")
Back to Journals » OncoTargets and Therapy » Volume 13
TP53INP2 Modulates Epithelial-to-Mesenchymal Transition via the GSK-3β/β-Catenin/Snail1 Pathway in Bladder Cancer Cells
Authors Zhou Z, Liu X, Li Y, Li J, Deng W, Zhong J, Chen L, Li Y, Zeng X, Wang G, Zhu J, Fu B
Received 28 February 2020
Accepted for publication 16 August 2020
Published 28 September 2020 Volume 2020:13 Pages 9587—9597
DOI https://doi.org/10.2147/OTT.S251830
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjay Singh
Zhengtao Zhou,1,2,* Xiaoqiang Liu,1,2,* Yulei Li,1 Junhua Li,3 Wen Deng,1 Jian Zhong,4 Luyao Chen,1 Yu Li,1 Xiantao Zeng,5 Gongxian Wang,1,2 Jingyu Zhu,3 Bin Fu1,2
1Department of Urology, The First Affiliated Hospital of Nanchang University, Nanchang, People’s Republic of China; 2Jiangxi Institute of Urology, Nanchang, People’s Republic of China; 3Department of Urology, Third Hospital of Hangzhou, Hangzhou, People’s Republic of China; 4Department of Surgery, Nankang District Chinese Medicine Hospital, Ganzhou, People’s Republic of China; 5Department of Urology, Zhongnan Hospital of Wuhan University, Wuhan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Bin Fu
Department of Urology, The First Affiliated Hospital of Nanchang University, 17 Yongwaizheng Street, Nanchang, Jiangxi 330006, People’s Republic of China
Tel +86 13879103861
Email [email protected]
Jingyu Zhu
Department of Urology, Third Hospital of Hangzhou, 38 Westlake Road, Hangzhou, Zhejiang 310009, People’s Republic of China
Tel +86 571 87827269
Email [email protected]
Background: The tumor protein p53-inducible nuclear protein 2 (TP53INP2), an autophagy protein, is essential for autophagosome formation. The deregulation of autophagy is associated with multiple human diseases, including cancer. The present study aims to explore the role of TP53INP2 in bladder cancer.
Materials and Methods: Quantitative real-time polymerase chain reaction was used to detect the mRNA level. Relative TP53INP2 protein expression was detected by immunohistochemistry and Western blot. The effect of TP53INP2 silencing on the proliferation, migration, and invasion of bladder cancer cells was investigated by CCK-8 detection kit and transwell assay. In addition, transfection and immunofluorescence were performed.
Results: In this study, we report that high expression of TP53INP2 is correlated with poor patient survival in bladder cancer. Results demonstrate that the depletion of TP53INP2 inhibits the migration, invasion, and epithelial-to-mesenchymal transition (EMT) of bladder cancer cells. The underlying mechanism was explored. Results show that the TP53INP2 knockdown suppresses EMT by inhibiting the active non-phosphorylated β-catenin and decreasing the Snail1 levels. Furthermore, the glycogen synthase kinase-3 beta (GSK-3β) inhibitor IM-12 abrogates the effect of TP53INP2 silencing. Interestingly, the induction of autophagy partially abrogates the TP53INP2 knockdown-induced decrease in active β-catenin and inhibition of migration and invasion in bladder cancer cells.
Conclusion: In summary, our results show that the downregulation of TP53INP2 inhibits EMT via the GSK-3β/β-catenin/Snail1 pathway in bladder cancer. The findings of this study uncover the novel role of TP53INP2 and offer new insights into bladder cancer clinical therapy.
Keywords: TP53INP2, β-catenin, Snail1, EMT, bladder cancer
Introduction
Bladder cancer (BC) is the fourth most common cancer in men and is the first cause of death among human urologic cancers in the United States, which has approximately 62,380 new cases and 12,520 deaths in 2018.1 BC is highly recurrent and progressive. Approximately 50% of patients with nonmuscle-invasive BC (Ta, T1) experience cancer recurrence and 20% of patients progress to muscle-invasive BC (MIBC).2 Patients diagnosed with MIBC have an unfavorable prognosis and a 5-year overall survival period of approximately 50%.3 Despite the crucial advances in BC research, the mortality rate of BC remains unchanged in decades due to the lack of specific targets.4
Epithelial-to-mesenchymal transition (EMT) is a basic process by which cells of epithelial lineage lose epithelial traits and undergo conversion to a mesenchymal lineage, thereby reducing intercellular adhesion and increasing cell motility. EMT plays a critical role in normal developmental processes and confers metastatic properties by promoting the mobility and invasion of cancer cells.5 EMT is also associated with the generation of cancer stem cells, which may explain BC recurrence and metastasis.6
The tumor protein p53-inducible nuclear protein 2 (TP53INP2), which is also called diabetes- and obesity-related gene (DOR), is a bifunctional protein that regulates transcription and enhances starvation-induced autophagy. TP53INP2 is a nuclear protein that regulates thyroid hormone-related gene expression by functioning as a transcriptional coactivator of the thyroid hormone receptor. TP53INP2 promotes rDNA transcription by facilitating the assembly of POLR1/RNA polymerase I preinitiation complex at rDNA promoters.7,8 Upon starvation and rapamycin treatment, nuclear TP53INP2 directly interacts with nuclear-deacetylated LC3 and relocates together with LC3 from the nucleus to the cytoplasm. TP53INP2 then interacts with the transmembrane protein VMP1 and contributes to autophagosome formation.9,10
TP53INP2 also regulates cell migration,11 but its underlying molecular mechanism remains unknown. In this study, we demonstrated that the downregulation of TP53INP2 inhibits migration, invasion, and EMT partly via the glycogen synthase kinase-3 beta (GSK-3β)/β-catenin/Snail1 pathway in BC. Furthermore, the TP53INP2 silencing-induced decrease of active β-catenin and inhibition of migration and invasion are dependent on autophagy.
