosome 22q12, was identified as an oncogene in tumorigenesis and was responsible for chemoresistance.14,15 Previous studies in UCB identified TUG1 associated with UCB progression. High expression of TUG1 has been documented to be correlated with enhanced UCB cell proliferation and matastasis.16 Additionally, ADM-resistant acute myeloid leukemia tissues and HL60/ADR cells have been shown to express high levels of TUG1, and its knockdown facilitated the sensitivity of HL60/ADR cells to ADM by epigenetically promoting miR-34a expression.17 A previous paper reported that TUG1 was responsible for the ADM resistance of bladder urothelial carcinoma.18 However, the upstream regulatory mechanism of TUG1-mediated progression and ADM resistance in UCB remains unknown. As the key transcription factor, Nrf2 has been demonstrated to control lncRNA expression in erythroid cells and mammary stem cells.19,20 Therefore, we speculated that Nrf2-mediated up-regulation of lncRNA TUG1 was crucial to the progression and ADM resistance in urothelial carcinoma of the bladder.
In this study, we hypothesized that aberrant expression of Nrf2 and TUG1 in UCB might drive a mechanism for ADM resistance, and found that Nrf2 induced the up-regulation of TUG1 to promote progression and ADM resistance in UCB.
Materials and methods
Patient samples
We obtained UCB tissues and paired normal tissues from 27 patients with histopathologically diagnosed UCB in Huaihe Hospital of Henan University. Clinicopathological features of patients with UCB were showed in Table 1. All participates did not accept any adjuvant therapy prior to surgery. This study was reviewed and approved by the Ethics Committee of Huaihe Hospital of Henan University, and all written informed consents were obtained.
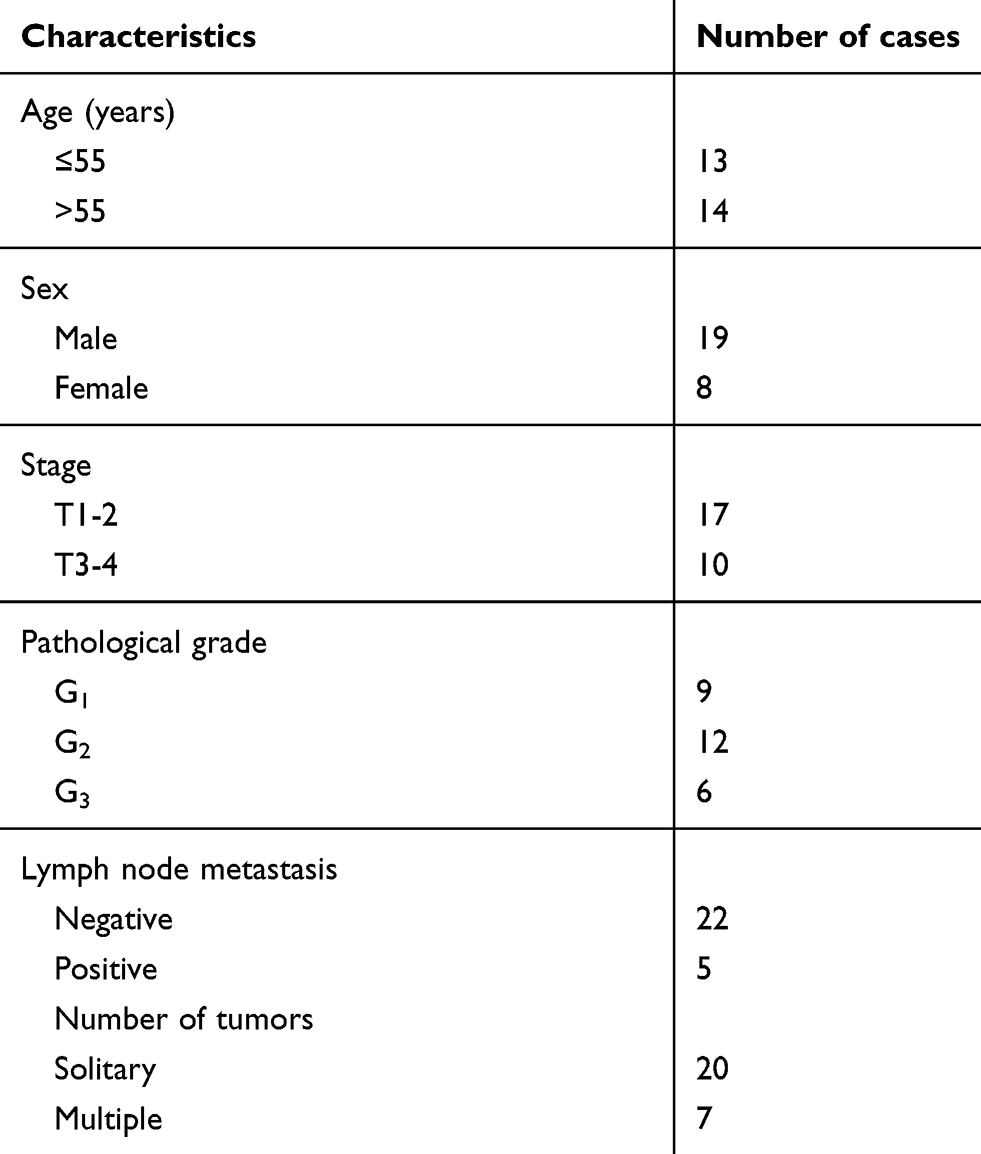 |
Table 1 Clinicopathological features of patients with urothelial carcinoma of the bladder (UCB) |
Cell culture
Human UCB cell lines (EJ-1, 5637 and T24) and normal human urothelial cells (SV-HUC-1) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Human UCB cell line BIU-87 was purchased from the Chinese Academy of Sciences Cell Bank (Shanghai, China). BIU-87/ADM and T24/ADM cells were established by stepwise exposure of BIU-87 and T24 cells to increasing concentrations of ADM (0.1, 0.2, 0.4, 0.6, 0.8, 1.0 μg/ml). Each round screened the surviving cells for the beginning of the next drug resistance concentration, until the cells surviving in 1 μg/ml were BIU-87/ADM and T24/ADM. Cells were incubated in RPMI-1640 medium (Solarbio, Beijing, China) supplemented with 10% fetal bovine serum (FBS; Solarbio), streptomycin (100 mg/ml; Solarbio) and penicillin (100 units/ml; Solarbio) and maintained in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
To knockdown Nrf2, BIU-87 and T24 cells were transfected with small interfering RNA (siRNA) specific for Nrf2 (si-Nrf2) or treated with ML385 (2 μM, a specific Nrf2 inhibitor). Similarly, si-TUG1 was used to knockdown TUG1. After 48 h of incubation, cells were collected to analyze the expression of Nrf2 and TUG1.
