")
Back to Journals » Journal of Experimental Pharmacology » Volume 12
Tranexamic Acid Improves Memory and Learning Abilities in Aging Mice
Authors Hiramoto K , Yamate Y, Matsuda K, Sugiyama D, Iizuka Y
Received 1 October 2020
Accepted for publication 26 November 2020
Published 18 December 2020 Volume 2020:12 Pages 653—663
DOI https://doi.org/10.2147/JEP.S284532
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Bal Lokeshwar
Keiichi Hiramoto,1 Yurika Yamate,1 Kazunari Matsuda,2 Daijiro Sugiyama,2 Yasutaka Iizuka2
1Department of Pharmaceutical Sciences, Suzuka University of Medical Science, Suzuka, Mie 513-8670, Japan; 2R&D Department, Daiichi Sankyo Healthcare Co., LTD, Shinagawa-ku, Tokyo 103-8234, Japan
Correspondence: Keiichi Hiramoto Department of Pharmaceutical Sciences
Suzuka University of Medical Science, 3500-3 Minamitamagakicho, Suzuka, Mie 513-8670, Japan
Tel +81-59-340-0575
Fax +81-59-368-1271
Email [email protected]
Purpose: Although the onset mechanism of Alzheimer’s disease, which co-occurs with aging, has been extensively studied, no effective methods that improve the decline in memory and learning abilities following aging have been developed. Tranexamic acid provided promising results for ameliorating photo-aging and extending the natural lifespan. However, it is unknown whether it affects the decline in memory and learning abilities due to aging. In this study, we examined the effect of tranexamic acid on memory and learning abilities of naturally aging mice.
Methods: ICR mice were orally administered with tranexamic acid (12 mg/kg/day) three times weekly for 2 years, and their memory and learning abilities were compared between the tranexamic acid-treated and non-treated groups.
Results: The decline in memory and learning abilities due to aging was ameliorated by tranexamic acid administration. The expression of plasmin and amyloid-β decreased following the treatment with tranexamic acid. Furthermore, the number of M1-type brain macrophages diminished and that of M2 macrophages increased. In addition, administration of tranexamic acid decreased the concentrations of interleukin (IL)-1β and tumor necrosis factor-α, while it increased the levels of IL-10 and transforming growth factor-α in the brain.
Conclusion: These results indicated that tranexamic acid suppressed the secretion of the inflammatory cytokines aging M1-type macrophages, thereby improving age-related memory and learning abilities.
Keywords: plasmin, chronic inflammation, M1-type macrophage, M2-type macrophage, TNF-α, IL-1β
Introduction
In animals, including human beings, vital body functions, such as muscle and nerve conduction velocity diminish with aging. With regard to the functional change in the brain or nervous system, the levels of dopamine D2 receptors and glutamate, which mediate neurotransmission in the hippocampus, decrease with aging, thus altering the motor function, memory, and learning abilities in mice.1–3
An aged mammal shows remarkable dysmnesia. In an aging brain, senile plaques are formed with the aggregation and deposition of amyloid β proteins, which are generated by the cleavage of amyloid precursor proteins, deposits besides a cell appears. Accumulation of these aggregates appears like neurofibrillary tangles. These neurofibrillary tangles are formed in the neuron by the accumulation of paired helical filaments, which primarily consist of a phosphorylated protein called tau.4 Senile plaques and neurofibrillary tangles are observed in parts of the brain, such as the cerebral cortex, a smell infield, and hippocampus, that are associated with memory and learning.5 In addition, aging results in altered expression of more than 100 genes in the brain of mice.6 In the human brain, the expression of genes associated with synaptic transmission or synaptic plasticity caused by aging is inhibited remarkably. Reactive oxygen species (ROS), which produce oxidative stress, tend to form lesions in the promoter region of such genes.7 Therefore, the increase in ROS with aging is considered to be one of the causes for suppression of memory and learning abilities. In this study, we suggest the possible association of age-related memory impairment with increased expression of pyruvate carboxylase, a metabolic enzyme that affects the mitochondria in the glial cells and not the neurons.8 Thus, glial cells play an important role in memory and learning, and research on glial cells is still progressing.
Alzheimer’s disease (AD) and vascular dementia that have cerebrovascular pathologies, along with aging in the brain, lead to dementia. AD is a progressive central-neural-degenerative disease that alters memory and learning abilities, and there are more onsets as an underlying disease of dementia than vascular dementia.9,10 In patients with AD, deposition of senile plaques and neurofibrillary tangles throughout the hippocampus and cerebral cortex was observed.11 The main components of the plaques are amyloid-β and tau proteins, which following aggregation lead to the onset of AD,12,13 according to the amyloid cascade hypothesis of AD.14 Additionally, the choline acetyltransferase activity is reduced in the cerebral cortex, and the number of cholinergic neurons in the nucleus basalis of Meynert and basal forebrain is remarkably reduced. Damage to cholinergic nuclei disrupts the cholinergic neurotransmission from the basal forebrain to the cerebral cortex or hippocampus. Thus, the theory that states that onset of AD occurs owing to an insufficiency in acetylcholine production is called the cholinergic hypothesis of AD.15 Furthermore, it was recently reported that the malfunction of the nervous system is closely related to neurodegenerative diseases such as AD or Parkinson’s disease. A malfunction in the nervous system caused by a chronic inflammatory condition results in continuous and accelerated production of inflammatory cytokines and free radicals by microglia and astrocytes resulting in histological changes.16–20
Although many reports regarding the onset mechanism of AD in aging patients have been published, there is a lack of effective treatments and prophylaxis for this condition. However, trans-4-(aminomethyl) cyclohexane carboxylic acid, also called tranexamic acid, is a known, safe, and traditional treatment with anti-plasmin activity. Plasmin inhibits arachidonic acid, increasing the level of prostaglandins (PGs), which is a metabolite of arachidonic acid.21 PG is associated with the induction of acute inflammation and the secretion of cytokines during chronic inflammation.21 Therefore, owing to its anti-plasmin function, tranexamic acid exhibits an anti-inflammatory effect.22 Previously, we reported the beneficial effects of tranexamic acid regarding the amelioration of skin photo-aging.23 Moreover, the drug has been reported to extend natural lifespan.24 However, the effect of tranexamic acid against the decline in memory and learning abilities due to aging has not been investigated.