Materials and Methods
Cell Culture and Treatment
The human BC cell lines 5637, EJ, BIU-87, UM-UC-3, and T24 and the human immortalized uroepithelium cell line SV-HUC-1 were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences, Shanghai Institute of Cell Biology (Shanghai, China). The 5637, EJ, and BIU87 cells were maintained in RPMI-1640 (Gibco). The T24 and UM-UC-3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco), and the SV-HUC-1 cells were cultured in the F-12K medium (Gibco). These media were supplemented with 10% heat-inactivated fetal bovine serum (FBS, Hyclone), 100 U/mL penicillin, and 100 µg/mL streptomycin and placed in a humidified incubator with 5% CO2 maintained at 37°C.
Transfection
The EJ and the BIU87 cells were seeded into 6-well plates at a density of 2 × 105 cells per well. The TP53INP2 siRNA and the negative control siRNA (siNC) were obtained from RiboBio (Guangzhou, China). The sense strand sequences of siRNAs were as follows: siTP53INP2-1#, 5′-GACGAGAGCUGGUUUGUUATT-3′; siTP53INP2-2#, 5′-CCUUACAUGUCUCACACUATT-3′; and siNC, 5′-UUCUCCGAACGUGUCACGUTT-3′. The EJ and the BIU87 cells were grown to 70–80% confluence and transfected using the Lipofectamine™ 2000 transfection reagent (Invitrogen, USA) and the Opti-MEM medium (Gibco) in accordance with the manufacturer’s instructions.
RNA Extraction and Real-Time Quantitative RT-PCR (qRT-PCR) Analysis
At 48 h after transfection, the total RNA was extracted from the EJ and the BIU87 cells by using the TRIzol reagent (Invitrogen). cDNA was synthesized using the Takara PrimeScript RT Reagent Kit (Takara). For qRT-PCR, 400 ng total RNA was used in the Takara PrimeScript RT reagent Kit (Takara) following the manufacturer’s instructions, and PCR was performed in the ABI PRISM 7500 real-time PCR system (Applied Biosystems). The expression level of β-actin was used as the internal control. All reactions were performed at least in triplicate. The oligonucleotide sequences of the qRT-PCR primers were TP53INP2 forward (5′-CGGCTGGCTCATCATTGAC-3′) and TP53INP2 reverse (5′-CAGCTCTCGTCCATCAAGGA-3′).
Western Blot
Protein extraction and Western blot were performed as previously described.12 Antibodies targeting GAPDH (Abcam, ab8245, 1:1000), β-actin (CST, 4970, 1:1000), TP531NP2 (Biovision, A1169-100, 1:500), Vimentin (CST, 5741, 1:1000), E-cadherin (CST, 3195, 1:1000), N-cadherin (CST, 13,116, 1:1000), β-catenin (CST, 8480, 1:1000), non-phospho (active) β-catenin (CST, 19,807, 1:1000), Snail1 (Affinity, AF6032, 1:500), and Histone H3 (Affinity, AF0863, 1:500) were used with the secondary antibody horseradish peroxidase-labeled goat antirabbit (CST, 7074, 1:4000) or anti-mouse (CST, 7076, 1:4000) and incubated at room temperature for 1 h. The protein bands were visualized using enhanced chemiluminescence.
Cell Viability Assay
The effects of siTP53INP2 on the viability of the EJ and the BIU87 cells were assessed using the CCK-8 detection kit (Keygen, Nanjing, China). The cells transfected with TP53INP2 siRNA for 72 h were seeded into a 96-well plate at a density of 3000 cells per well. The cells were incubated at 37°C for 24, 48, 72, and 96 h. The cells were then added with the CCK-8 solution and incubated at 37°C for 2 h. Absorbance was measured at 450 nm.
Immunohistochemistry
For immunohistochemical analysis, 16 bladder cancer samples and paired adjacent non-tumor tissues were formalin-fixed and paraffin-embedded. The immunohistochemical staining of paraffin-embedded bladder cancer tissue sections using primary antibodies against TP53INP2 (Bioworld, BS61399) was performed as previously described.13 Briefly, after dewaxing, rehydration, antigen retrieval, and blocking, the sections were incubated overnight at 4°C with the TP53INP2 antibody. Color detection was performed by DAB and hematoxylin counterstaining. Stained tumor and adjacent nontumor tissues were classified into two groups (low and high) in accordance with the staining intensity of each tissue.
Immunofluorescence
At 24 h following transfection, the cells cultured on glass coverslips were fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.01% Triton X-100 for 20 min at room temperature, blocked with 5% goat serum for 1 h at 37°C, and incubated with primary antibodies against non-phospho (active) β-catenin (CST, 19,807, 1:100) and Snail1 (Affinity, AF6032, 1:100) overnight at 4°C. The secondary antibodies were Alexa 488-conjugated goat anti-mouse or anti-rabbit IgG (Abcam). 4′,6-diamidino-2-phenylindole (10 µg/mL, 32,670; Sigma-Aldrich) was used to counterstain the nuclei. Fluorescent images were obtained under a microscope (Carl Zeiss, Germany) and processed using Photoshop software (Adobe).
Cell Migration and Invasion Assay
The EJ and the BIU87 cells seeded into 6-well plates were transfected with siTP53INP2 or nontarget control for 48 h and then seeded into the upper chamber of the transwell system. For the migration assay, the prepared cells were seeded into the upper chamber with serum-free medium. The medium of the lower chamber was supplemented with 10% FBS as a chemoattractant. The cells were incubated for 24 h and fixed with 4% formaldehyde. The cells that did not migrate through the pores were removed using a cotton swab. The cells that migrated to the lower surface of the membrane in the transwell system were stained with crystal violet. The migrated cells on the underside of the membrane were counted. For the invasion assay, the upper chamber of the insert was precoated with 0.1 mL Matrigel matrix (300 μg/mL, Corning) following the above procedures. Each experiment was conducted in triplicate. Finally, five representative fields at 100× magnification were randomly imaged.