Cell transfection
The coding sequence of Nrf2 was amplified and subcloned into pcDNA3.1 to generate pcDNA-Nrf2. si-Nrf2, si-TUG1, sh-Nrf2, sh-TUG1 and matched controls were synthesized by Genechem (Shanghai, China). Cell transfection was performed using Lipofectamine 2,000 (Invitrogen, Carlsbad, CA, USA), in accordance with the manufacturer’s specifications.
Transwell invasion assay
After treatment, BIU-87 and T24 cells in serum-free medium were seeded in the upper part of the transwell chamber (Corning, Steuben County, New York, USA), which was precoated with Matrigel (Franklin Lakes, NJ, USA). Directional invasion was induced through addition of 10% FBS-containing RPMI-1640 medium to the lower part of the transwell chamber and cells were allowed to invade for 24 h in a 5% CO2 incubator at 37°C. Remaining cells on the inner side were gently removed with a cotton swab and cells adherent to the outer side were fixed with 4% paraformaldehyde (Solarbio), followed by staining with 0.1% crystal violet (Solarbio) for 15 min. The number of invaded cells was counted in six random fields under a light microscope.
Detection of cell proliferation capacity
Cells were seeded in a 96-well plate at a density of 1×105 cells per well and transfected with si-Nrf2, si-TUG1 or siRNA, followed by incubation with different concentrations (0, 5, 10, 20 and 40 μg/ml) of ADM. CCK-8 reagent (10 μl; Beyotime, Shanghai, China) was added into each well. After 2 h of incubation, the absorbance value (OD value) at 450 nm was detected using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA).
Flow cytometry
The apoptosis of BIU-87 and T24 cells was evaluated using the Annexin V-APC/7-AAD Apoptosis kit (MultiSciences, Shanghai, China), following the manufacturer’s direction. Briefly, BIU-87 and T24 cells were collected, washed with PBS, and resuspended in 1× binding buffer after transfection. Thereafter, cells were incubated with Annexin V-FITC and PI for 15 min at 37°C in darkness. After addition of 1 × binding buffer, flow cytometry was utilized to evaluate the apoptosis of CAL-27 and TSCCA cells by measuring the mean fluorescent intensity.
Western blot analysis
Total protein was prepared from UCB tissues and cells using RIPA buffer, and protein quantification was conducted by a spectrophotometer (Thermo Fisher Scientific). Protein extracts were subjected to 14% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer to polyvinylidene fluoride membranes (Millipore, Bradford, MA, USA). After blocking with 5% nonfat milk, membranes were probed with primary antibodies at 4°C overnight, followed by incubation with horseradish peroxidase-conjugated secondary antibody (Boster, Wuhan, China) for 1 h at room temperature. Primary antibodies were as follows: anti-Nrf2 (R&D Systems, Minneapolis, MN, USA), anti-Ki-67 (Boster), anti-matrix metalloproteinase (MMP)-2 (R&D Systems), anti-MMP-9 (R&D Systems), anti-cleaved caspase 3 (R&D Systems), anti-p-glycoprotein (p-gp; Abcam, Cambridge, MA, USA) and anti-β-actin (Boster). Immunoblots were developed by ECL reagents (Pierce, Rockford, IL, USA) and quantified by the Image J software (National Institutes of Health, NY, USA).
Quantitative reverse transcription polymerase chain reaction
RNA isolation was performed using TRIzol reagent (Life Technologies, Carlsbad, CA) and the cDNA was prepared from 1 μg of RNA using PrimeScript™ RT reagent Kit (Takara, Dalian, China). Quantitative reverse transcription polymerase chain reaction (RT-PCR) was carried out to analyze the expression of TUG1, Nrf2 and MDR1 using SYBR® Premix Ex Taq kit (Takara) in the ABI prism 7900 sequence detection system (Life Technologies). Relative expression levels of TUG1, Nrf2 and MDR1 were normalized to β-actin using the 2−ΔΔCt method. Primer sequences were listed as follows: TUG1 forward, 5ʹ-TAG CAG TTC CCC AAT CCT TG-3ʹ and reverse, 5ʹ-CAC AAA TTC CCA TCA TTC CC-3ʹ; Nrf2 forward, 5ʹ-ACA CGG TCC ACA GCT CAT C-3ʹ and reverse, 5ʹ-TGT CAA TCA AAT CCA TGT CCT G-3ʹ; MDR1 forward, 5ʹ-GCT GTC AAG GAA GCC AAT GCC T-3ʹ and reverse, 5ʹ-TGC AAT GGC GAT CCT CTG CTT C-3ʹ; β-actin forward, 5ʹ-TCC CTG GAG AAG AGC TAC GA-3ʹ and reverse, 5ʹ-AGC ACT GTG TTG GCG TAC AG-3ʹ.
Xenograft tumor assay
Animal protocols were in strict accordance with the guiding principles of institutional animal ethics committee. All animal experiments were reviewed and approved by the Experimental Animal Ethical Committee of the Huaihe Hospital of Henan University. Four- to six-week-old male Balb/c-nude mice were purchased from Slac Laboratory (Shanghai, China). T24/ADM cells stably expressing sh-Nrf2, sh-TUG1 or sh-NC (negative control) were subcutaneously injected into the flank of nude mice. Subsequently, mice were intraperitoneally injected with ADM or saline. At the 32th day after inoculation, all mice were anaesthetized and decapitated, and the tumor masses were resected, pictured and weighed. Tumor diameters were measured every 4 days with calipers, and tumor volume was calculated by the following formula: volume =0.5 × length × width2.
Statistical analysis
Data were given as the mean ± standard deviation of the mean (SD) from 3 independent experiments. Statistical analysis was done using SPSS 20.0 software and the significance of differences between relevant data sets was analyzed with student’s t test or one-way analysis of variance. A probability value of P<0.05 was designated as the level of significance.
