")
Back to Journals » Drug Design, Development and Therapy » Volume 14
Tranilast Inhibits Pulmonary Fibrosis by Suppressing TGFβ/SMAD2 Pathway
Authors Kato M , Takahashi F, Sato T , Mitsuishi Y, Tajima K, Ihara H , Nurwidya F , Baskoro H, Murakami A, Kobayashi I, Hidayat M , Shimada N, Sasaki S, Mineki R, Fujimura T, Kumasaka T, Niwa SI, Takahashi K
Received 11 June 2020
Accepted for publication 23 September 2020
Published 29 October 2020 Volume 2020:14 Pages 4593—4603
DOI https://doi.org/10.2147/DDDT.S264715
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Motoyasu Kato,1,2 Fumiyuki Takahashi,1,2 Tadashi Sato,1,2 Yoichiro Mitsuishi,1,2 Ken Tajima,1,2 Hiroaki Ihara,1,2 Fariz Nurwidya,1,2 Hario Baskoro,1,2 Akiko Murakami,1,2 Isao Kobayashi,1,2 Moulid Hidayat,1,2 Naoko Shimada,1– 3 Shinichi Sasaki,1,2 Reiko Mineki,4 Tsutomu Fujimura,5 Toshio Kumasaka,6 Shin-ichiro Niwa,7 Kazuhisa Takahashi1– 3
1Department of Respiratory Medicine, Juntendo University Graduate School of Medicine, Tokyo, Japan; 2Research Institute for Diseases of Old Ages, Juntendo University Graduate School of Medicine, Tokyo, Japan; 3Leading Center for the Development and Research of Cancer Medicine, Juntendo University Graduate School of Medicine, Tokyo, Japan; 4Laboratory of Proteomics and Biomolecular Science, Research Support Center, Juntendo University Graduate School of Medicine, Tokyo, Japan; 5Laboratory of Bioanalytical Chemistry, Tohoku Medical and Pharmaceutical University, Sendai, Miyagi, Japan; 6Department of Pathology, Japanese Red Cross Medical Center, Tokyo, Japan; 7Link Genomics, Incorporated, Tokyo, Japan
Correspondence: Motoyasu Kato
Department of Respiratory Medicine, Juntendo University, Graduate School of Medicine, Bunkyo-Ku, Tokyo 113-8431, Japan
Tel +81-3-5802-1063
Fax +81-3-5802-1617
Email [email protected]
Purpose: Idiopathic pulmonary fibrosis (IPF) is characterized by the accumulation of extracellular matrix (ECM) protein in the lungs. Transforming growth factor (TGF) β-induced ECM protein synthesis contributes to the development of IPF. Tranilast, an anti-allergy drug, suppresses TGFβ expression and inhibits interstitial renal fibrosis in animal models. However, the beneficial effects of tranilast or its mechanism as a therapy for pulmonary fibrosis have not been clarified.
Methods: We investigated the in vitro effect of tranilast on ECM production and TGFβ/SMAD2 pathway in TGFβ 2-stimulated A549 human alveolar epithelial cells, using quantitative polymerase chain reaction, Western blotting, and immunofluorescence. In vitro observations were validated in the lungs of a murine pulmonary fibrosis model, which we developed by intravenous injection of bleomycin.
Results: Treatment with tranilast suppressed the expression of ECM proteins, such as fibronectin and type IV collagen, and attenuated SMAD2 phosphorylation in TGFβ 2-stimulated A549 cells. In addition, based on a wound healing assay in these cells, tranilast significantly inhibited cell motility, with foci formation that comprised of ECM proteins. Histological analyses revealed that the administration of tranilast significantly attenuated lung fibrosis in mice. Furthermore, tranilast treatment significantly reduced levels of TGFβ, collagen, fibronectin, and phosphorylated SMAD2 in pulmonary fibrotic tissues in mice.
Conclusion: These findings suggest that tranilast inhibits pulmonary fibrosis by suppressing TGFβ/SMAD2-mediated ECM protein production, presenting tranilast as a promising and novel anti-fibrotic agent for the treatment of IPF.
Keywords: tranilast, idiopathic pulmonary fibrosis, TGFβ, SMAD2
Background
Idiopathic pulmonary fibrosis (IPF) is a chronic pulmonary disorder of unknown etiology and is characterized by progressive deposition of extracellular matrix (ECM) proteins such as collagen and fibronectin.1 Pathological findings of IPF include the formation of fibroblastic foci rich in ECM proteins in lung parenchyma of patients, and the number of fibroblastic foci is closely associated with progression and poor prognosis of the disease.2 Transforming growth factor (TGF) β plays a crucial role in the induction of pulmonary fibrosis.3 TGFβ can induce epithelial–mesenchymal transition (EMT) in human alveolar epithelial cells through the activation of SMAD2,4 and has been implicated in ECM synthesis by activated mesenchymal cells, resulting in the progression of fibrosis.5
Recently, much interests have been focused on the attenuation of the TGFβ pathway as a treatment strategy for IPF. The antibiotic and tetracycline family member, methacycline, reportedly inhibited the expression of TGFβ-induced ECM proteins, such as collagen and fibronectin, in human alveolar epithelial cell line A549 cells, and attenuated bleomycin-induced pulmonary fibrosis in vivo.6 Therefore, attenuation of TGFβ pathway may result in the inhibition of pulmonary fibrosis, and the agent that directly blocks the TGFβ pathway may have potential therapeutic application in IPF.
Tranilast (N-3,4-dimethoxycinnamoyl-anthranilic acid) is an anti-allergic drug used for asthma as well as atopic and fibrotic pathologies, including keloid and scleroderma.7,8 Tranilast has been shown to inhibit renal and cardiovascular fibrosis in experimental models.9,10 Tranilast attenuated renal fibrosis induced by unilateral ureteral obstruction in rats through the reduction of TGFβ and phospho-SMAD2 expression and modulation of EMT.11 Although tranilast reportedly inhibited the progression of pulmonary fibrosis in mice, the mechanism of tranilast inhibition of pulmonary fibrosis has not been elucidated in detail.
In this study, we examined the effect of tranilast on TGFβ-induced ECM synthesis and foci formation in A549 cells in vitro and BLM-induced pulmonary fibrosis in vivo.