In this study, we examined the effect of tranexamic acid on memory and learning abilities in an aging mouse model. Here, we continuously administered tranexamic acid to mice for 2 years and compared memory and learning abilities between treated and untreated mice. In addition, we investigated the relationship of memory and learning abilities with inflammation.
Materials and Methods
Animal Experiments
Eight-week-old male ICR mice (SLC, Hamamatsu, Shizuoka, Japan) were maintained in individual cages in an air-conditioned room at 23 ± 1 °C under specific-pathogen-free conditions with a 12-h light/dark cycle. Mice were distributed among the following groups (six mice per group; total of 18 mice): control (non-treatment), solvent, and tranexamic acid groups. All the experiments were performed in triplicate. Blood and brain (cerebrum) samples were collected 2 years after the initiation of the study (Figure 1). All the experiments were blinded in this study. This study was performed in strict accordance with the Guide for the Care and Use of Laboratory Animals of Suzuka University of Medical Science and was approved by the Animal Experiment Ethics Committee of the Suzuka University of Medical Science on September 25, 2014 (approval number: 34). All surgeries were performed under anesthesia using pentobarbital (50 mg/kg; Nacalai Tesque, Kyoto, Japan), and complete efforts were made to minimize the suffering of the mice.
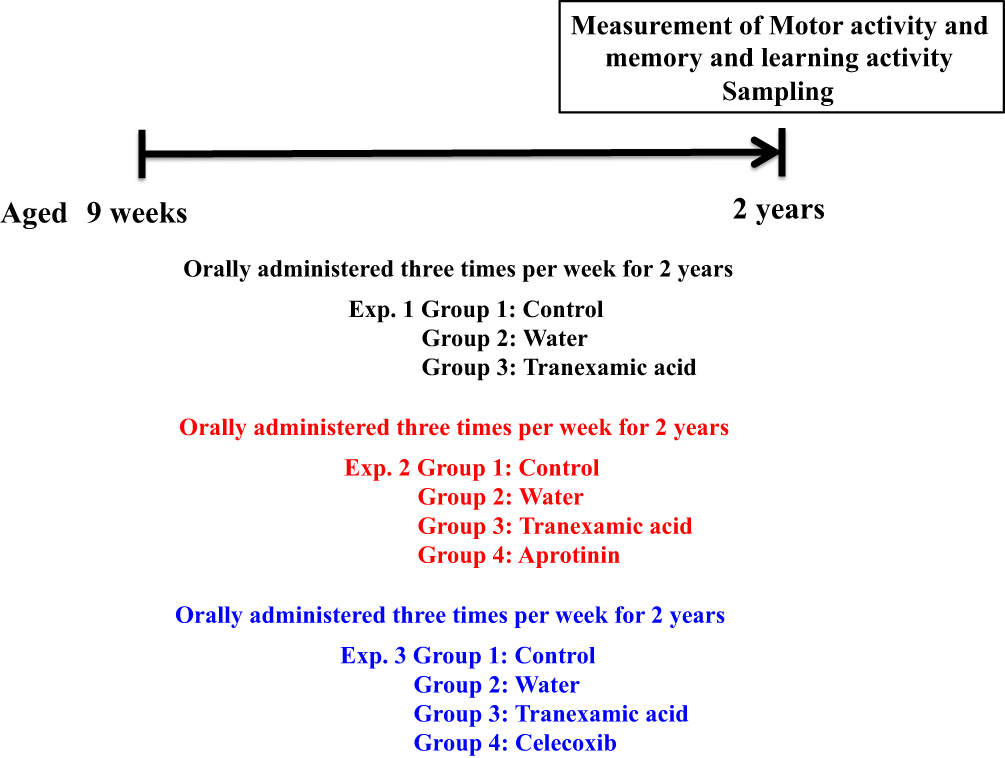 |
Figure 1 Schematic representation of the experiment. |
Treatment with Tranexamic Acid
Approximately 12 mg/kg of tranexamic acid (Daiichi Sankyo Healthcare Co., Ltd., Tokyo, Japan) in distilled water was orally administered to each mouse three times per week for 2 years, whereas the mice in the solvent group received distilled water only.25 This dose of 12 mg/kg/day for mice was derived from that used in humans (750 mg/60 kg). In addition, toxicity was not observed with long-term tranexamic acid administration.
Treatment with Plasmin Inhibitor (Aprotinin)
Approximately 100 KIU/kg of aprotinin (Cayman, Ann Arbor, MI, USA) in saline solution was intraperitoneally injected into ICR mice three times per week for 2 years. The mice in the solvent group received saline solution.26,27
Treatment with a COX-2-Inhibitor (Celecoxib)
Approximately 10 mg/kg of celecoxib (Tocris Bioscience, Avonmouth, Bristol, UK) in 0.25% dimethyl sulfoxide (DMSO) was orally administered into each ICR mouse three times per week for 2 years. Mice in the solvent group received 0.25% DMSO solution.28
Open Field Test
The open field area (50 x 50 x 40 cm3) was made of plastic. Motor activity (distance [cm] moved by mice) was measured over a 15 min period using a video-tracking system (Smart2, Bio Research Center, Nagoya, Japan), and the data obtained was graphed.