Results
Increased expression of Nrf2 and TUG1 in UCB tissues
We first evaluated the expression of Nrf2 and TUG1 in UCB tissues and then, in particular, investigated the correlation between Nrf2 and TUG1 expression in UCB tissues. RT-PCR analysis showed that the expression levels of Nrf2 and TUG1 were markedly higher in UCB tissues than those in paired normal tissues (Figure 1A and B). In parallel, a positive correlation between Nrf2 and TUG1 expression was discovered in UCB tissues (Figure 1C).
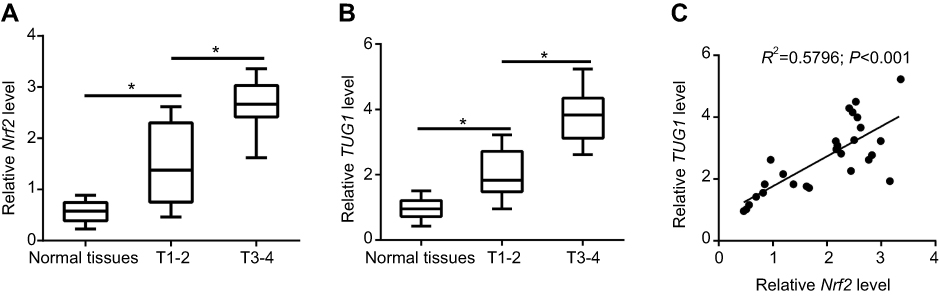 |
Figure 1 Increased expression of Nrf2 and TUG1 in urothelial carcinoma of the bladder (UCB) tissues. (A and B) 27 pairs of UCB tissues and paired normal tissues were collected and analyzed for the expression of Nrf2 and TUG1 by RT-PCR. (C) Correlation analysis of Nrf2 and TUG1 expression in UCB tissues. *P<0.05. |
Up-regulation of Nrf2 and TUG1 in ADM-resistant cells
We validated the differential expression of Nrf2 and TUG1 in UCB cell lines (EJ-1, 5637, BIU-87 and T24) by RT-PCR and Western blot. As a result, the expression of Nrf2 was markedly upregulated in UCB cell lines, especially in BIU-87 and T24 cells, as compared to normal human urothelial cells (SV-HUC-1) (Figure 2A and B). To understand the role of Nrf2 and TUG1 in chemoresistance, we compared the expression levels of Nrf2 and TUG1 in UCB cells (BIU-87 and T24) and the ADM-resistant UCB cells (BIU-87/ADM and T24/ADM). The results of Western blot demonstrated that the expression of Nrf2 was remarkably increased in BIU-87/ADM and T24/ADM cells as compared to BIU-87 and T24 cells (Figure 2C). Similarly, the expression of TUG1 was increased in UCB cell lines, especially in BIU-87 and T24 cells, compared with SV-HUC-1 cells (Figure 2D). Meanwhile, the expression of TUG1 was higher in BIU-87/ADM and T24/ADM cells than that in BIU-87 and T24 cells, as evidenced by RT-PCR (Figure 2E).
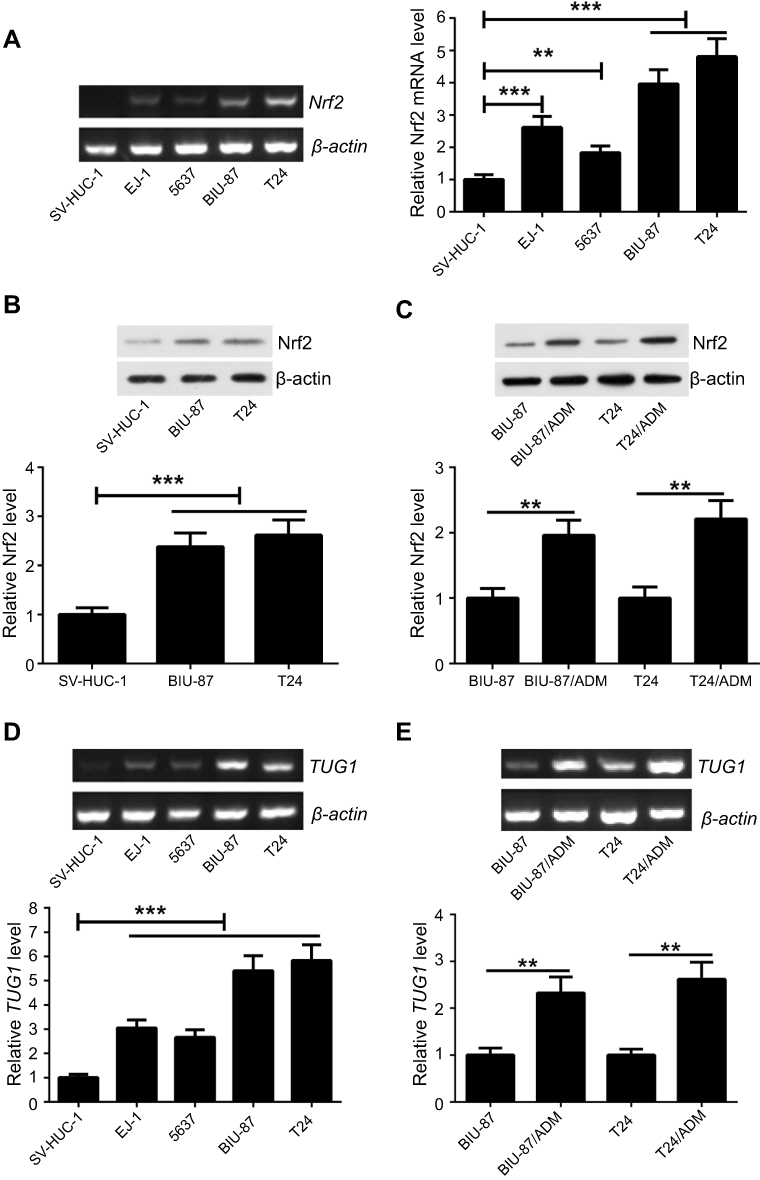 |
Figure 2 Up-regulation of Nrf2 and TUG1 in ADM-resistant cells. (A and B) Nrf2 expression was measured in UCB cell lines (EJ-1, 5637, BIU-87 and T24) and normal human urothelial cells (SV-HUC-1) by RT-PCR and Western blot. (C) Western blot analysis of Nrf2 expression showed increased expression of Nrf2 in ADM-resistant cells (BIU-87/ADM and T24/ADM). (D) TUG1 expression was measured in UCB cell lines (EJ-1, 5637, BIU-87 and T24) and SV-HUC-1 cells by RT-PCR. (E) RT-PCR analysis of TUG1 expression indicated increased expression of TUG1 in BIU-87/ADM and T24/ADM cells. **P<0.01 and ***P<0.001. |
Nrf2 positively regulates the expression of TUG1 in UCB cells
To understand the basis for higher Nrf2 and TUG1 expression in UCB cells, we investigated the relationship between Nrf2 and TUG1 expression in BIU-87 and T24 cells. Nrf2 expression was restored by transfecting BIU-87 and T24 cells with pcDNA-Nrf2, while Nrf2 level was knockdown in BIU-87 and T24 cells using si-Nrf2 or ML385 (Figure 3A and B). Overexpression of Nrf2 obviously increased the expression of TUG1 in BIU-87 and T24 cells (Figure 3C and D). Conversely, knockdown of Nrf2 by siRNA caused a marked decrease in TUG1 expression in BIU-87 and T24 cells (Figure 3E and F). Similarly, treatment of BIU-87 and T24 cells with ML385 resulted in a dose-dependent reduction in TUG1 expression (Figure 3G and H).