Methods
Cells and Reagents
A549 cells are human lung cancer cell lines that maintain the characteristics of type II alveolar epithelial cells and were obtained from the RIKEN Bioresource Center (Tokyo, Japan). Tranilast was purchased from Sigma-Aldrich, Inc. (St. Louis, MO, USA), while TGFβ2 and tumor necrosis factor alpha (TNFα) were obtained from eBioscience (San Diego, CA, USA). TGFβ receptor inhibitor (SB431542) was purchased from TOCRIS Bioscience (Woburn, MA, USA). Details of cell culture and reagents are provided under Supplementary Materials and Methods.
In vitro Proliferation Assay
Details of the method are provided under Supplementary Materials and Methods.
Quantitative Polymerase Chain Reaction
Quantitative polymerase chain reaction conditions and primer sequences used for the detection of transcripts are described under Supplementary Materials and Methods and Supplementary Table 1.
Western Blot
Details of the method and antibodies are provided under Supplementary Materials and Methods.
Immunofluorescence
A549 cells were treated with TGFβ2 and TNFα and immunofluorescence for fibronectin was performed as described under Supplementary Materials and Methods.
Cell Wound Healing Assay
Details of this method are provided under Supplementary Materials and Methods.
Fibrotic Foci Assay
A549 cells were cultured for 48 h in a 24-well chamber, followed by incubation with 5 and 100 ng/mL of TGFβ2 and TNFα, respectively, for 48 h. Foci formation following treatment with TGFβ2 and TNFα were observed as described previously.12 Fibrotic foci ratio (FFR) was calculated by dividing areas of foci with the area of the target field.
Bleomycin-Induced Pulmonary Fibrosis Model
Male 12-week-ICR mice, weighing about 35 g, were obtained from Japan Oriental Kobo (Tokyo, Japan). Mice were housed in a room under controlled temperature (25ºC), humidity, and lighting (12/12-h light-dark cycle). Mice were categorized into four groups (A-D) at random, and mice in two groups (A and B) were injected with 10 mg/kg/day of bleomycin (BLM, Nippon Kayaku, Tokyo, Japan) dissolved in 200 µL normal saline for 5 consecutive days,13 followed by treatment with tranilast 200 mg/kg. Mice in the other two groups (C and D) were injected with normal saline alone. Mice were administered either tranilast (200 mg/kg in 200 µL 5% carboxymethyl cellulose [CMC]; groups A and C) or 5% CMC alone (groups B and D) by oral gavage twice daily from day 8 to 20 (Supplementary Table 2). All animals were sacrificed by intraperitoneal injection of pentobarbital at day 22 from the initiation of BLM injection. Both lungs were removed and frozen immediately in liquid nitrogen. Whole lungs were fixed in buffered 10% formalin solution (Wako Pure Chemical Industries, Tokyo, Japan) for histological examination. Fixed paraffin sections (3 μm in size) were cut and stained with hematoxylin-eosin (HE) or Masson trichrome (MT) for the microscopic visualization of fibrotic lesions.
Fibrosis Score
Lung specimens were obtained from all five lobes, and all available specimens were reviewed. Images of sections stained with HE and MT were obtained using a microscope (Axiophoto; Carl Zeiss Corporation; Oberkochen, Germany) with a CCD camera (SPOT; Diagnostic Instruments; Sterling Heights, MI).14 Fibrotic changes were scored by Ashcroft score.15,16 In addition, two physicians (MK and FT) independently reviewed the same samples to evaluate the interobserver correlation of our method.
Collagen Content
The level of collagen in lung tissues was assessed with the Sircol collagen assay (Biocolor Ltd., Carrickfergus, Northern Ireland, UK) following the manufacturer’s instructions.
Enzyme-Linked ImmunoSorbent Assay (ELISA) for Anti-TGFβ1 Antibody Detection
TGFβ1 concentration in BAL fluid was analyzed using ELISA sandwich, specifically developed with antibodies against canine TGFβ1 (Mouse/Rat/Porcine/Canine TGFβ1, MB100B, R&D Systems, Minneapolis, USA). ELISA was performed for each sample following the manufacturer’s instructions. Reading was performed at 450 nm by using a microplate reader (Bio-Rad, Richmond, USA).
Immunohistochemistry
Immunohistochemical staining of mice lungs for fibronectin and phospho-SMAD2 was performed as described in Supplementary Materials and Methods.
Ethics
All animal experiments were carried out in accordance with the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology (Notice No. 71, 2006) and were approved by the Committee for Animal Experimentation of Juntendo University with the Approval No. 290,031.
Statistics
Statistical analysis was carried out using analysis of variance (ANOVA). The difference between the means was considered to be statistically significant at p < 0.05.
Results
Tranilast Inhibits TGFβ2-Induced ECM and Mesenchymal Protein Expression in A549 Cells
We initially examined the effect of various concentrations of tranilast on proliferation of A549 cells. At a concentration below 200 μM, tranilast did not influence the growth of A549 cells (Figure S1). Subsequently, A549 cells were treated with various concentrations of tranilast in the presence or absence of 5 ng/mL TGFβ2. As shown in Figure 1A, qPCR analysis revealed that TGFβ2 stimulation alone resulted in the upregulation of mesenchymal markers, including N-cadherin, fibronectin, and collagen type IV alpha 1 chain (COL4A1); however, tranilast significantly inhibited TGFβ2-induced mRNA expression of these mesenchymal factors (Figure 1A). Further, Western blotting analysis showed that TGFβ2 stimulation led to the loss of the epithelial marker E-cadherin and increased the expression of mesenchymal markers such as fibronectin, type IV collagen, and N-cadherin (Figure 1B). Treatment with tranilast suppressed TGFβ2-induced expression of the mesenchymal markers and restored the expression of E-cadherin in a dose-dependent manner (Figure 1C). These findings suggest that tranilast inhibits TGFβ2-induced ECM and mesenchymal protein expression in A549 cells.