Statistical Analysis
All data are presented as mean ± SD of at least three replicates per group. Statistical analysis was performed to determine the significance of the difference between groups by using ANOVA or Student’s t-test. All statistical analyses were performed using the GraphPad/Prism software for Windows. p < 0.05 was considered statistically significant.
Results
TP53INP2 is Overexpressed in the Human BC Tissue and Cell Lines
To evaluate the involvement of TP53INP2 in BC, we analyzed the TP53INP2 expression levels in 16 pairs of BC tissues and adjacent nontumor tissues by using qRT-PCR. As shown in Figure 1A, the relative expression levels of TP53INP2 were increased in the BC samples. Next, we tested the TP53INP2 level in 16 BC tissues and paired 16 adjacent non-tumor tissues by immunohistochemistry. As presented in Figure 1B, TP53INP2 was highly expressed in 10/16 (62.5%) BC and 4/16 (25%) adjacent nontumor tissues (p < 0.05). Subsequently, we detected the TP53INP2 expression in the BC cell lines. Consistently, the TP53INP2 expression was significantly upregulated in the invasive BC cell lines (EJ, BIU87, UM-UC-3, and T24 cells) relative to the normal urothelial cell line SV-HUC-1, but unchanged in the non-invasive bladder cancer cell line (5637) (Figure 1C). To further investigate whether TP53INP2 expression levels were correlated with the prognosis of patients with BC, we searched The Cancer Genome Atlas (TCGA) database by using Gene Expression Profiling Interactive Analysis (GEPIA) and found that the high TP53INP2 expression was correlated with the poor prognosis of patients with BC (Figure 1D).14
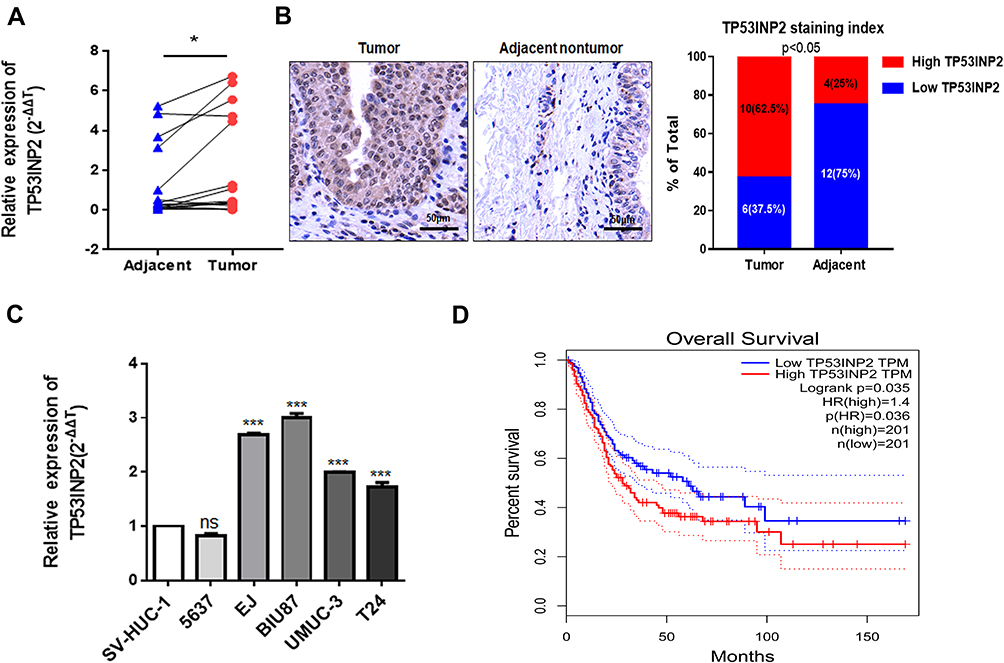 |
Figure 1 TP53INP2 expression in bladder cancer tissues and cells. Notes: (A) The relative expression quantities of TP53INP2 in 16 pairs of bladder cancer tissues and paired adjacent nontumor tissues were detected using qRT-PCR. (B) IHC staining of TP53INP2 in 16 BC tissues and paired 16 adjacent non-tumor tissues obtained from patients with bladder cancer. Scale bar, 50 μm. (C) The TP53INP2 expression levels were detected in the human normal urothelial cell line SV-HUC-1 and in five types of human bladder cancer cell lines, including 5637, EJ, BIU87, UM-UC-3, and T24, via qRT-PCR. (D) Overall survival of the 402 bladder cancer patients was analyzed in accordance with the TP53INP2 expression level in the TCGA database by GEPIA. **** p < 0.001; * p < 0.05. Abbreviations: qRT-PCR, real-time quantitative PCR; IHC, immunohistochemistry; BC, bladder cancer; TCGA, the cancer genome atlas; GEPIA, gene expression profiling interactive analysis; ns, no statistical significance. |
TP53INP2 Plays a Role in the Migration and Invasion of BC Cells
To investigate the role of TP53INP2 in BC cells, we knocked down TP53INP2 in two BC cell lines, namely, BIU87 and EJ (Figure 2A). Subsequently, we examined the role of TP53INP2 in the proliferation, migration, and invasion of BC cells by conducting cell viability and transwell assays. However, as shown in Figure 2B, when TP53INP2 was silenced, the growth of the BIU87 and EJ cells remained unchanged. However, the TP53INP2 knockdown significantly inhibited cell migration and invasion (Figure 2C and D).