Behavioral Assessment
The water maze test was adapted from the Morris method.29 Mice were given two blocks of training with four trials, every day during the test week. Each mouse remained on the platform for a 60 s interval. To evaluate the memory and learning ability of mice, we measured the swimming time (goal latency) from being placed in the water to reaching the platform in each trial. We measured the time taken to arrive at this platform and set the average of six mice to the mean escape latency.
Western Blot Analysis of the Brain Samples
Cerebrum samples were collected from all mice 2 years after treatment and were homogenized in lysis buffer (Kurabo, Osaka, Japan) and centrifuged at 8000 × g for 10 min. Western blot analysis was performed using previously described methods.30 The membranes were incubated at room temperature for 1 h with primary antibodies against amyloid precursor protein (1:1000; Abcam, Cambridge, MA, USA), amyloid-β (1:1000; Rockland Immunochemicals Inc., Gilbertsville, PA, USA), ionized calcium-binding adaptor molecule 1 (Iba1, marker of microglial and macrophage, 1:1000; Wako, Osaka, Japan), chemokine receptor 7 (CCR7, 1:1000; Abcam), CD80 (1:1000; Bioss Antibodies Inc., Woburn, MA, USA), CD163 (1:1000; Abcam), CD206 (1:1000; Abcam), plasmin (1:1000; Abgent, San Diego, CA, USA), urokinase-type plasminogen activator (uPA, 1:500; Abcam), and β-actin protein (1:5000; Sigma-Aldrich, St. Louis, MO, USA). The protein bands on the membranes were visualized following the incubation of membranes with horseradish peroxidase-conjugated secondary antibody (Novex, Frederick, MD, USA); band intensities were detected using the ImmunoStar Zeta regent (Wako, Osaka, Japan). The images of protein bands were acquired using the multi-grade software program (Fuji-film, Greenwood, SC, USA).
Quantification of IL-1β, IL-10, Tumor Necrosis Factor (TNF)-α, and Transforming Growth Factor (TGF)-α Proteins Using Enzyme-Linked Immunosorbent Assay (ELISA)
The levels of IL-1β, IL-10, TNF-α, and TGF-α in the brain were determined using commercial ELISA kits (IL-1β and IL-10: Abcam; TNF-α: RayBiotech Inc., Norcross, GA, USA; TGF-β: MyBioSource, San Diego, CA, USA) according to the respective manufacturer’s instructions.
Statistical Analysis
All the data were presented as means ± standard deviations. Results were statistically analyzed using one-way analysis of variance using Microsoft Excel 2010, followed by Tukey’s post hoc test performed using SPSS, version 20 (IBM, Armonk, NY, USA). Differences with p values <0.05 were considered statistically significant.
Results
Effect of Tranexamic Acid on the Behavior of Aging Mice
According to the open field test, after 2 years of treatment with tranexamic acid, the motor activity of mice increased (Figure 2A), while the mean escape latency in the Morris water maze decreased (Figure 2B).
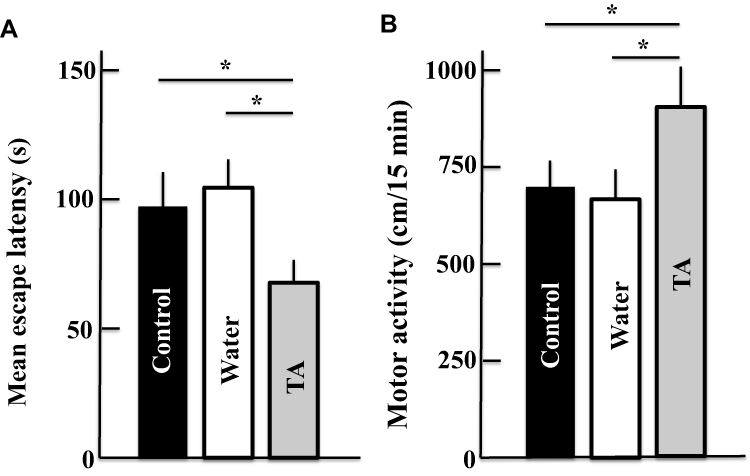 |
Figure 2 Effect of tranexamic acid administration on memory, learning, and motor activities of aging mice as determined by Morris water maze (A) and open field tests (B). The values are presented as means ± standard deviations using samples from six mice; *p < 0.05. Abbreviation: TA, tranexamic acid. |
Effect of Tranexamic Acid on the Expression of Plasmin and uPA in Aging Mice
The protein levels of plasmin, a target of tranexamic acid, and uPA decreased in tranexamic acid-treated aging mice (Figure 3).