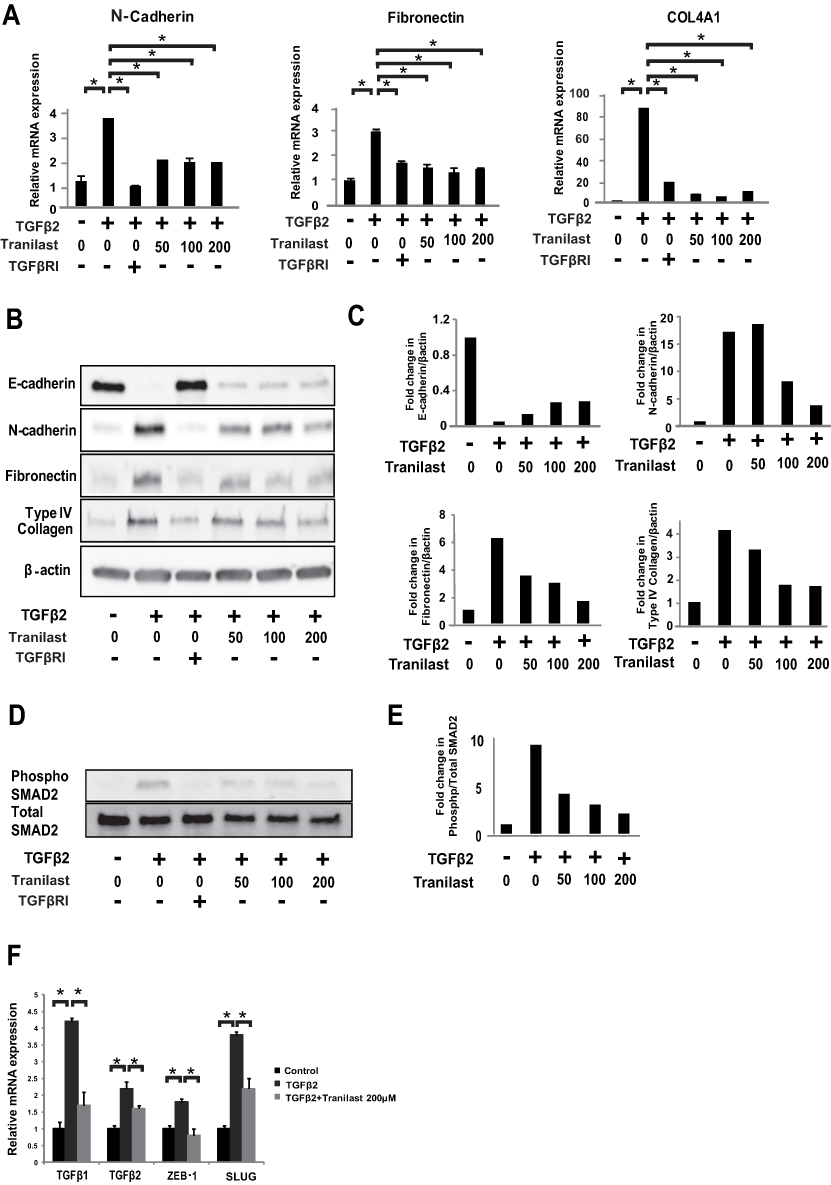 |
Figure 1 Tranilast inhibited TGFβ2-induced expression of mesenchymal markers and ECM synthesis via TGFβ/SMAD2 pathway in A549 cells. A549 cells were treated with various concentrations (50, 100, and 200 μM) of tranilast in presence or absence of 5 ng/mL TGFβ2 for 72 h. (A) Expression levels of N-cadherin, fibronectin, and COL4A1 mRNA was analyzed by quantitative polymerase chain reaction (qPCR). TGFβRI: TGFβ receptor inhibitor. (B) Expression of proteins E-cadherin, N-cadherin, fibronectin, and COL4A1 was analyzed by Western blotting. (C) Fold changes were calculated by setting the ratios of the any protein/β-actin band intensities. (D) A549 cells were treated with tranilast in presence or absence of 5 ng/mL TGFβ2 for evaluation of SMAD2. The phosphorylation level of Smad2 was analyzed by Western blotting. (E) Fold changes were calculated by setting the ratios of the phospho protein/total protein band intensities. (F) A549 cells with or without TGFβ2 stimulation were treated with tranilast for evaluation of TGFβ and transcriptional factors. Expressions of mRNA levels for TGFβ1, TGFβ2, ZEB-1, and Slug were evaluated by qPCR. Each bar represents mean ± SD of three independent experiments. *p < 0.01. |
Tranilast Suppresses TGFβ/SMAD2 Pathway in A549 Cells
We examined the effect of tranilast on TGFβ/SMAD2 pathway. A549 cells were treated with different concentrations of tranilast in the presence of TGFβ2 for 72 h. Western blotting analysis revealed that tranilast attenuated TGFβ2-induced phosphorylation of SMAD2 (Figure 1D and E). As expected, TGFβ2 stimulation upregulated the expression of TGFβ1, TGFβ2, zinc finger E-box binding homeobox 1 (ZEB1), and SLUG, which are involved in TGFβ pathway; however, tranilast treatment suppressed the expression of these genes (Figure 1F).
Tranilast Attenuates TGFβ-Induced Cell Motility in A549 Cells
A wound healing assay was performed to evaluate the effect of tranilast on cell motility. A549 cells were treated with various concentrations of tranilast with or without 5 ng/mL TGFβ2, and the cell monolayer was scratched following a 48 h incubation. Migration of the cells into the wound area was observed 48 h after scratching, and wound healing ratio (WHR) was evaluated in each well. Tranilast significantly attenuated TGFβ2-induced cell motility in A549 cells (Figure 2A).