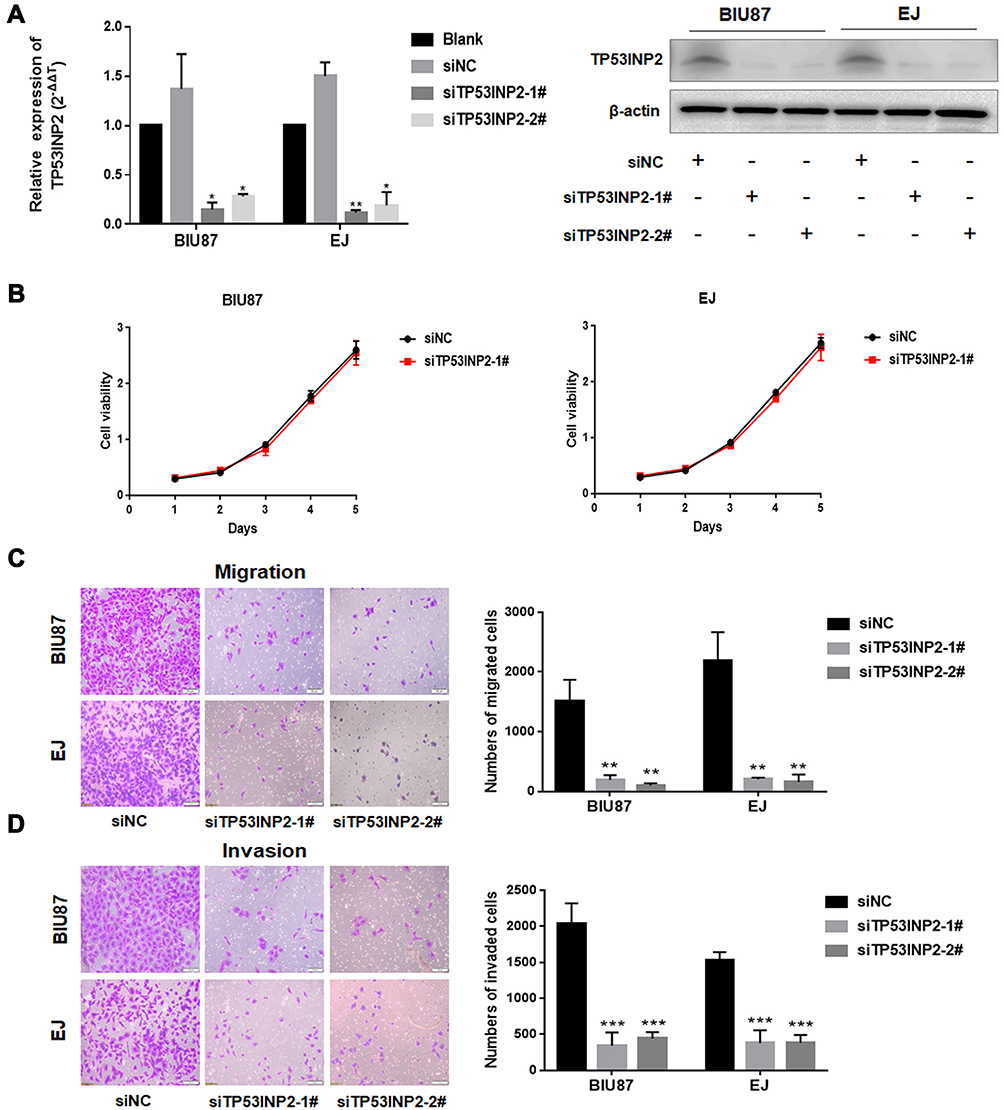 |
Figure 2 Effects of TP53INP2 knockdown on the proliferative, migrative, and invasive capabilities of bladder cancer cells. Notes: (A) TP53INP2 siRNA were transiently transfected into the BIU87 and EJ human bladder cancer cells. The cells were incubated for 48 h and subjected to qRT-PCR, and Western blot was performed to detect the efficiency of interference. (B) The CCK-8 assay was conducted to detect cell viability. (C, D) The transwell assay was performed to detect the changes in bladder cell migrative and invasive capabilities after interference in TP53INP2. Scale bar, 50 μm. *** p < 0.001; ** p < 0.01; * p < 0.05.Abbreviations: qRT-PCR, real-time quantitative PCR; CCK-8, cell counting kit-8. |
TP53INP2 Downregulation Inhibits EMT via the β-Catenin/Snail1 Pathway in BC Cells
EMT plays an important role in the invasion and metastasis of many tumor types.5 We speculated that TP53INP2 knockdown affected EMT and contributed to the reduction of BC cell migration and invasion. We examined the effect of TP53INP2 knockdown on EMT. As shown in Figure 3A, TP53INP2 knockdown increased the E-cadherin level and decreased the Snail1, N-Cadherin, and Vimentin levels. These results suggested that TP53INP2 knockdown inhibited the EMT of BC cells.