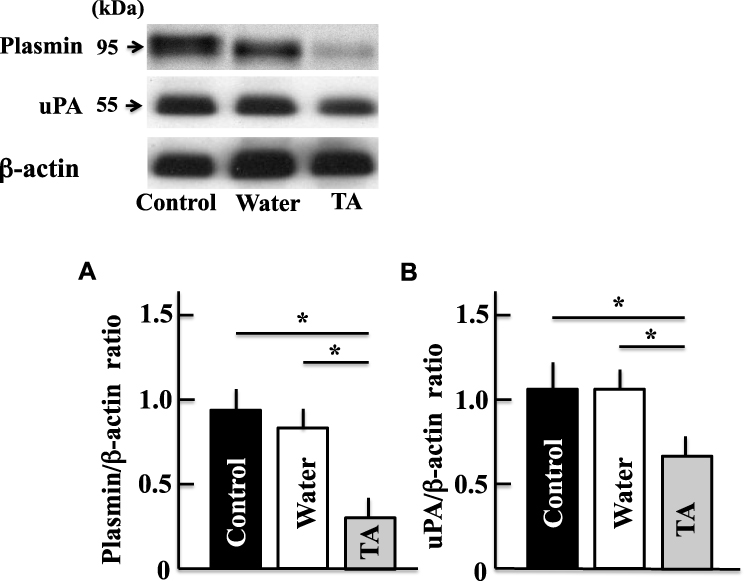 |
Figure 3 Effects of tranexamic acid on the expression of plasmin (A) and urokinase-type plasminogen activator (B). Two years after treatment, we measured the levels of plasmin and urokinase-type plasminogen activator in the mice brains by Western blotting analysis. The values are expressed as means ± standard deviations derived from the samples from six mice. *p < 0.05. Abbreviation: TA, tranexamic acid; uPA, urokinase-type plasminogen activator. |
Effect of Tranexamic Acid on the Expression of Amyloid Precursor and Amyloid-β Proteins in Aging Mice
The levels of amyloid precursor and amyloid-β proteins in the brains of mice treated with tranexamic acid decreased compared with those in the brains of untreated mice or mice treated with the solvent (Figure 4).
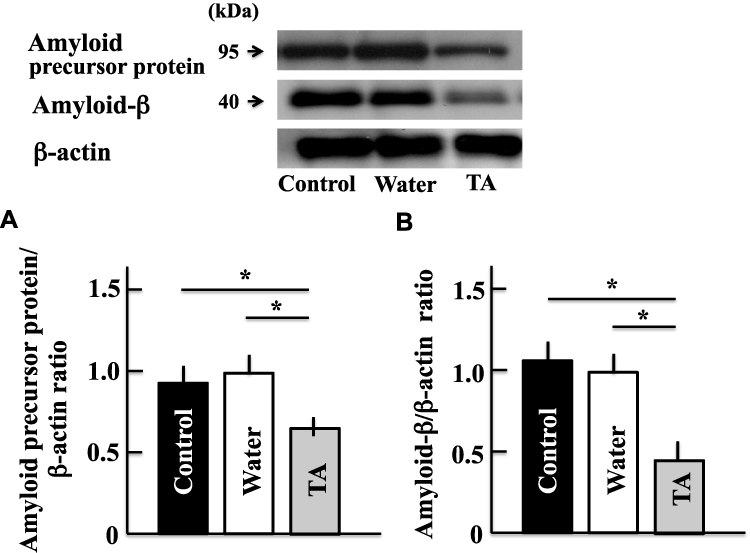 |
Figure 4 Effects of tranexamic acid on the expression of amyloid precursor (A) and amyloid-β (B) proteins. Two years after treatment, we measured the expression of amyloid precursor and amyloid-β proteins in the mice brains by Western blotting analysis. Values are expressed as means ± standard deviations derived from the samples from six mice. *p < 0.05. Abbreviation: TA, tranexamic acid. |
Effect of Tranexamic Acid on the Expression of Iba1, CCR7, CD80, CD163, and CD206 in Aging Mice
Although the expression of Iba1, which is a microglial (microglia are a type of brain macrophage) marker, did not change following the administration of tranexamic acid (Figure 5A), the levels of CCR7 and CD80, which are markers of M1-type macrophages, decreased in the treatment group than in the control group (Figure 5B and C). However, the expression of CD163 and CD206, which are markers of M2-type macrophages, increased in mice from the tranexamic acid group compared with that in mice from the control group (Figure 5D and E).
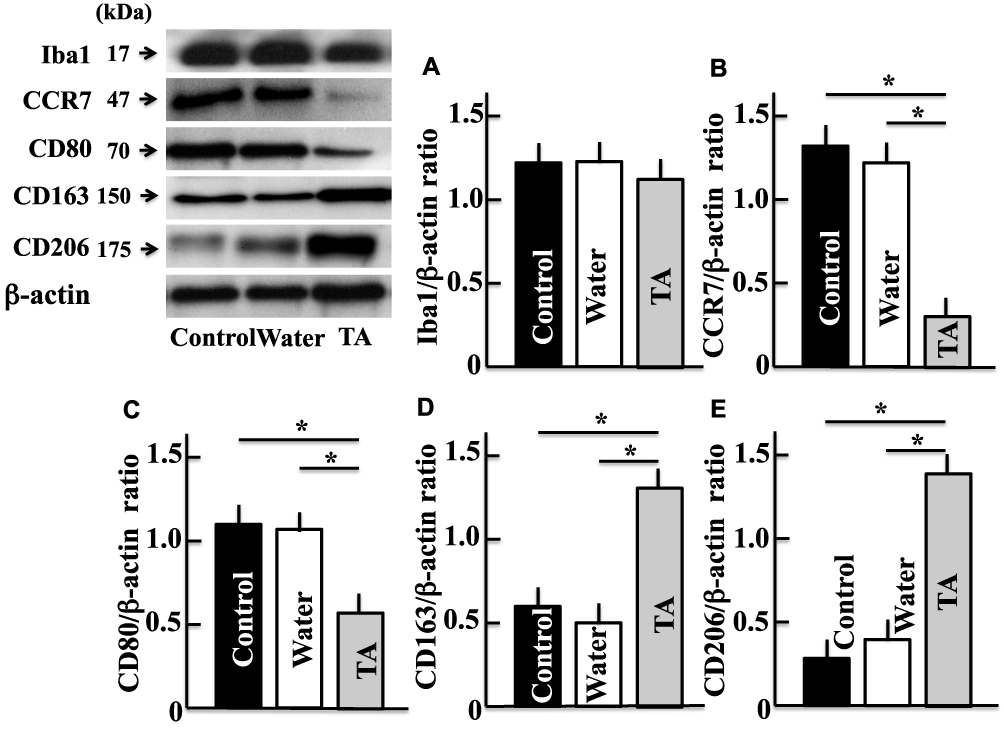 |
Figure 5 Effects of tranexamic acid on the expression of IbA microglia marker (A), CCR7 (B), CD80 (C), CD163 (D), and CD206 (E). Two years after treatment, we measured the levels of IbA, CCR7, CD80, CD163, and CD206 in mice brains by Western blotting analysis. Values are expressed as means ± standard deviations derived from the samples from six mice. *p < 0.05. Abbreviation: TA, tranexamic acid. |
Effect of Tranexamic Acid on the Levels of IL-1β, TNF-α, IL-10, and TGF-β in the Brains of Aging Mice
In the brains of mice treated with tranexamic acid, the levels of inflammatory cytokines such as IL-1β and TNF-α (Figure 6A and B) decreased, while those of the anti-inflammatory cytokines such as IL-10 and TGF-β increased (Figure 6C and D), when compared to those in the brains of mice from the control group.