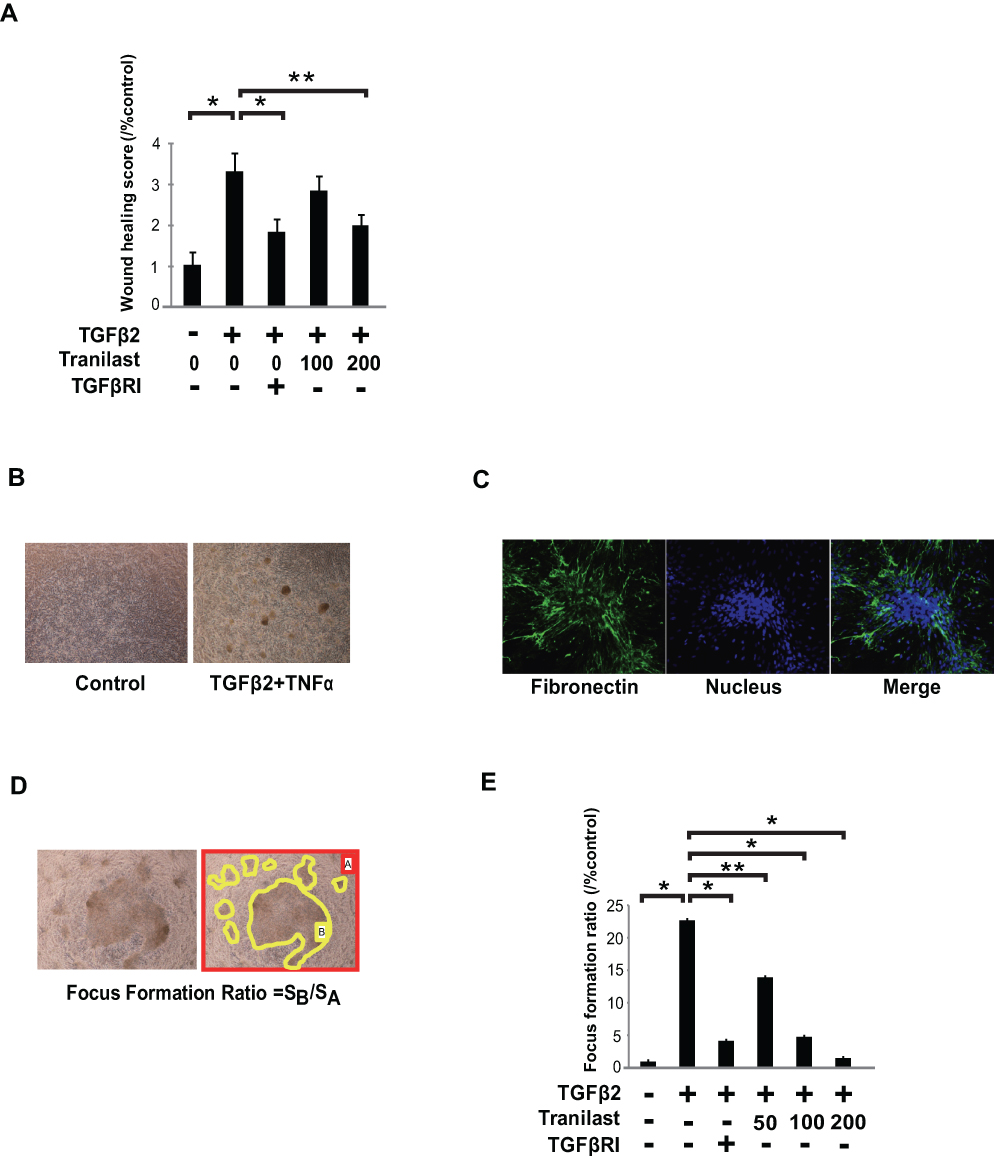 |
Figure 2 Tranilast attenuated cell migration and foci formation in A549 cells. (A) A549 cells with or without TGFβ2 (5 ng/mL) stimulation were incubated with 100 or 200 μM tranilast for 48 h and a wound was created by scratching the cell monolayer to assess cell migration; the wound area was evaluated. Photos of sections were taken under 200 × magnification using a microscope (Olympus, Japan) with a CCD camera. Wound areas were measured by ImageJ. Wound healing ratio in each well was calculated by dividing the wound areas. (B) To evaluate foci formation, A549 cells were incubated in presence of TGFβ2 (5 ng/mL) and TNFα (100 ng/mL) in 24-well chambers. Following 48 h stimulation, photos of sections were taken using a microscope fitted with a CCD camera. (C) Foci were strongly immunostained with fibronectin antibody. (D) Fibrotic foci ratio (FFR) was calculated by dividing the areas of foci with the area of the target field at six randomly selected wells using ImageJ. (E) The effect of several concentrations of tranilast (50, 100, and 200 μM) on foci formation in A549 cells. These cells in each group were observed after TGFβ2 stimulation with or without tranilast. The average of FFR was calculated for each group. Each bar represents mean ± SD of three independent experiments. *p < 0.01, **p < 0.05. |
Tranilast Suppresses TGFβ2 and TNFα-Induced Foci Formation in A549 Cells
Fibroblastic foci comprised of ECM proteins are characteristic features in the lungs of IPF patients, and the number of fibroblastic foci correlates with progression of IPF.17 To evaluate the effect of tranilast on the foci formation of human alveolar epithelial cells, A549 cells were cultured in the medium with 5 ng/mL of TGFβ2 and 100 ng/mL of TNFα for 48 h. As shown in Figure 2B, formation of cell foci was only induced by the stimulation with TGFβ2 and TNFα, but not by TGFβ2, TGFβ1, TNFα, or a combination of TGFβ1 and TNFα to (data not shown). These foci were strongly immunostained with fibronectin antibody, indicating that they were comprised of ECM proteins including fibronectin (Figure 2C). We calculated the focus formation ratio (FFR) by dividing the areas of foci with the area of the target field at six randomly selected fields (Figure 2D). Tranilast significantly suppressed foci formation in a dose-dependent manner (Figure 2E).
Tranilast Attenuates BLM-Induced Pulmonary Fibrosis in Mice
To investigate the anti-fibrotic effects of tranilast in vivo, we evaluated murine pulmonary fibrosis model induced by BLM injection. Lung tissues were stained with HE and MT stains (Figure 3A). Oral administration of tranilast significantly attenuated pulmonary fibrosis in mice treated with BLM, as evident from quantitative histological analysis (Figure 3B). Tranilast alone had no effect on lung tissues of control mice.