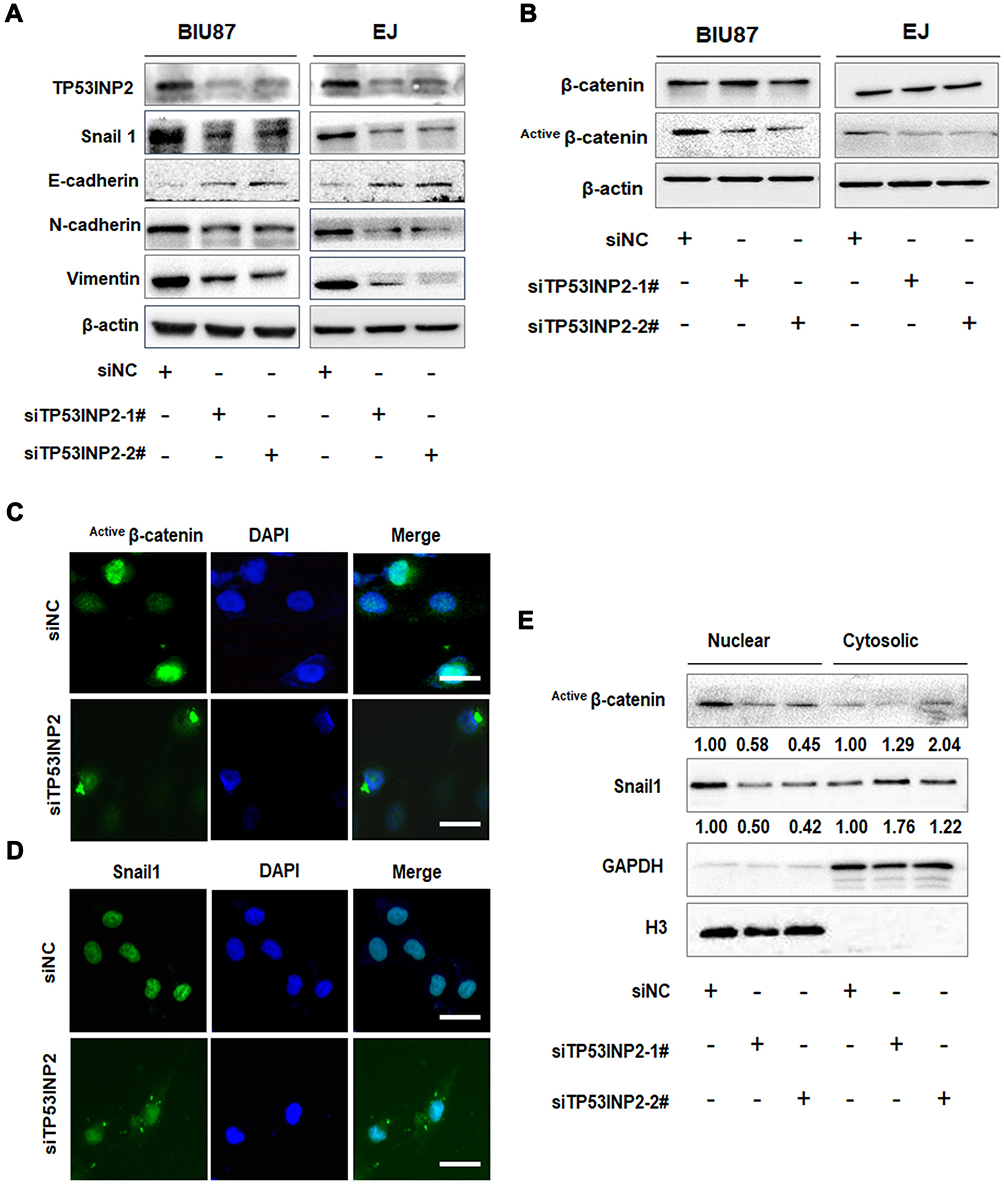 |
Figure 3 Effects of TP53INP2 knockdown on EMT and β-catenin. Notes: (A, B) The negative control or siTP53INP2 were transfected into the BIU87 and EJ cells. The cells were incubated for 48 h, and the changes in the expressions of EMT molecular markers and β-catenin were detected by Western blot. β-actin was used as an internal control. (C, D) The cellular location of active β-catenin and Snail1 in EJ cells after interference in TP53INP2. Scale bar, 50 μm. (E) The abundance of the active forms of β-catenin and Snail1 in nuclear and cytosolic fractions were detected by Western blot. GAPDH and H3 were used as cytosolic or nuclear controls.Abbreviations: EMT, epithelial-to-mesenchymal transition; GAPDH, glyceraldehyde 3-phosphate dehydrogenase. |
We then investigated the molecular mechanism by which TP53INP2 knockdown inhibited EMT in BC cells. TP53INP2 activates β-catenin through the sequestration of GSK-3β into late endosomes in an autophagy-dependent manner.15 The Wnt/β-catenin pathway plays a key role in EMT. Thus, we hypothesized that the GSK-3β/β-catenin pathway was involved in the EMT regulated by TP53INP2. Interestingly, we found that TP53INP2 knockdown decreased the level of the non-phospho active β-catenin but not total β-catenin (Figure 3B).
The Snail1 zinc-transcription factor is a key regulator of EMT that can be phosphorylated by GSK-3β. GSK-3β phosphorylates Snail1 at two consensus motifs: phosphorylation of the second motif causes Snail1 to localize in the cytoplasm, whereas phosphorylation of the first motif causes Snail1 to undergo degradation by β-TrCP-mediated ubiquitination.16,17 The sequestration of GSK-3β into a late-endosomal compartment causes β-catenin cytosolic accumulation and subsequent nuclear translocation.18 Therefore, we tested whether TP53INP2 knockdown affected the subcellular localization of β-catenin and Snail1. As expected, TP53INP2 knockdown increased the amount of active β-catenin and Snail1 in the cytoplasm (Figure 3C–E).
GSK-3β Inhibition Abrogates the TP53INP2 Knockdown-Mediated EMT Inhibition
We then determined whether GSK-3β was required for the TP53INP2 knockdown-mediated inhibition of EMT. As shown in Figure 4A–C, the administration of IM-12, an inhibitor of GSK-3β, abrogated the reduction of migration and invasion caused by TP53INP2 knockdown in the BIU87 and EJ cells. In addition, the TP53INP2 knockdown-induced decrease in non-phospho (active) β-catenin and Snail1 were blocked by IM-12 (Figure 4D). The TP53INP2 knockdown-induced EMT suppression was also abrogated by the administration of IM-12, indicating that the effect on EMT mediated by TP53INP2 knockdown was dependent on GSK-3β (Figure 4D).