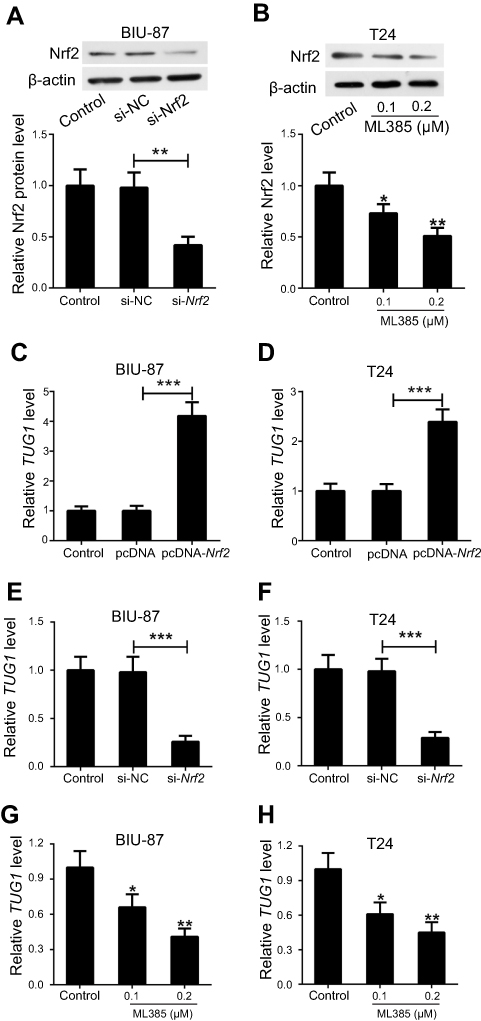 |
Figure 3 Nrf2 positively regulates the expression of TUG1 in UCB cells. (A) BIU-87 and T24 cells were transfected with pcDNA-Nrf2, si-Nrf2 or matched controls, and the transfection efficiency was identified by Western blot. (B) BIU-87 and T24 cells were treated with different doses (0.1 and 0.2 μM) of ML385, and Nrf2 levels were determined by Western blot. (C and D) RT-PCR analysis of TUG1 expression indicated increased expression of TUG1 in BIU-87 and T24 cells transfected with pcDNA-Nrf2. (E and F) RT-PCR analysis of TUG1 expression indicated decreased expression of TUG1 in BIU-87 and T24 cells transfected with si-Nrf2. (G and H) RT-PCR analysis of TUG1 expression showed down-regulation of TUG1 in BIU-87 and T24 cells treated with ML385. *P<0.05, **P<0.01 and ***P<0.001. |
Knockdown of either Nrf2 or TUG1 inhibits the progression of UCB in vitro
Since Nrf2 and TUG1 were upregulated in UCB, we knockdown Nrf2 and TUG1 to evaluate their functional roles in UCB cell proliferation, invasion and apoptosis. si-Nrf2, si-TUG1 or si-NC was transfected into BIU-87 and T24 cells, respectively. CCK-8 assay showed that the viability of BIU-87 and T24 cells was strikingly reduced in Nrf2-silenced cells and TUG1-silenced cells as compared to control cells (Figure 4A). Moreover, decreased levels of Ki-67 expression were noticed in Nrf2-silenced cells and TUG1-silenced cells (Figure 4B and C). Meanwhile, knockdown of Nrf2 remarkably inhibited the invasion of BIU-87 and T24 cells and decreased the expression of MMP-2 and MMP-9 in BIU-87 and T24 cells. Intriguingly, TUG1 knockdown led to similar functional effects as those of Nrf2 knockdown in BIU-87 and T24 cells (Figure 4D–G). In parallel, Nrf2 knockdown conspicuously promoted the apoptosis of BIU-87 and T24 cells. Also, TUG1 knockdown caused an increased rate of apoptotic cells in BIU-87 and T24 cells (Figure 4H and I). Furthermore, a pronounced elevation in cleaved caspase-3 expression was found in Nrf2-silenced cells and TUG1-silenced cells as seen by Western blot (Figure 4J and K).