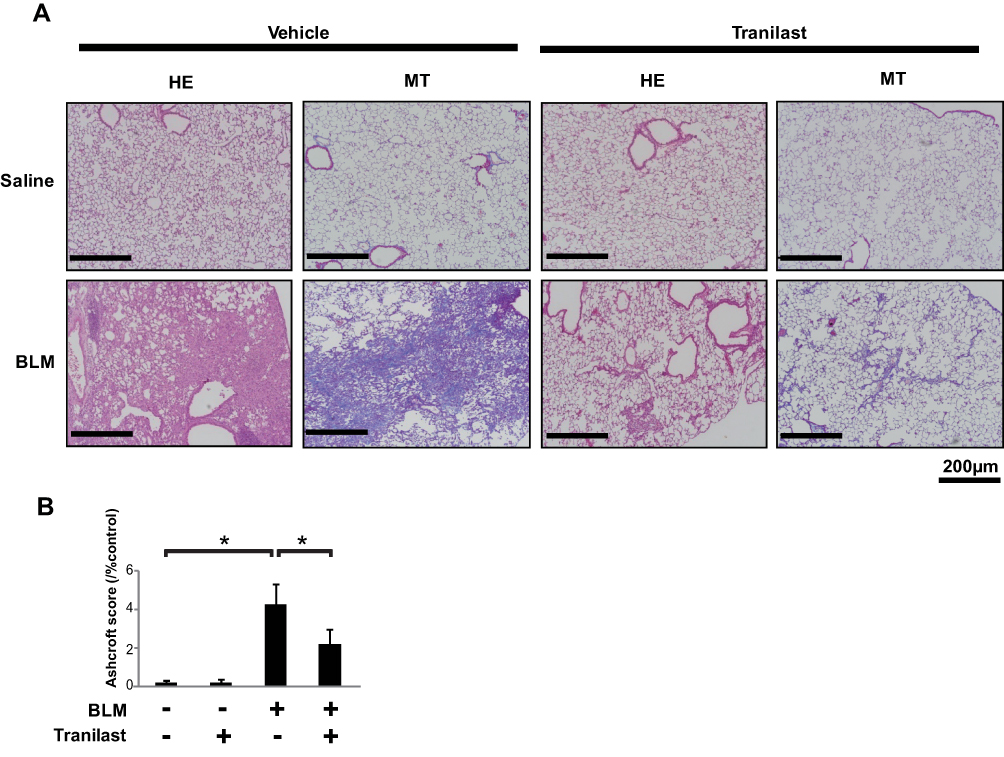 |
Figure 3 Tranilast attenuated BLM-induced pulmonary fibrosis in mice. (A) Histological examination was performed by hematoxylin-eosin (HE) as well as Masson trichrome (MT) staining. Bar = 200 µm. (B) The grade of pulmonary fibrosis was evaluated by the method described by Ashcroft and coworkers.19 Treatment groups were composed of saline-treated with vehicle (n = 10), saline-treated with tranilast (n = 10), BLM-treated with vehicle (n = 9), and BLM-treated with tranilast (n = 9). More than 30 randomized fields of each of the five lung lobes in mice were examined. Fibrotic score is presented as mean ± SD of all the fields examined in each group. *p < 0.01. |
We evaluated the inhibitory effect of tranilast on TGFβ1 and ECM protein production, as well as on SMAD2 phosphorylation in lungs of murine pulmonary fibrosis model. BLM-treated mice exhibited an increased TGFβ1 level in bronchoalveolar lavage (BAL) fluid. However, enzyme-linked immunosorbent assay (ELISA) revealed that tranilast treatment significantly decreased TGFβ1 levels (Figure 4A). The Sircol collagen assay showed that tranilast-treated mice displayed significantly lower collagen content in the whole lung tissues than those treated with vehicle (Figure 4B).
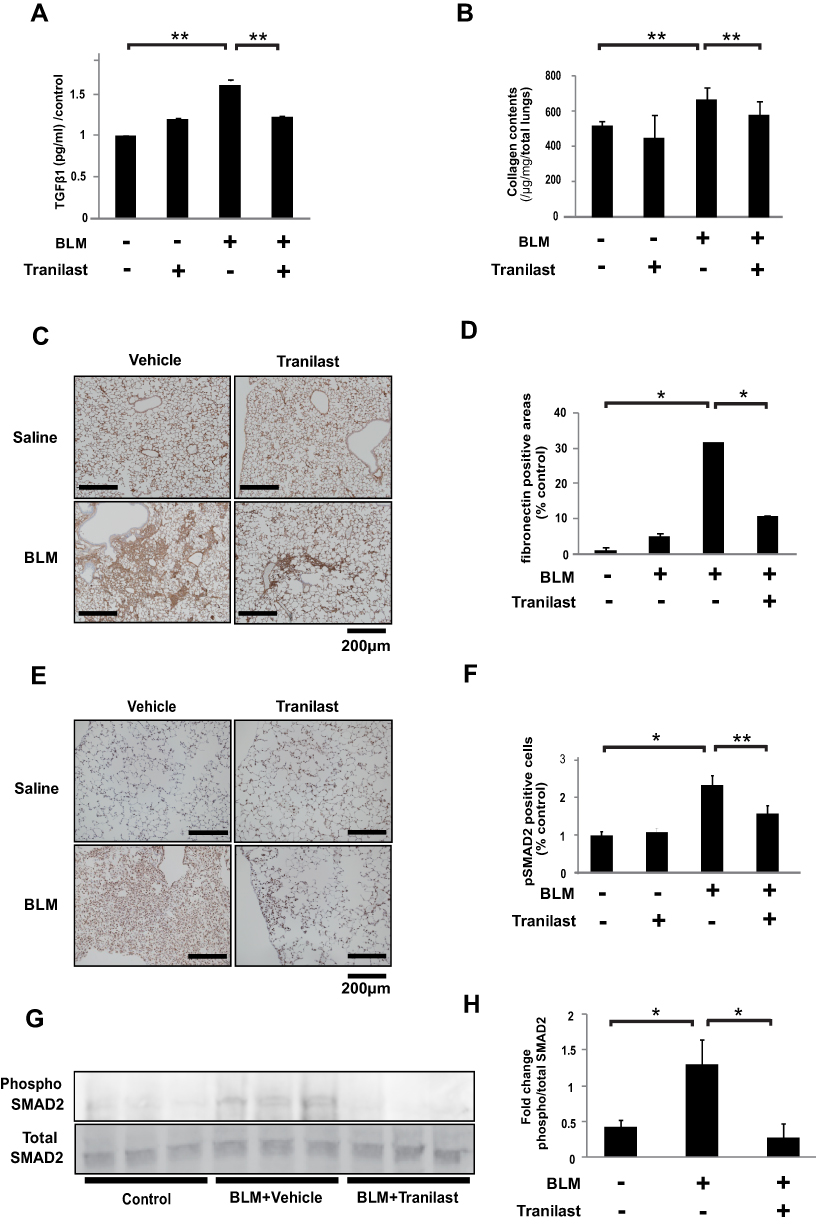 |
Figure 4 Tranilast attenuated BLM-induced ECM production and phosphorylation of SMAD2 in mice. (A) TGFβ1 concentration in BAL fluid was evaluated by ELISA. (B) Effects of tranilast on collagen contents. Data are presented as mean ± SD in each group of 10 mice. **p < 0.05. (C) Effect of tranilast on expression of fibronectin in the murine lungs by immunohistochemistry (IHC). Bar = 200 µm. (D) The average of the percentage of fibronectin positive ratio in each of the four groups was calculated by dividing the average of each group with that of the control group. Data are presented as mean ± SD in each group of 10 mice. *p < 0.01. (E) IHC staining for phospho-SMAD2 in the murine lungs. (F) The phospho-Smad2-positive cells were counted in 10 fields at × 200 magnification. Bar = 200 µm. The average of the percentage of phospho-SMAD2-positive cells in each of the four groups was calculated by dividing the average of each group with that of the control group. Data are presented as mean ± SD in each group of 10 mice. *p < 0.01, **p < 0.05. (G) Levels of phospho- and total-SMAD2 in the whole lung lysates of mice by Western blotting analysis. (H) Fold changes were analyzed by setting the ratios of the phospho/total protein band intensities. |
Expression of fibronectin (Figure 4C) and phospho-SMAD2 (Figure 4E) was evaluated by immunohistochemical analysis. BLM treatment upregulated the expression of fibronectin in murine lungs; however, tranilast administration significantly suppressed fibronectin expression (Figure 4D). In comparison to mice treated with BLM and vehicle, those treated with BLM and tranilast exhibited lower number of cells positive for phospho-SMAD2 in murine lungs (Figure 4F). Tranilast significantly attenuated BLM-induced phosphorylation of SMAD2, as revealed by Western blotting analysis (Figure 4G and H). These findings suggest that tranilast inhibited TGFβ1, ECM production, and SMAD2 phosphorylation in the lungs of a murine pulmonary fibrosis model.
Discussion
Our study highlights four main findings: (A) tranilast suppressed ECM protein expression and attenuated SMAD2 phosphorylation in TGFβ2-stimulated human alveolar epithelial cells (A549 cell line); (B) tranilast inhibited cell motility and foci formation in A549 cells; (C) tranilast attenuated BLM-induced pulmonary fibrosis in mice; (D) tranilast suppressed TGFβ1, ECM production, and SMAD2 phosphorylation in the lungs of mice treated with BLM.