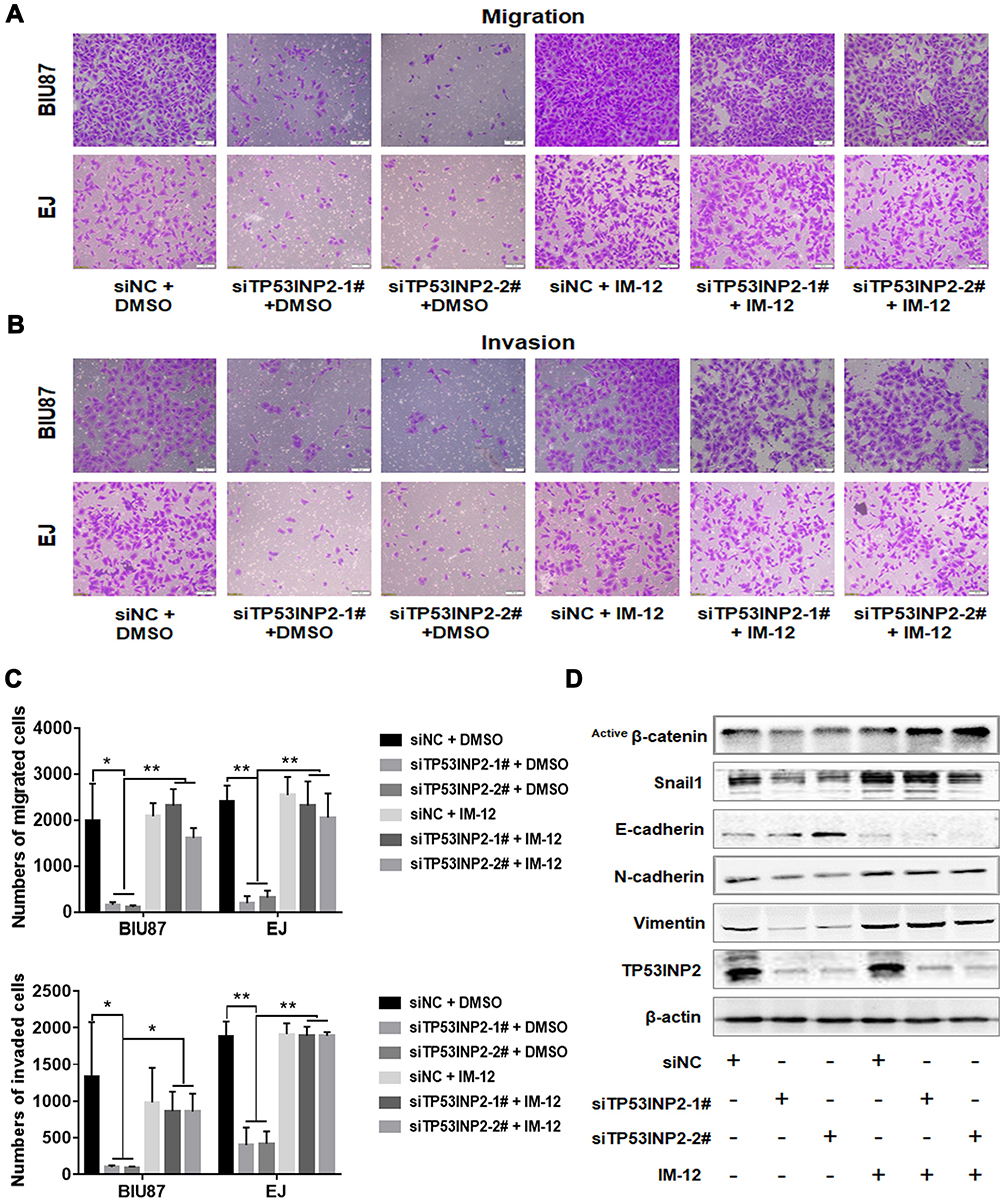 |
Figure 4 TP53INP2 regulates bladder cancer cell migration and invasion via the GSK-3β/β-catenin pathway. Notes: (A–C) The BIU87 and the EJ bladder cancer cells were treated with the GSK-3β inhibitor IM-12 (3 µM) after interference in TP53INP2 for 6 h. The cells were incubated for 48 h, and the transwell assay was performed to detect the changes in bladder cell migrative and invasive capabilities. Scale bar, 50 μm. (D) Changes in the expressions of EMT molecular markers and active β-catenin were detected by Western blot. β-actin was used as an internal control. ** p < 0.01; * p < 0.05.Abbreviation: EMT, epithelial-to-mesenchymal transition. |
Autophagy Induction Partially Abrogates the Effect of TP53INP2 Knockdown
Autophagy is involved in EMT regulation.19 TP53INP2 contributes to basic autophagy by regulating autophagosome formation.10,20 As shown in Figure 5A, TP53INP2 knockdown decreased the conversion ratio of LC3B from type I to type II. We subsequently determined whether TP53INP2 knockdown-induced EMT inhibition affected by autophagy. We found that the induction of autophagy with rapamycin partly rescued the decrease in active β-catenin induced by the TP53INP2 knockdown (Figure 5B). Rapamycin also blocked TP53INP2 knockdown-induced inhibition of migration and invasion (Figure 5C).