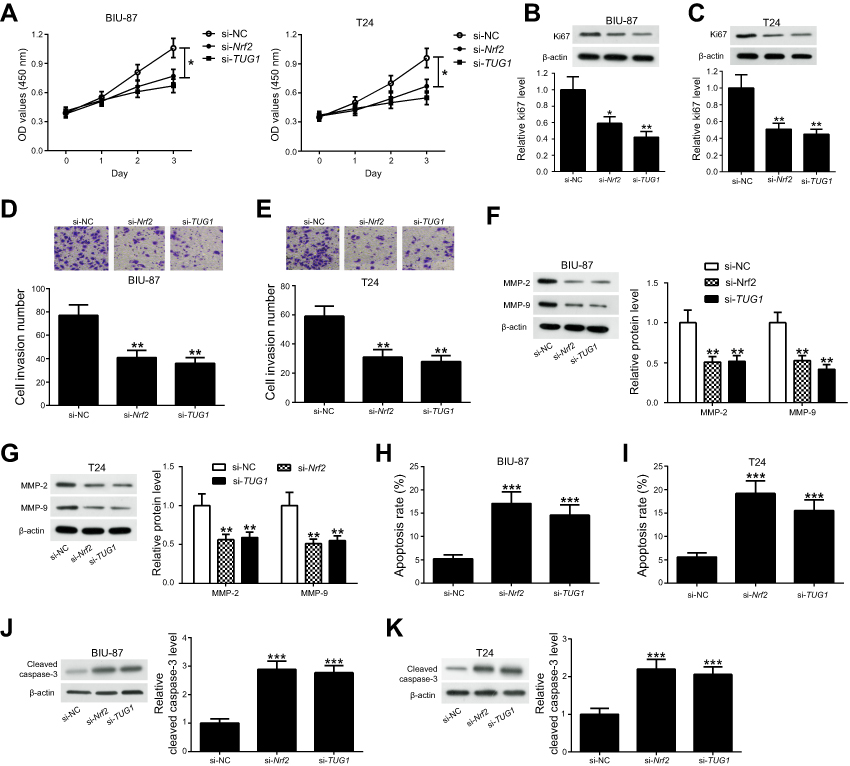 |
Figure 4 Knockdown of either Nrf2 or TUG1 inhibits the progression of UCB in vitro. BIU-87 and T24 cells were transfected with si-Nrf2, si-TUG1 or si-NC. (A) Cell viability was analyzed at the indicated time-points (0, 1, 2, 3 days after transfection) by CCK-8 assay. (B and C) Ki-67 expression was measured 48 h post-transfection by Western blot. (D and E) The invasion ability of BIU-87 and T24 cells was determined 48 h post-transfection by transwell invasion assay. (F and G) The protein levels of MMP-2 and MMP-9 were examined by Western blot. (H and I) Cell apoptosis was analyzed 48 h post-transfection by flow cytometry. (J and K) Cleaved caspase-3 levels were evaluated in BIU-87 and T24 cells transfected with si-Nrf2 or si-TUG1 by Western blot.*P<0.05, **P<0.01 and ***P<0.001. |
Knockdown of either Nrf2 or TUG1 enhances the chemosensitivity of ADM-resistant UCB cells to ADM
Previous studies documented that multidrug resistance 1 gene (MDR1) was implicated in the chemoresistance mechanisms of bladder cancer.21 Here, we detected the expression of MDR1 mRNA and its encoded protein P-glycoprotein (p-gp). Identical conclusions were obtained in our study, the expression levels of p-gp protein and MDR1 mRNA were higher in BIU-87/ADM and T24/ADM cells as compared to BIU-87 and T24 cells (Figure 5A and B).To examine whether knockdown either Nrf2 or TUG1 restores the sensitivity of BIU-87/ADM and T24/ADM cells to ADM, BIU-87/ADM and T24/ADM cells were transfected with si-Nrf2, si-TUG1 or si-NC, followed by stimulation with increasing doses (0, 5, 10, 20 and 40 μg/ml) of ADM. The results of CCK-8 assay revealed that down-regulation of Nrf2 markedly inhibited the viability of BIU-87/ADM and T24/ADM cells in the presence of ADM. Also, the reduced viability of BIU-87/ADM and T24/ADM cells was observed in the si-TUG1 group in comparison with the si-NC group in the presence of ADM (Figure 5C). In addition, Blocking either Nrf2 or TUG1 obviously downregulated the expression of p-gp in BIU-87/ADM and T24/ADM cells (Figure 5D and E).
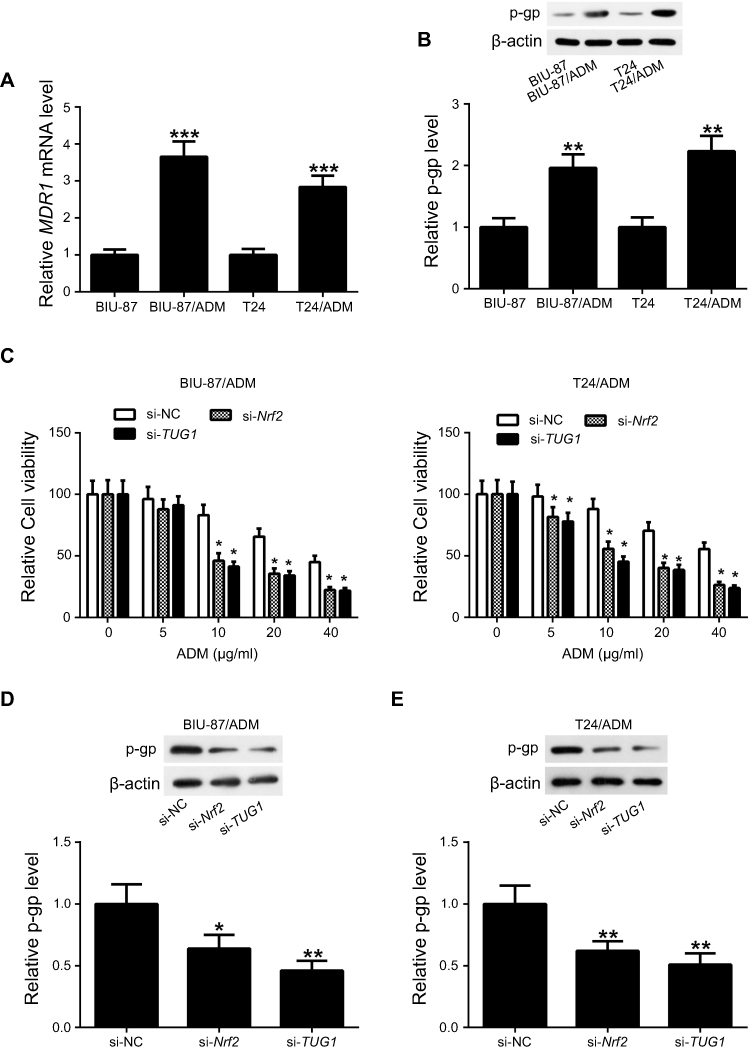 |
Figure 5 Knockdown of either Nrf2 or TUG1 enhances the chemosensitivity of ADM-resistant UCB cells to ADM. (A) RT-PCR analysis of MDR1 mRNA expression indicated up-regulation of MDR1 in BIU-87/ADM and T24/ADM cells. (B) Western blot analysis of p-gp expression showed elevated expression of p-gp in BIU-87/ADM and T24/ADM cells. (C) BIU-87/ADM and T24/ADM cells were transfected with si-Nrf2, si-TUG1 or si-NC, followed by exposure to indicated doses (0, 5, 10, 20 and 40 μg/ml) of ADM. Cell viability was determined using CCK-8 assay. (D and E) BIU-87/ADM and T24/ADM cells were transfected with si-Nrf2, si-TUG1 or si-NC. At 48 h post transfection, the expression of p-pg was evaluated by Western blot. *P<0.05, **P<0.01 and ***P<0.001. |
Knockdown of either Nrf2 or TUG1 enhances the sensitivity of UCB cells to ADM in vivo