TGFβ contributes to the pathogenesis of IPF by inducing EMT in alveolar epithelial cells, leading to the formation of fibroblastic foci and abnormal accumulation of ECM proteins.18 EMT is characterized by the loss of the epithelial marker E-cadherin, gain of mesenchymal markers such as fibronectin, and enhanced cell motility. EMT is often associated with cancer invasion and metastasis;19 of note, ECM was also reported to be involved in the pathogenesis of IPF.20,21 In fact, several research studies on pulmonary fibrosis focused on EMT. However, at least one concluded that EMT is not involved in the pathogenesis of BLM-induced lung fibrosis and human IPF.22 Therefore, whether EMT associates with IPF pathogenesis is still controversial.23 Although lung fibroblasts are undoubtedly related to the pathogenesis of IPF and are usually used for the research of lung fibrosis, the pathogenesis of IPF multifactorial.24 In fact, the first step of lung fibrosis is alveolar epithelial damage caused by several factors; thus, type II alveolar epithelial cells will also have a role in the pathogenesis of IPF. Of note, several studies supporting an association between EMT and lung fibrosis have been published.25–30
TGFβ is known as one of the pro-fibrotic key mediators in the pathogenesis of IPF and associates with EMT, fibroblast to myofibroblast transition, and senescence. Among the TGFβ family, TGFβ1 is an important key mediator of lung fibrosis; in fact, it is usually used as a biomarker for the analysis of lung fibrosis. TGFβ2 is also often used for the evaluation of lung fibrosis.27,31 In our research, we focused on the association between fibroblastic foci and EMT, as per a previous report.21 Therefore, we established an alveolar epithelial cells-induced foci assay system using an alveolar epithelial cell line. We tried to develop EMT-induced foci using several mediators; however, we could only establish foci via the stimulation with both TGFβ2 and TNFα. Of note, we used TGFβ2 in all of the assays in this study, precisely because we used TGFβ2 with TNFα for foci development.
Fibroblastic foci are one of the important pathological features associated with the progression of fibrosis in IPF.17,32,33 Therefore, we focused on fibroblastic foci as targets for the treatment of IPF. We designed an assay of fibroblastic foci formation using A549 cells stimulated with TGFβ2 and TNFα. Foci formation in our A549 in vitro model was associated with EMT; of note, foci comprised fibronectin. In theory, if a candidate drug suppresses the development of foci formation, it will attenuate the progression of lung fibrosis in IPF. In our study, tranilast treatment suppressed TGFβ-induced EMT as well as the production of mesenchymal and ECM proteins such as collagen and enhanced cell motility. Tranilast treatment also significantly inhibited foci formation in A549 cells.
Tranilast effectively prevents the progression of fibrosis in various organs, including inhibiting TGFβ1-induced ECM production in the proximal tube cells of the kidney and modulating renal fibrosis induced by unilateral ureteral obstruction in rats.11 In addition, tranilast inhibited TGFβ1 expression, prevented atrial remodeling, and suppressed atrial fibrillation development in a canine model of cardiovascular fibrosis.34 Moreover, tranilast was reported to inhibit pulmonary fibrosis by suppressing alveolar macrophage activation in alveolar macrophage (MAC-1 positive) cells and suppressing the production of reactive oxygen species in a BLM-induced lung fibrosis model.35,36 In addition, tranilast reportedly inhibits lung fibrosis by suppressing BLM-induced activation of neutrophils and alveolar macrophages in mast cell-deficient WBB6F1-W/Wv mice.37 Our current study focused on the TGFβ/SMAD pathway in epithelial cells and also shows that tranilast inhibits ECM protein production, which is consistent with previous reports.