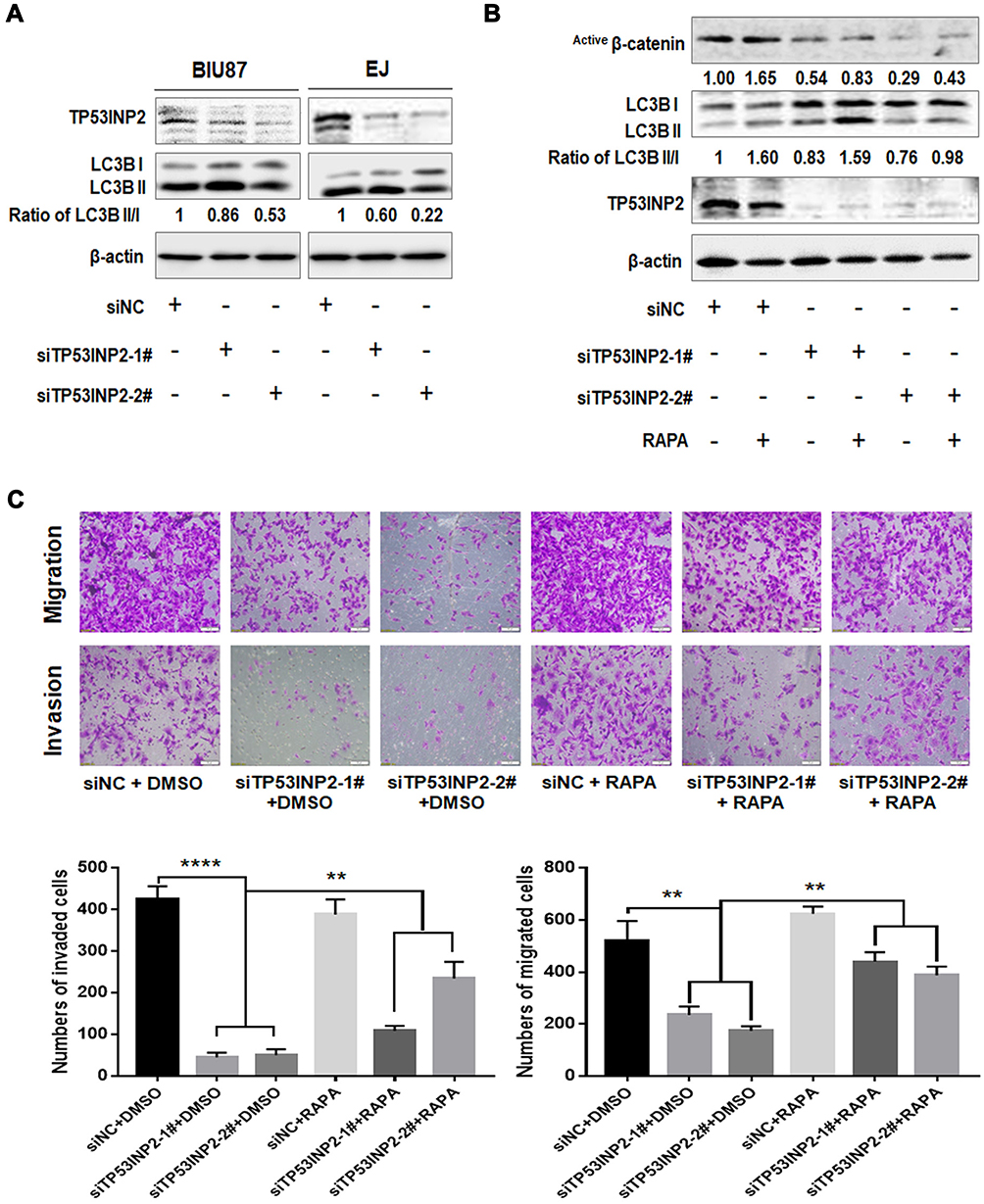 |
Figure 5 Autophagy was involved in the TP53INP2-induced regulation of active β-catenin, migration, and invasion. Notes: (A) The changes in the expression of autophagy molecular marker LC3 were detected using Western blot after interference in TP53INP2. (B) Active β-catenin expression was detected by Western blot after the cells were subjected to rapamycin treatment (340 nM) and interference in TP53INP2. (C) Transwell assay of EJ cells treated with TP53INP2 siRNA or rapamycin. Scale bar, 50 μm. ** p < 0.01, **** p < 0.001. |
Discussion
The Wnt/β-catenin pathway plays a crucial role in development, tissue regeneration, and stem cells and cancers.21 In the absence of Wnt signal (off state), the cytoplasmic β-catenin forms a complex with Axin, casein kinase 1 (CK1), GSK-3β, and adenomatous polyposis coli protein (APC), thereby mediating the phosphorylation of β-catenin. CK1 and GSK-3β sequentially phosphorylate β-catenin at N-terminal Ser and Thr residues, resulting in its ubiquitylation and proteasomal degradation by the SCFβ–TrCP ubiquitin ligase. Upon exposure to the Wnt ligand (on state), GSK-3β is displaced from the APC/Axin complex through endosome sequestration.18 The APC/Axin complex is then inactivated, and the hypophosphorylated β-catenin is stabilized by escaping degradation. The stabilized β-catenin translocates to the nucleus and binds to TCF/LEF-1 transcription factors to transactivate the expression of Wnt response genes.22 The Wnt/β-catenin pathway plays a key role in EMT. Several key transcription factors regulating the E-cadherin expression, such as Twist, Snail1, and ZEB1, are direct or indirect transcriptional targets of the Wnt pathway.23 In the current study, we have demonstrated that TP53INP2 is overexpressed in BC tissues and predicts the poor prognosis of patients with BC. The TP53INP2 knockdown inhibits the invasion, migration (Figure 2C and D), and EMT (Figure 3A and B) of BC cells by decreasing the expression of non-phospho (active) β-catenin and Snail1.
Here we report that TP53INP2 was significantly upregulated in the invasive BC lines (BIU87, EJ, UM-UC-3, and T24) but unchanged in the non-invasive bladder cancer cell line (5637) (Figure 1C), which indicated that the overexpression of TP53INP2 might be specifically correlated to the invasive capability of BC. We then knocked down TP53INP2 by siRNA and found that TP53INP2 silencing can inhibit the invasive and migratory capabilities and EMT of BC cells. The key to canonical Wnt signaling is GSK-3β inhibition, which results in hypophosphorylated or nonphosphorylated β-catenin, which has increased stability.24 TP53INP2 activates β-catenin through the sequestration of GSK-3β into late endosomes in an autophagy-dependent manner.15 We then speculate that the GSK-3β/β-catenin pathway may be involved in the EMT regulated by TP53INP2. Interestingly, we have found that the TP53INP2 knockdown decreases the level of non-phospho (active) β-catenin (Figure 3B) and relocates active β-catenin from the nucleus to the cytoplasm (Figure 3C). However, in contrast with a previous study,15 the current study has revealed thatTP53INP2 knockdown does not decrease the level of total β-catenin. This result can be explained by the short-term knockdown of TP53INP2, which decreases GSK-3β sequestration. This result leads to a decrease in non-phospho (active) β-catenin but does not significantly change the total β-catenin levels. Moreover, GSK-3β can phosphorylate Snail1 and marks it for degradation.25 In the current study, the TP53INP2 knockdown decreased the level of Snail1 and increased the amount of Snail1 in the cytoplasm (Figure 3B and D). Furthermore, the inhibition of GSK-3β by IM-12 abrogates the TP53INP2 knockdown-mediated inhibition of EMT. These observations suggest that the TP53INP2 knockdown inhibits EMT through the GSK-3β/β-catenin/Snail1 pathway.