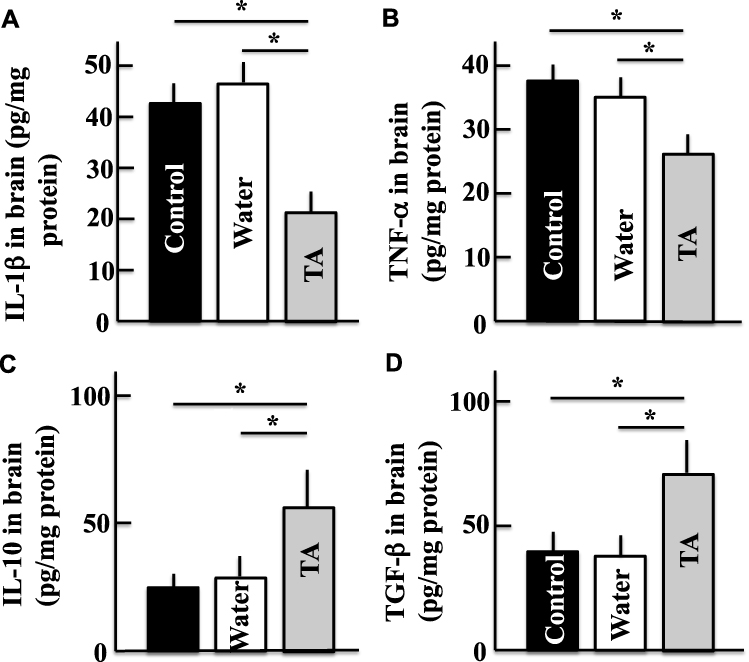 |
Figure 6 Effects of tranexamic acid on the levels of IL-1β (A), TNF-α (B), IL-10 (C), and TGF-β (D) in the mice brains. Two years after treatment, we measured the concentrations of IL-1β, TNF-α, IL-10, and TGF-β in the mice brains using ELISA kits. Values are expressed as means ± standard deviations derived from the samples from 10 mice. *p < 0.05. Abbreviation: TA, tranexamic acid. |
Effect of Aprotinin on the Expression of Amyloid-β and on the Memory and Learning Abilities of Aging Mice
After the administration of aprotinin, the decline in memory and learning abilities in aging mice was ameliorated to levels similar to those observed following treatment with tranexamic acid (Figure 7A). Similar to mice from the tranexamic acid-treated group, the aprotinin-treated group had reduced expression of amyloid-β protein in the brain compared with the control group (Figure 7B).
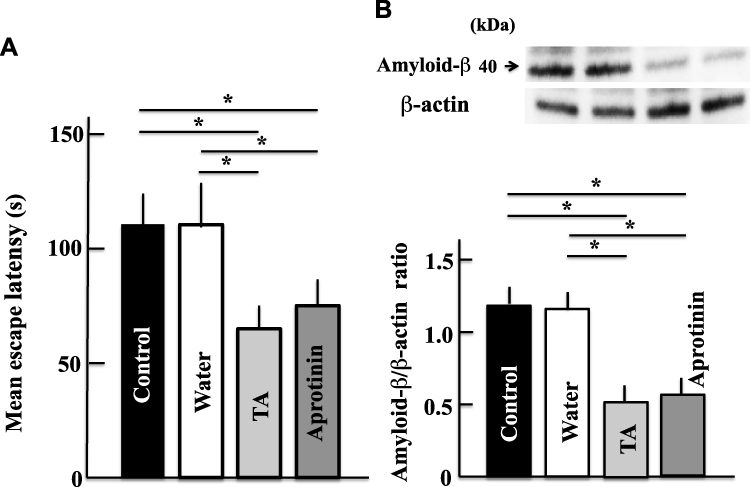 |
Figure 7 Effects of aprotinin (plasmin inhibitor) on memory and learning abilities (A) and expression of amyloid-β (B) in aging mice. Two years after treatment, we measured the memory and learning abilities and levels of amyloid-β in the brain by the Morris water maze test and Western blot analysis, respectively. Values are expressed as means ± standard deviations derived from measurements from 10 mice. *p < 0.05. Abbreviation: TA, tranexamic acid. |
Effect of Celecoxib on the Expression of Amyloid-β and on the Memory and Learning Abilities of Aging Mice
Memory and learning abilities as well as the expression of amyloid-β were not significantly different between the celecoxib group and control group (Figure 8A). However, a statistically significant increase in these parameters were noted in the celecoxib-treated mice compared with that in the tranexamic acid-treated mice (Figure 8B).