Our study has several limitations. First, we did not assess the effect of lung fibrosis attenuation/prevention by anti-fibrotic agents, in comparison with that of tranilast. Of note, almost all of the previous studies also did not compare the effects of the candidate medicine studied with those of approved drugs. For instance, one of the existing anti-fibrotic agents, nintedanib, was not compared pre-clinically with another existing anti-fibrotic agent, pirfenidone, in the context of prevention of lung fibrosis.31 Of note, the effect of tranilast on lung fibrosis is still low; this said, we still need to compare the efficacy of tranilast on lung fibrosis (in comparison with positive control drugs, including nintedanib or pirfenidone) in the future. Second, we used the A549 cell line as representative of type II alveolar epithelial cells. The A549 cell line is a human alveolar epithelial lung cancer cell line with a KRAS mutation.38 Therefore, these cells have characteristics of cancer cells such as fast cell proliferation and immortalization. Ideally, we should have used primary type II alveolar epithelial cells for the analysis of lung fibrosis. However, it is difficult to culture primary human alveolar epithelial cells. To perform some experiments, particularly for the evaluation of EMT using primary alveolar epithelial cells is also quite challenging. Thus, many researchers, ourselves included, evaluate EMT and epithelium in IPF using A549 cells instead of human primary type II alveolar epithelial cells.4,6,39–43 This said, we should have in mind the different cell characteristics of A549 and primary human alveolar epithelial cells in the analysis of the experimental results.
Conclusions
Our results showed that tranilast attenuates pulmonary fibrosis through the inhibition of SMAD2 phosphorylation and suppression of TGFβ-mediated ECM protein production in mesenchymal cells.
Abbreviations
IPF, idiopathic pulmonary fibrosis; ECM, extracellular matrix; TGF, transforming growth factor; EMT, epithelial–mesenchymal transition; BLM, bleomycin; CMC, carboxymethylcellulose; HE, hematoxylin-eosin; MT, Masson trichrome; FFR, focus formation ratio; qPCR, quantitative polymerase chain reaction; TGFβRI, TGFβ receptor inhibitor.
Data Sharing Statement
Not applicable.
Consent for Publication
Not applicable.
Ethics Approval and Consent to Participate
The Institutional Review Board at the Juntendo University School of Medicine approved the procedures. (No. 290,031).
Acknowledgments
This study was supported by Japan Society for the Promotion of Science KAKENHI Grant Number 26461200. This study was partially supported by Ministry of Health, Labour and Welfare of Japan awarded to the Study Group on Diffuse Pulmonary Disorders, Scientific Research/Research on intractable diseases, Grant for Cross-disciplinary Collaboration, Grant from Institute for Environmental & Gender-specific Medicine, and President’s Grant for Interfaculty Collaboration, Juntendo University.
Funding
This study was supported by Research Grants from the Satoshi Okamoto Memorial Foundation of Pulmonary Fibrosis, Japan research foundation for clinical pharmacology, Mochida Memorial Foundation for Medical and Pharmaceutical Research, and Yokoyama Foundation for Clinical Pharmacology.
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345:517–525. doi:10.1056/NEJMra003200.
2. Flaherty KR, Colby TV, Travis WD, et al. Fibroblastic foci in usual interstitial pneumonia: idiopathic versus collagen vascular disease. Am J Respir Crit Care Med. 2003;167:1410–1415. doi:10.1164/rccm.200204-373OC.
3. Kolosova I, Nethery D, Kern JA. Role of Smad2/3 and p38 MAP kinase in TGF-beta1-induced epithelial-mesenchymal transition of pulmonary epithelial cells. J Cell Physiol. 2011;226:1248–1254. doi:10.1002/jcp.22448.
4. Kasai H, Allen JT, Mason RM, et al. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir Res. 2005;6:56. doi:10.1186/1465-9921-6-56.
5. Chapman HA. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol. 2011;73:413–435. doi:10.1146/annurev-physiol-012110-142225.
6. Xi Y, Tan K, Brumwell AN, et al. Inhibition of epithelial-to-mesenchymal transition and pulmonary fibrosis by methacycline. Am J Respir Cell Mol Biol. 2014;50:51–60. doi:10.1165/rcmb.2013-0099OC.
7. Yamada H, Tajima S, Nishikawa T. Tranilast inhibits collagen synthesis in normal, scleroderma and keloid fibroblasts at a late passage culture but not at an early passage culture. J Dermatol Sci. 1995;9:45–47. doi:10.1016/0923-1811(94)00355-i.
8. Suzuki H, Tanaka K, Kaneshige H, et al. The effects of long term Tranilast administration on bronchial hypersensitivity in asthmatics. Panminerva Med. 1989;31:88–93.
9. Qi W, Chen X, Twigg S, et al. Tranilast attenuates connective tissue growth factor-induced extracellular matrix accumulation in renal cells. Kidney Int. 2006;69:989–995. doi:10.1038/sj.ki.5000189.
10. Betge S, Kunz C, Figulla H, et al. Late onset oral treatment with tranilast following large myocardial infarction has no beneficial effects on cardiac remodeling and mortality in rats. Exp Ther Med. 2014;8:1789–1796. doi:10.3892/etm.2014.2003.
11. Kaneyama T, Kobayashi S, Aoyagi D, et al. Tranilast modulates fibrosis, epithelial-mesenchymal transition and peritubular capillary injury in unilateral ureteral obstruction rats. Pathology. 2010;42:564–573. doi:10.3109/00313025.2010.508784.
12. Takahashi E, Nagano O, Ishimoto T, et al. Tumor necrosis factor-alpha regulates transforming growth factor-beta-dependent epithelial-mesenchymal transition by promoting hyaluronan-CD44-moesin interaction. J Biol Chem. 2010;285:4060–4073. doi:10.1074/jbc.M109.056523.
13. Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol. 2008;590:400–408. doi:10.1016/j.ejphar.2008.06.046.
14. Enomoto N, Suda T, Kato M, et al. Quantitative analysis of fibroblastic foci in usual interstitial pneumonia. Chest. 2006;130:22–29. doi:10.1016/j.ejphar.2008.06.046.
15. Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41:467–470. doi:10.1136/jcp.41.4.467.
16. Takahashi F, Takahashi K, Okazaki T, et al. Role of osteopontin in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2001;24:264–271. doi:10.1165/ajrcmb.24.3.4293.