Given the significance of Nrf2 and TUG1 in vitro, we further characterized whether knockdown either Nrf2 or TUG1 enhances the sensitivity of UCB cells to ADM in vivo. T24/ADM cells stably expressing sh-Nrf2, sh-TUG1 or sh-NC were subcutaneously injected into nude mice. Subsequently, mice were intraperitoneally injected with ADM or saline. The results revealed that xenograft tumors from sh-Nrf2 or sh-TUG1 transfected T24/ADM cells grew slower than the tumors from sh-NC-transfected T24/ADM cells. Moreover, the tumor growth was slower in tumors from sh-Nrf2 or sh-TUG1 transfected T24/ADM cells than the tumors from sh-NC-transfected T24/ADM cells in the presence of ADM (Figure 6A). The tumor weight was prominently lighter in the sh-Nrf2 group and the sh-TUG1 group than that in the sh-NC group under ADM administration (Figure 6B). The expression level of Nrf2 protein in the sh-Nrf2 group was lower than that in the sh-NC group (Figure 6C).The expression levels of TUG1, MDR1 mRNA and p-gp protein were obviously decreased in tumors from sh-Nrf2 or sh-TUG1 transfected T24/ADM cells as compared to the tumors from sh-NC-transfected T24/ADM cells (Figure 6D–F).
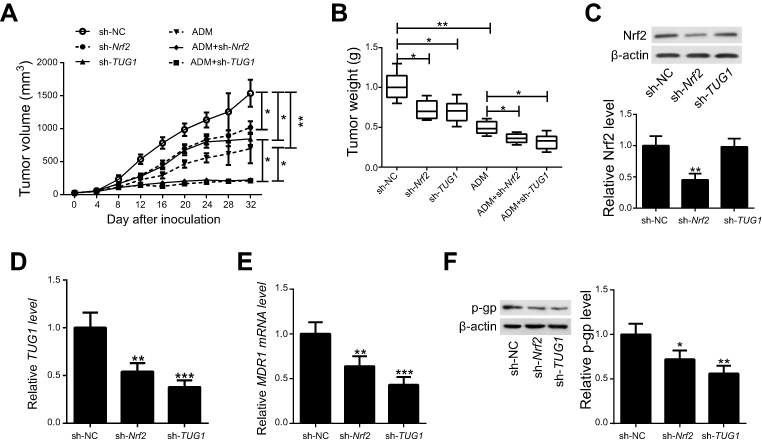 |
Figure 6 Knockdown of either Nrf2 or TUG1 enhances the sensitivity of UCB cells to ADM in vivo. T24/ADM cells stably expressing sh-Nrf2, sh-TUG1 or sh-NC were subcutaneously injected into nude mice. Subsequently, mice were intraperitoneally injected with ADM or saline. (A) Tumor growth curves of T24/ADM cells transfected with sh-Nrf2, sh-TUG1 or sh-NC and treated with ADM or saline in vivo. (B) The mean tumor weight of the six groups. (C) Western blot analysis was performed to examine expression of Nrf2 protein in xenografted tumors. (D and E) RT-PCR analysis of TUG1 and MDR1 mRNA expression in xenografted tumors. (F) Western blot analysis of p-gp protein expression in xenografted tumors. *P<0.05, **P<0.01 and ***P<0.001. |
Discussion
Prior studies have noted the significance of Nrf2. It participates in regulating cellular redox homeostasis, thus exerting as a vital player in chemoresistance. Nrf2 has been considered as a potential therapeutic target for chemoresistance.22 For instance, inhibition of Nrf2 has been postulated to enhance the chemosensitivity of THP-1 cells to proteasome inhibitors.23 In addition, cisplatin-resistant RT112 cells have been shown to express high levels of Nrf2, and its knockdown partially restored the chemosensitivity to cisplatin.24 Studies in 253J tumor cell lines panel suggested that Nrf2 was upregulated in cisplatin resistant tumor cells, and down-regulation of Nrf2 enhanced the chemosensitivity to cisplatin and reduced the migration of 253J cells.25 In vitro and in vivo experiments revealed that knockdown of Nrf2 increased the sensitivity of human lung cancer A549 cells to cisplatin, vinorelbine and carboplatin, as well as inhibited the growth of xenograft tumor, suggesting that overexpression of Nrf2 is a central contributor in the development of chemoresistance.26 However, there are limit studies suggesting the biological role of Nrf2 in mediating ADM resistance in UCB. Our study identified up-regulation of Nrf2 in ADM resistant UCB cells, and found that ADM resistant UCB cells were resensitized upon knockdown of Nrf2, fitting the established notion of Nrf2 as a key regulator in the development of chemoresistance and as a promising target to restore chemosensitivity.