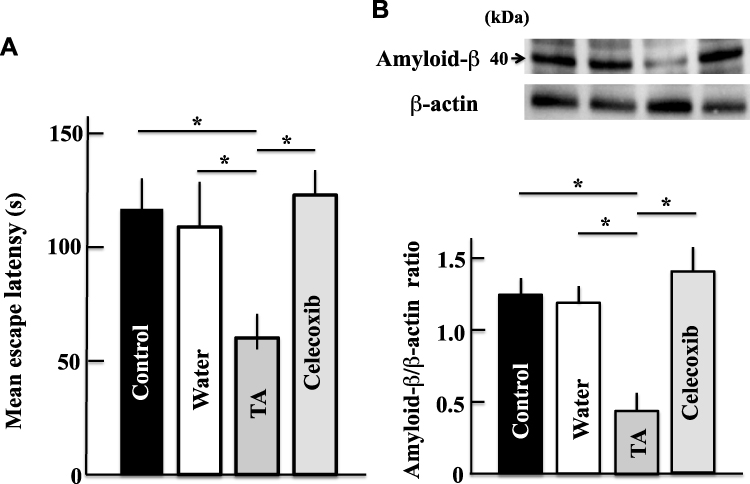 |
Figure 8 Effects of celecoxib (COX-2 inhibitor) on the memory and learning abilities (A) and expression of amyloid-β (B) in aging mice. Two years after treatment, we measured the memory and learning abilities and levels of amyloid-β using the Morris water maze test and Western blot analysis, respectively. Values are expressed as means ± standard deviations derived from measurements from 10 mice. *p < 0.05. Abbreviation: TA, tranexamic acid. |
Discussion
In this study, treatment of mice with tranexamic acid for 2 years ameliorated aging-induced decline in memory and learning abilities. We also observed that the expression of amyloid-β proteins decreased in tranexamic acid-treated mice. However, the expression of CCR7, CD80, IL-1β, and TNF-α in mice brains decreased following tranexamic acid administration, while that of CD163, CD206, IL-10, and TGF-β increased. Amelioration of aging-induced decline in memory and learning abilities in mice was also observed after aprotinin administration, as opposed to that after celecoxib administration.
Neurodegenerative diseases such as AD and Parkinson’s disease alter the normal physiological conditions of the immune system.31 A continuous accelerated production of inflammatory cytokines or free radicals from microglia due to the malfunction of the immune system is observed during chronic inflammatory conditions. In the nervous system, microglia, a type of brain macrophages,32 are activated by interferon-γ when the inflammatory cytokines such as amyloid-β and free radicals are produced.33–35 When the inflammatory cytokines secreted by the microglia affect the neuronal tissues by inducing a histological change,17,36,37 the hypothalamic-pituitary-adrenal system gets activated. If high corticoid levels are maintained in the blood for a long period, a decrease in the level of brain-derived neurotropic factor and the inhibition of hippocampal neurogenesis may occur.38,39 Thus, inflammatory cytokines are involved in the decline in memory and learning abilities.
Chronic inflammatory conditions are induced by continuous ROS secretion during aging.40 Moreover, it was reported that aging considerably increased the IL-6 and TNF-α levels in cells.41 Thus, ROS, IL-6, and TNF-α play important roles in chronic inflammatory conditions or aging. Tranexamic acid inhibits plasmin production.42 Plasmin processes the cytokines via matrix metalloproteinase (MMP)-9 and activates inflammatory reactions.43 Furthermore, plasmin has an important role in migration and infiltration of inflammatory cells after the degradation of the extracellular matrix by MMP activation.44 Thus, the increase in the levels of inflammatory cytokines induced by plasmin promotes the accumulation of amyloid-β. However, tranexamic acid inhibits the production of inflammatory cytokines by inhibiting the production of plasmin, thereby eliciting protective effects against the accumulation of amyloid-β.
Additionally, plasmin is directly involved in the activation of macrophages.45 Here, as cytokine secretion by brain microglia was modulated by tranexamic acid administration, we suggest that plasmin activates the microglia. Furthermore, in this study, the expression of M1 macrophage markers (CCR7 and CD80) was reduced with the administration of tranexamic acid. M1 macrophages secrete inflammatory cytokines, such as TNF-α, IL-6, and IL-12, thereby promoting inflammation.46,47 Conversely, the expression of M2 macrophage markers (CD163 and CD206) increased after tranexamic acid administration. M2 macrophages secrete anti-inflammatory cytokines, such as TGF-β and IL-10, thereby inhibiting inflammation.48 Importantly, we hypothesized that by altering the M1/M2 macrophage balance, the denaturation of neurons could be controlled. The decline in memory and learning abilities due to aging was suppressed by the inhibitory effect of tranexamic acid on the actions of plasmin. Recently, Ni et al reported that the conversion of M1/M2 polarization was regulated by a lysosomal enzyme, cathepsin E, and its switch, cathepsin B.49 However, the plasmin-mediated regulation of M1/M2 conversion requires further research.
Furthermore, plasmin inhibits 14-arachidonic acid and induces the production of prostaglandin E2.50 However, in our study, tranexamic acid exerted an anti-inflammatory effect by inhibiting the production of the prostaglandin E2. Moreover, mice administered with celecoxib, an anti-inflammatory agent, inhibited COX-2-mediated prostaglandin E2 production, impaired memory and learning abilities, and favored amyloid-β accumulation. A similar effect, as observed with tranexamic acid administration, was seen in the experiment of plasmin inhibition after aprotinin administration. Therefore, we suggest that the anti-inflammatory effect caused by COX-2 inhibition was not related to the improvement in memory and learning abilities caused by tranexamic acid.