17. Harada T, Watanabe K, Nabeshima K, et al. Prognostic significance of fibroblastic foci in usual interstitial pneumonia and non-specific interstitial pneumonia. Respirology. 2013;18:278–283. doi:10.1111/j.1440-1843.2012.02272.x.
18. Lomas NJ, Watts KL, Akram KM, et al. Idiopathic pulmonary fibrosis: immunohistochemical analysis provides fresh insights into lung tissue remodelling with implications for novel prognostic markers. Int J Clin Exp Pathol. 2012;5:58–71.
19. Christine LC, Robert AW. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi:10.1126/science.1203543.
20. Kevin KK, Matthias CK, Paul JW, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A. 2006;103:13180–13185. doi:10.1073/pnas.0605669103.
21. Yamaguchi M, Hirai S, Tanaka Y, et al. Fibroblastic foci, covered with alveolar epithelia exhibiting epithelial-mesenchymal transition, destroy alveolar septa by disrupting blood flow in idiopathic pulmonary fibrosis. Lab Invest. 2017;97:232–242. doi:10.1038/labinvest.2016.135.
22. Jason RR, Christina EB, Michael JC, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108(52):E1475–83. doi:10.1073/pnas.1117988108
23. Salton F, Volpe MC, Confalonieri M. Epithelial⁻mesenchymal transition in the pathogenesis of idiopathic pulmonary fibrosis. Medicina (Kaunas). 2019;55:83. doi:10.3390/medicina55040083
24. Sgalla G, Iovene B, Calvello M, et al. Idiopathic pulmonary fibrosis: pathogenesis and management. Respir Res. 2018;19:32. doi:10.1186/s12931-018-0730-2.
25. Qian W, Cai X, Qian Q, et al. Astragaloside IV modulates TGF -β1-dependent epithelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. J Cell Mol Med. 2018;22:4354–4365. doi:10.1111/jcmm.13725
26. Kanemaru R, Takahashi F, Kato M, et al. Dasatinib suppresses TGFβ-mediated epithelial-mesenchymal transition in alveolar epithelial cells and inhibits pulmonary fibrosis. Lung. 2018;196:531–541. doi:10.1007/s00408-018-0134-6
27. Ihara H, Mitsuishi Y, Kato M, et al. Nintedanib inhibits epithelial-mesenchymal transition in A549 alveolar epithelial cells through regulation of the TGF-β/Smad pathway. Respir Investig. 2020;58:275–284. doi:10.1016/j.resinv.2020.01.003
28. Lin L, Han Q, Xiong Y, et al. Krüpple-like-factor 4 attenuates lung fibrosis via inhibiting epithelial-mesenchymal transition. Sci Rep. 2017;7:15847. doi:10.1038/s41598-017-14602-7.
29. Han Q, Lin L, Zhao B, et al. Inhibition of mTOR ameliorates bleomycin-induced pulmonary fibrosis by regulating epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2018;500:839–845. doi:10.1016/j.bbrc.2018.04.148
30. Goldmann T, Zissel G, Watz H, et al. Human alveolar epithelial cells type II are capable of TGFβ-dependent epithelial-mesenchymal-transition and collagen-synthesis. Respir Res. 2018;19:138. doi:10.1186/s12931-018-0841-9.
31. Chaudhary NI, Roth GJ, Hilberg F, et al. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29:976–985. doi:10.1183/09031936.00152106
32. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44–e68. doi:10.1164/rccm.201807-1255ST
33. Nicholson AG, Fulford LG, Colby TV, et al. The relationship between individual histologic features and disease progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2002;166:173–177. doi:10.1164/rccm.2109039.
34. Nakatani Y, Nishida K, Sakabe M, et al. Tranilast prevents atrial remodeling and development of atrial fibrillation in a canine model of atrial tachycardia and left ventricular dysfunction. J Am Coll Cardiol. 2013;61:582–588. doi:10.1016/j.jacc.2012.11.014.
35. Mori H, Tanaka H, Kawada K, et al. Suppressive effects of tranilast on pulmonary fibrosis and activation of alveolar macrophages in mice treated with bleomycin: role of alveolar macrophages in the fibrosis. Jpn J Pharmacol. 1995;67:279–289. doi:10.1254/jjp.67.279
36. Aoki Y, Kojo Y, Yamada S, et al. Respirable dry powder formulation of bleomycin for developing a pulmonary fibrosis animal model. J Pharm Sci. 2012;101:2074–2081. doi:10.1002/jps.23102.
37. Mori H, Kawada K, Zhang P, Uesugi Y, Sakamoto O, Koda A. Bleomycin-induced pulmonary fibrosis in genetically mast cell-deficient WBB6F1-W/Wv mice and mechanism of the suppressive effect of tranilast, an antiallergic drug inhibiting mediator release from mast cells, on fibrosis. Int Arch Allergy Appl Immunol. 1991;95:195–201. doi:10.1159/000235429
38. Singh A, Greninger P, Rhodes D, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500. doi:10.1016/j.ccr.2009.03.022.
39. Namba T, Tanaka K-I, Ito Y, et al. Induction of EMT-like phenotypes by an active metabolite of leflunomide and its contribution to pulmonary fibrosis. Cell Death Differ. 2010;17:1882–1895. doi:10.1038/cdd.2010.64
40. Kyung SY, Kim DY, Yoon JY, et al. Sulforaphane attenuates pulmonary fibrosis by inhibiting the epithelial-mesenchymal transition. BMC Pharmacol Toxicol. 2018;19:13. doi:10.1186/s40360-018-0204-7.
41. Liao K, Yong CW, Hua K, et al. SB431542 inhibited cigarette smoke extract induced invasiveness of A549 cells via the TGF-β1/Smad2/MMP3 pathway. Oncol Lett. 2018;15:9681–9686. doi:10.3892/ol.2018.8556
42. Pattarayan D, Sivanantham A, Krishnaswami V. Tannic acid attenuates TGF-β1-induced epithelial-to-mesenchymal transition by effectively intervening TGF-β signaling in lung epithelial cells. J Cell Physiol. 2018;233:2513–2525. doi:10.1002/jcp.26127
43. Zhang C, Zhu X, Hua Y, et al. YY1 mediates TGF-β1-induced EMT and pro-fibrogenesis in alveolar epithelial cells. Respir Res. 2019;20:249. doi:10.1186/s12931-019-1223-7.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.