The significance of TUG1 in chemoresistance has been widely studied. TUG1 has been shown to serve as an important role in drug resistance, whereas, its role might vary in different cancers. Tang et al have been identified the down-regulation of TUG1 in triple negative breast cancer. They showed that overexpression of TUG1 markedly augmented the sensitivity of MDA-MB-231 and BT549 cells to cisplatin via miR-197/nemo-like kinase axis through inhibiting WNT signaling.27 Conversely, a recent report investigated the role of TUG1 in osteosarcoma, and found that TUG1 was overexpressed in osteosarcoma. Moreover, TUG1 knockdown suppressed glucose consumption, lactate production and cell viability in osteosarcoma cells through up-regulation of hexokinase-2.28 Also, TUG1 was overexpressed in small cell lung cancer (SCLC). Knockdown of TUG1 impaired cell proliferation, migration and invasion, promoted cell apoptosis and cell cycle arrest, and enhanced SCLC cell sensitivity to anti-cancer drugs by regulating LIM domain kinase 2b via enhancer of zeste homolog 2.29 In addition, up-regulation of TUG1 was also discovered in pancreatic cancer tissue and cells. functional studies in pancreatic ductal adenocarcinoma cells showed that up-regulation of TUG1 promoted cell viability, migration and invasion, suppressed cell apoptosis, as well as reduced the gemcitabine chemosensitivity.30 However, the role and mechanism of TUG1 in chemoresistance in UCB remain to be completely elucidated. In this study, our results demonstrated that TUG1 functioned as an important tumor promoting factor in UCB growth and chemoresistance, contributing to promote tumorigenesis and enhance chemoresistance. A positive correlation between Nrf2 and TUG1 expression was discovered in UCB tissues, more importantly, Nrf2 positively regulated the expression of TUG1 in UCB cells, indicating the functional interaction between Nrf2 and TUG1 in UCB tumorigenesis and ADM resistance. Therapeutic drug resistance is regarded as a dominant hindrance toward curative cancer treatment.31 The occurrence of chemoresistance is considered the result of multiple factors, including altered expression of drug influx and efflux transporters, alterations in drug targets and increased antioxidant defense systems.32 p-gp, also known as ATP-binding cassette sub-family B member 1 (ABCB1), belongs to the superfamily of ATP-binding cassette (ABC) transporters and is an ATP-dependent drug efflux pump that leads to reduced intracellular drug accumulation in drug-resistant cells.33 Notably, high expression of p-gp encoded by MDR1 is mainly responsible for multidrug resistance.34 As an example, up-regulation of ABCB1 was shown to contribute to the development of nab-paclitaxel resistance, and suppression of ABCB1 by cabozantinib and crizotinib sensitized ABCB1-overexpressing urothelial bladder cancer cells to nab-paclitaxel, suggesting that targeting MDR1 appears to be an effective approach for overcoming therapeutic drug resistance.35 Nrf2 has been shown to be an important inducer of p-gp upregulation.36 Nrf2-dependent upregulation of xCT modulates the sensitivity of T24 cells to proteasome inhibition.37 TUG1 depletion repressed cell proliferation and promoted cell apoptosis in BIU87 cells under radiation.38 But more importantly, whether p-gp is involved in Nrf2 and TUG1-mediated chemoresistance in UCB remains unclear. In our study, up-regulation of p-gp and MDR was identified by us in ADM resistant UCB cells, and blocking either Nrf2 or TUG1 could downregulated the expression of p-gp, raising the possibility that targeting Nrf2 or TUG1 may be an effective approach for overcoming ADM resistance in UCB.
Conclusion
In summary, our results demonstrated that Nrf2 and TUG1 were upregulated in UCB tissues and cells, as well as ADM-resistant UCB cells. Functionally, Nrf2 induces the up-regulation of lncRNA TUG1 to promote progression and ADM resistance in UCB. This study indicates that Nrf2-mediated TUG1 acts as a key player in the development of ADM resistance in UCB and may constitute an ideal target to combat ADM resistance in UCB.
Ethics
The study was reviewed and approved by the Ethics Committee of Huaihe Hospital of Henan University. All patients gave their written informed consent, in compliance with the principles of Declaration of Helsinki.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chen C, Lu L, Yan S, et al. Autophagy and doxorubicin resistance in cancer. Anticancer Drugs. 2018;29(1):1–9. doi:10.1097/CAD.0000000000000572
2. Smolensky D, Rathore K, Cekanova M. Phosphatidylinositol- 3-kinase inhibitor induces chemosensitivity to a novel derivative of doxorubicin, AD198 chemotherapy in human bladder cancer cells in vitro. BMC Cancer. 2015;15:927. doi:10.1186/s12885-015-1584-3
3. Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. doi:10.1056/NEJMoa1114287
4. Tseng CY, Wang JS, Chao MW. Causation by diesel exhaust particles of endothelial dysfunctions in cytotoxicity, pro-inflammation, permeability, and apoptosis induced by ROS generation. Cardiovasc Toxicol. 2016;17(4):384–392.
5. Rushworth SA, Macewan DJ. The role of nrf2 and cytoprotection in regulating chemotherapy resistance of human leukemia cells. Cancers. 2011;3(2):1605–1621. doi:10.3390/cancers3021605
6. Singh A, Wu H, Zhang P, Happel C, Ma J, Biswal S. Expression of ABCG2 (BCRP) is regulated by Nrf2 in cancer cells that confers side population and chemoresistance phenotype. Mol Cancer Ther. 2010;9(8):2365–2376. doi:10.1158/1535-7163.MCT-10-0338
7. Bao L, Wu J, Dodson M, et al. ABCF2, an Nrf2 target gene, contributes to cisplatin resistance in ovarian cancer cells. Mol Carcinog. 2017;56(6):1543–1553. doi:10.1002/mc.22615
8. S A R, Zaitseva L, M Y M, et al. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-κB and underlies its chemo-resistance. Blood. 2012;120(26):5188–5198. doi:10.1182/blood-2012-04-422121
9. Chen J, Solomides C, Simpkins F, Simpkins H. The role of Nrf2 and ATF2 in resistance to platinum-based chemotherapy. Cancer Chemother Pharmacol. 2017;79(2):369–380. doi:10.1007/s00280-016-3225-1
10. Chakravarty D, Sboner A, Nair SS, et al. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat Commun. 2014;5:5383. doi:10.1038/ncomms5972
11. Huang MD, Chen WM, Qi FZ, et al. Long non-coding RNA ANRIL is upregulated in hepatocellular carcinoma and regulates cell apoptosis by epigenetic silencing of KLF2. J Hematol Oncol. 2015;8:50. doi:10.1186/s13045-015-0153-1
12. Li Z, Zhao X, Zhou Y, et al. The long non-coding RNA HOTTIP promotes progression and gemcitabine resistance by regulating HOXA13 in pancreatic cancer. J Transl Med. 2015;13:84. doi:10.1186/s12967-015-0541-x
13. Chen X, Xie R, Gu P, et al. Long noncoding RNA LBCS inhibits self-renewal and chemoresistance of bladder cancer stem cells through epigenetic silencing of SOX2. Clin Cancer Res. 2018;25(4):1389–1403.