Conclusions
In this study, we observed that tranexamic acid ameliorated aging-induced decline in memory and learning abilities of aging mice by inhibiting the synthesis of plasmin and decreasing the levels of chronic inflammation and activity of microglia (M2 macrophages) in the brain. Therefore, the origin of aging is related to chronic inflammation. As tranexamic acid suppresses chronic inflammation, it may suppress neurodegeneration, which occurs owing to aging. Thus, it may be effective against the decline in memory and learning abilities associated with aging (Figure 9). Further studies should be conducted to elucidate the relationship between plasmin inhibitors and switching of M1 to M2 by microglia, which could lead to the development of novel therapeutic methods against neurodegenerative conditions associated with aging.
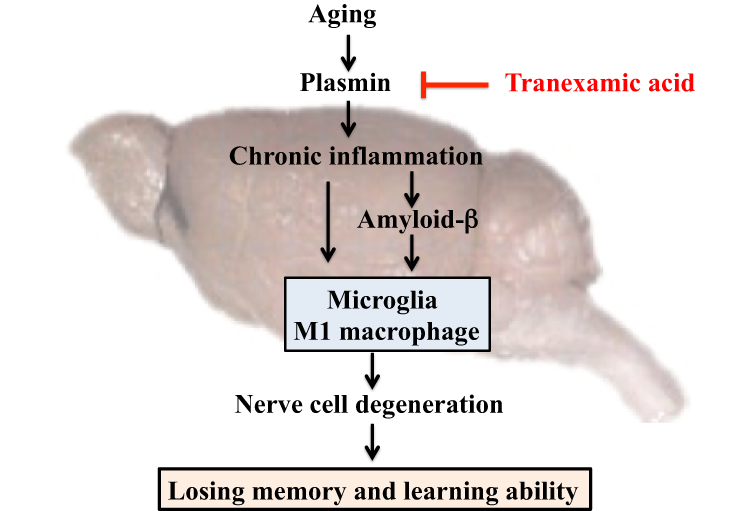 |
Figure 9 Mechanism of the effect of tranexamic acid on the decline of aging-induced memory and learning abilities in mice. |
Acknowledgments
This study was supported by the grants obtained from JSPS KAKENHI (grant number: 18K11085) and Daiichi Sankyo Healthcare Co., Ltd.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest associated with this study.
References
1. McEntee WJ, Crook TH. Glutamate: its role in learning, memory, and the aging brain. Psychopharmacology. 1993;111(4):391–401.
2. Papenberg G, Li S-C, Nagel IE, et al. Dopamine and glutamate receptor genes interactively influence episodic memory in old age. Neurobiol Aging. 2014;35(5):1213. doi:10.1016/j.neurobiolaging.2013.11.014
3. Nyberg L, Karalija N, Salami A, et al. Dopamine D2 receptor availability is linked to hippocampal-caudate functional connectivity and episodic memory. Proc Natl Acad Sci U S A. 2016;113(28):7918–7923.
4. Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278:412–419.
5. Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60:1495–1500.
6. Jiang CH, Tsien JZ, Schultz PG, Hu Y. The effects of aging on gene expression in the hypothalamus and cortex of mice. Proc Natl Acad Sci USA. 2001;98(4):1930–1934.
7. Lu T, Pan Y, Kao A-Y, et al. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429(6994):883–891.
8. Yamazaki D, Horiuchi J, Ueno K, et al. Glial dysfunction causes age-related memory impairment in Drosophila. Neuron. 2014;84(4):753–763.
9. Becher E, Orellana Rios CL, Lahmann C, et al. Anxiety as a risk factor of Alzheimer’s disease and vascular dementia. Br J Psychiatry. 2018;213(5):654–660.
10. Shabir O, Berwick J, Francis SE. Neurovascular dysfunction in vascular dementia, Alzheimer’s and atherosclerosis. BMC Neurosci. 2018;19(1):62. doi:10.1186/s12868-018-0465-5
11. Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368(9533):387–403. doi:10.1016/S0140-6736(06)69113-7
12. Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Aβ accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging. 2005;26(9):1235–1244. doi:10.1016/j.neurobiolaging.2005.05.022
13. Holtzman DM, Morris JC, Goate AM. Alzheimer’s Disease: the Challenge of the Second Century. Sci Trans Med. 2011;3(77):77sr1. doi:10.1126/scitranslmed.3002369
14. Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10(9):698–712.
15. Francis PT, Palmer AM, Snape M, Wilcock GK. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66(2):137–147. doi:10.1136/jnnp.66.2.137
16. Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J Immunol. 1993;151(4):2132–2141.
17. Li J, Baud O, Vartanian T, Volpe JJ, Rosenberg PA. Peroxynitrite generated by inducible nitric oxide synthase and NADPH oxidase mediates microglial toxicity to oligodendrocytes. Proc Natl Acad Sci U S A. 2005;102(28):9936–9941. doi:10.1073/pnas.0502552102