14. Katsushima K, Natsume A, Ohka F, et al. Targeting the Notch-regulated non-coding RNA TUG1 for glioma treatment. Nat Commun. 2016;7:13616. doi:10.1038/ncomms13616
15. Xu C, Guo Y, Liu H, Chen G, Yan Y, Liu T. TUG1 confers cisplatin resistance in esophageal squamous cell carcinoma by epigenetically suppressing PDCD4 expression via EZH2. Cell Biosci. 2018;8:61. doi:10.1186/s13578-018-0260-0
16. Han Y, Liu Y, Gui Y, Cai Z. Long intergenic non-coding RNA TUG1 is overexpressed in urothelial carcinoma of the bladder. J Surg Oncol. 2013;107(5):555–559. doi:10.1002/jso.23264
17. Li Q, Song W, Wang J. TUG1 confers Adriamycin resistance in acute myeloid leukemia by epigenetically suppressing miR-34a expression via EZH2. Biomed Pharmacother. 2019;109:1793–1801. doi:10.1016/j.biopha.2018.11.003
18. Xie D, Zhang H, Hu X, Shang C. Knockdown of long non-coding RNA Taurine Up-Regulated 1 inhibited doxorubicin resistance of bladder urothelial carcinoma via Wnt/β-catenin pathway. Oncotarget. 2017;8(51):88689–88696. doi:10.18632/oncotarget.v8i51
19. Gao M, Zhao B, Chen M, et al. Nrf-2-driven long noncoding RNA ODRUL contributes to modulating silver nanoparticle-induced effects on erythroid cells. Biomaterials. 2017;130:14–27. doi:10.1016/j.biomaterials.2017.03.027
20. Zhang Y, Xia J, Li Q, et al. NRF2/Long noncoding RNA ROR signaling regulates mammary stem cell expansion and protects against estrogen genotoxicity. J Biol Chem. 2014;289(45):31310–31318. doi:10.1074/jbc.M114.604868
21. Sun Y, Guan Z, Liang L, et al. HIF-1alpha/MDR1 pathway confers chemoresistance to cisplatin in bladder cancer. Oncol Rep. 2016;35(3):1549–1556. doi:10.3892/or.2015.4536
22. Catanzaro E, Calcabrini C, Turrini E, Sestili P, Fimognari C. Nrf2: a potential therapeutic target for naturally occurring anticancer drugs? Expert Opin Ther Targets. 2017;21(8):781–793. doi:10.1080/14728222.2017.1351549
23. Rushworth SA, Bowles KM, MacEwan DJ. High basal nuclear levels of Nrf2 in acute myeloid leukemia reduces sensitivity to proteasome inhibitors. Cancer Res. 2011;71(5):1999–2009. doi:10.1158/0008-5472.CAN-10-3018
24. Hayden A, Douglas J, Sommerlad M, et al. The Nrf2 transcription factor contributes to resistance to cisplatin in bladder cancer. Urol Oncol. 2014;32(6):806–814. doi:10.1016/j.urolonc.2014.02.006
25. Ciamporcero E, Daga M, Pizzimenti S, et al. Crosstalk between Nrf2 and YAP contributes to maintaining the antioxidant potential and chemoresistance in bladder cancer. Free Radic Biol Med. 2018;115:447–457. doi:10.1016/j.freeradbiomed.2017.12.005
26. Bialk P, Wang Y, Banas K, Kmiec EB. Functional gene knockout of NRF2 increases chemosensitivity of human lung cancer A549 cells in vitro and in a xenograft mouse model. Mol Ther Oncolytics. 2018;11:75–89. doi:10.1016/j.omto.2018.10.002
27. Tang T, Cheng Y, She Q, et al. Long non-coding RNA TUG1 sponges miR-197 to enhance cisplatin sensitivity in triple negative breast cancer. Biomed Pharmacother. 2018;107:338–346. doi:10.1016/j.biopha.2018.07.076
28. Han X, Yang Y, Sun Y, Qin L, Yang Y. LncRNA TUG1 affects cell viability by regulating glycolysis in osteosarcoma cells. Gene. 2018;674:87–92. doi:10.1016/j.gene.2018.06.085
29. Niu Y, Ma F, Huang W, et al. Long non-coding RNA TUG1 is involved in cell growth and chemoresistance of small cell lung cancer by regulating LIMK2b via EZH2. Mol Cancer. 2017;16(1):5. doi:10.1186/s12943-016-0575-6
30. Yang F, Li X, Zhang L, Cheng L, Li X. LncRNA TUG1 promoted viability and associated with gemcitabine resistant in pancreatic ductal adenocarcinoma. J Pharmacol Sci. 2018;137(2):116–121. doi:10.1016/j.jphs.2018.06.002
31. Fraczek N, Bronisz I, Pietryka M, et al. An outline of main factors of drug resistance influencing cancer therapy. J Chemother. 2016;28(6):457–464. doi:10.1080/1120009X.2016.1218158
32. Gottesman MM, Lavi O, Hall MD, Gillet JP. Toward a better understanding of the complexity of cancer drug resistance. Annu Rev Pharmacol Toxicol. 2016;56:85–102. doi:10.1146/annurev-pharmtox-010715-103111
33. Shaffer BC, Gillet JP, Patel C, Baer MR, Bates SE, Gottesman MM. Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resist Updat. 2012;15(1–2):62–69. doi:10.1016/j.drup.2012.02.001
34. Silva R, Vilas-Boas V, Carmo H, et al. Modulation of P-glycoprotein efflux pump: induction and activation as a therapeutic strategy. Pharmacol Ther. 2015;149:1–123. doi:10.1016/j.pharmthera.2014.11.013
35. Vallo S, Kopp R, Michaelis M, et al. Resistance to nanoparticle albumin-bound paclitaxel is mediated by ABCB1 in urothelial cancer cells. Oncol Lett. 2017;13(6):4085–4092. doi:10.3892/ol.2017.5986
36. M R S, Jeddi F, Soozangar N, et al. Nrf2/P–glycoprotein axis is associated with clinicopathological characteristics in colorectal cancer. Biomed Pharmacother. 2018;104:458–464. doi:10.1016/j.biopha.2018.05.062
37. Ye P, Mimura J, Okada T, et al. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol. 2014;34(18):3421–3434. doi:10.1128/MCB.00221-14
38. Jiang H, Hu X, Zhang H, Li WB. Down-regulation of LncRNA TUG1 enhances radiosensitivity in bladder cancer via suppressing HMGB1 expression. Radiat Oncol. 2017;12(1):65. doi:10.1186/s13014-017-0802-3
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.