18. Anisman H. Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J Psychiatry Neurosci. 2009;34(1):4–20.
19. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732–741. doi:10.1016/j.biopsych.2008.11.029
20. Dowlati Y, Herrmann N, Swardfager W, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67(5):446–457. doi:10.1016/j.biopsych.2009.09.033
21. Okunishi K, Sisson TH, Huang SK, et al. Plasmin Overcomes Resistance to Prostaglandin E 2 in Fibrotic Lung Fibroblasts by Reorganizing Protein Kinase A Signaling. J Biol Chem. 2011;286(37):32231–32243. doi:10.1074/jbc.M111.235606
22. Isseroff RR, Rifkin DB. Plasminogen is present in the basal layer of the epidermis. J Invest Dermatol. 1983;80(4):297–299. doi:10.1111/1523-1747.ep12534677
23. Hiramoto K, Yamate Y, Sugiyama D, Matsuda K, Iizuka Y, Yamaguchi T. Tranexamic acid inhibits the plasma and non-irradiated skin markers of photoaging induced by long-term UVA eye irradiation in female mice. Biomed Pharmacother. 2018;107:54–58. doi:10.1016/j.biopha.2018.07.146
24. Hiramoto K, Yamate Y, Sugiyama D, et al. Effect of tranexamic acid in improving the lifespan of naturally aging mice. Inflammopharmacology. 2019;27(6):1319–1323.
25. Boudreau RM, Johnson M, Veile R, et al. Impact of tranexamic acid on coagulation and inflammation in murine models of traumatic brain injury and hemorrhage. J Surg Res. 2017;215:47–54.
26. Asimakopoulos G, Thompson R, Nourshargh S, et al. An anti-inflammatory property of aprotinin detected at the level of leukocyte extravasation. J Thorac Cardiovasc Surg. 2000;120(2):361–369.
27. Reichel CA, Lerchenberger M, Uhl B, et al. Plasmin inhibitors prevent leukocyte accumulation and remodeling events in the postischemic microvasculature. PLoS One. 2011;6(2):e17229.
28. Yoshino T, Kimoto A, Kobayashi S, et al. Pharmacological profile of celecoxib, a specific cyclooxygenase-2 inhibitor. Arzneimittelforschung. 2005;55(7):394–402.
29. Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60.
30. Hiramoto K, Sugiyama D, Takahashi Y, Mafune E. The amelioration effect of tranexamic acid in wrinkles induced by skin dryness. Biomed Pharmacother. 2016;80:16–22.
31. Monji A. The neuroinflammation hypothesis of psychiatric disorders. Psychiatria Et Neurologia Japonica. 2012;114(2):124–133.
32. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394.
33. Hwang J, Zheng LT, Ock J, et al. Inhibition of glial inflammatory activation and neurotoxicity by tricycle antidepressants. Neuropharmacology. 2008;55(5):826–834.
34. Lim CM, Kim SW, Park JY, et al. Fluoxetine affords robust neuroprotection in the postischemic brain via its anti-inflammatory effect. J Neurosci Res. 2009;87(4):1037–1045.
35. O’Sullivan JB, Ryan KM, Curtin NM, Harkin A, Connor TJ. Noradrenaline reuptake inhibitors limit neuroinflammation in rat cortex following a systemic inflammatory challenge: implications for depression and neurodegeneration. Int J Neuropsychopharmacol. 2009;12(5):687–699.
36. Buntinx M, Moreels M, Vandenabeele F, et al. Cytokine-induced cell death in human oligodendroglial cell lines: I. Synergistic effects of IFN-gamma and TNF-alpha on apoptosis. J Neurosci Res. 2004;76(6):834–845.
37. Buntinx M, Gielen E, Van Hummelen P, et al. Cytokine-induced cell death in human oligodendroglial cell lines. II: alterations in gene expression induced by interferon-gamma and tumor necrosis factor-alpha. J Neurosci Res. 2004;76(6):846–861.
38. Leonard BE. Inflammation, depression and dementia: are they connected? Neurochem Res. 2007;32(10):1749–1756.
39. Caraci F, Copani A, Nicoletti F, Depression DF. Alzheimer’s disease: neurobiological links and common pharmacological targets. Eur J Pharmacol. 2010;626(1):64–71.
40. Shimizu T, Marusawa H. Role of inflammation in the development of gastrointestinal cancers. Nihon Shokakibyo Gakkai Zasshi. 2011;108(8):1374–1382.
41. Neurath MF, Finotto SIL. 6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev. 2011;22(2):83–89.
42. Ng WC, Jerath A, Wasowicz M. Tranexamic acid: a clinical review. Anaesthesiol Intensive Ther. 2015;47(4):339–350.
43. Tsuji K, Aoki T, Tejima E, et al. Tissue plasminogen activator promotes matrix metalloproteinase-9 upregulation after focal cerebral ischemia. Stroke. 2005;36(9):1954–1959.
44. Gong Y, Hart E, Shchurin A, Hoover-Plow J. Inflammatory macrophage migration requires MMP-9 activation by plasminogen in mice. J Clin Invest. 2008;118(9):3012–3024.
45. Li Q, Ke F, Zhang W, et al. Plasmin plays an essential role in amplification of psoriasiform skin inflammation in mice. PLoS One. 2011;6(2):e16483.
46. Kawanokuchi J, Mizuno T, Takeuchi H, et al. Production of interferon-gamma by microglia. Mult Scler. 2006;12(5):558–564.
47. Takeuchi H, Jin S, Wang J, et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281(30):21362–21368.
48. Takeda A, Yasuda T, Miyata T, et al. Advanced glycation end products co-localized with astrocytes and microglial cells in Alzheimer’s disease brain. Acta Neuropathol. 1998;95(6):555–558.
49. Ni J, Wu Z, Peterts C, et al. The critical role of proteolytic relay through cathepsins B and E in the phenotypic change of microglia/macrophage. J Neurosci. 2015;35(36):12488–12501.
50. Nakano T, Fujita H, Kikuchi N, Arita H. Plasmin converts pro-form of group I phospholipase A2 into receptor binding, active forms. Biochem Biophys Res Commun. 1994;198(1):10–15.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.