Autophagy, which is a cellular self-degradative process removing damaged organelles and misfolded proteins and mobilizing intracellular nutrient resources, plays a key role in cell survival under stress conditions.26 Although autophagy induction triggers a molecular switch from a mesenchymal phenotype to an epithelial-like one in glioblastoma cellular models27 and EMT promotes autophagy of potentially metastatic cancer cells through immune surveillance escape of potentially metastatic cancer cells,28 the relationship between EMT and autophagy is still a matter of debate. TP53INP2 contributes to basic autophagy by regulating autophagosome formation.10,20 TP53INP2 knockdown inhibits the autophagy caused by bortezomib.29 In the current study, we demonstrated that TP53INP2 silencing inhibits autophagy. And we have investigated whether the TP53INP2 knockdown induces the inhibition of EMT affected by autophagy. As expected, the induction of autophagy with rapamycin has partially rescued the TP53INP2 knockdown-induced decrease in active β-catenin and inhibition of migration and invasion in BC cells (Figure 5B and C). This finding suggests that TP53INP2 knockdown-induced inhibition of EMT is dependent on autophagy and unveils the role of TP53INP2 in autophagy/EMT crosstalk.
Conclusion
Our results uncover the new role of TP53INP2 in EMT regulation. We have demonstrated that the TP53INP2 knockdown inhibits migration, invasion, and EMT via the GSK-3β/β-catenin/Snail1 pathway in BC (Figure 6). Our findings show that TP53INP2 can serve as a promising new target in BC therapy.
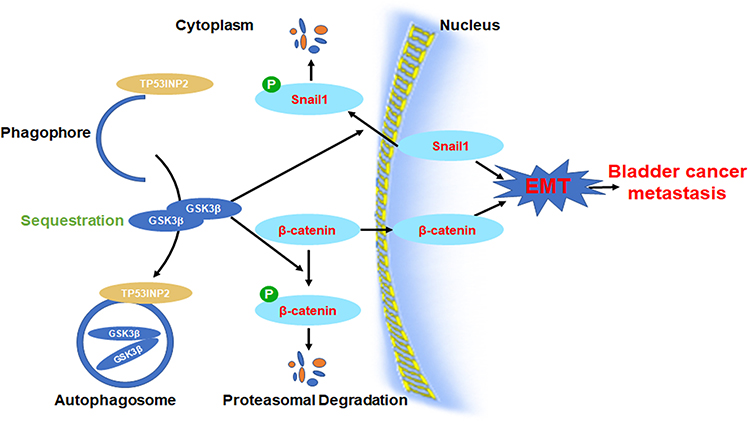 |
Figure 6 The diagram illustrates the hypothesis that TP53INP2 modulates cell migration, invasion, and EMT via the GSK-3β/β-catenin/Snail1 pathway in bladder cancer. Abbreviation: EMT, epithelial-to-mesenchymal transition. |
Author Contributions
ZHENGTAO ZHOU and XIAOQIANG LIU performed the experiments and generated data. XIAOQIANG LIU, YULEI LI, WEN DENG, JUNHUA LI, LUYAO CHEN and YU LI analyzed data. XIANTAO ZENG, GONGXIAN WANG and BIN FU designed experiments. ZHENGTAO ZHOU, XIAOQIANG LIU, and BIN FU wrote the manuscript. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of P.R. China (grant no. 81560419 and no. 81960512), the Natural Science Foundation of Jiangxi (grant no. 20151BAB205047 and 20202BAB216006) and the Jiangxi Province Infrastructure Facilities for Scientific Research Institutes (grant no. 20142BBA13038 and 20151BBA13047).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
2. Burger M, Catto JWF, Dalbagni G, et al. Epidemiology and risk factors of urothelial bladder cancer. Eur Urol. 2013;63(2):234–241. doi:10.1016/j.eururo.2012.07.033
3. Abdollah F, Gandaglia G, Thuret R, et al. Incidence, survival and mortality rates of stage-specific bladder cancer in United States: a trend analysis. Cancer Epidemiol. 2013;37(3):219–225. doi:10.1016/j.canep.2013.02.002
4. Kobayashi T, Owczarek TB, McKiernan JM, Abate-Shen C. Modelling bladder cancer in mice: opportunities and challenges. Nat Rev Cancer. 2015;15(1):42–54. doi:10.1038/nrc3858
5. Mittal V. Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol Mech Dis. 2018;13(1):395–412. doi:10.1146/annurev-pathol-020117-043854
6. Kobayashi T. Understanding the biology of urothelial cancer metastasis. Asian J Urol. 2016;3(4):211–222. doi:10.1016/j.ajur.2016.09.005
7. Baumgartner BG, Orpinell M, Duran J, et al. Identification of a novel modulator of thyroid hormone receptor-mediated action. PLoS One. 2007;2(11):1–13. doi:10.1371/journal.pone.0001183
8. Xu Y, Wan W, Shou X, et al. TP53INP2/DOR, a mediator of cell autophagy, promotes rDNA transcription via facilitating the assembly of the POLR1/RNA polymerase I preinitiation complex at rDNA promoters. Autophagy. 2016;12(7):1118–1128. doi:10.1080/15548627.2016.1175693
9. Nowak J, Archange C, Tardivel-Lacombe J, et al. The TP53INP2 protein is required for autophagy in mammalian cells. Mol Biol Cell. 2009;20(3):870–881. doi:10.1091/mbc.e08-07-0671.
10. Huang R, Xu Y, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57(3):456–466. doi:10.1016/j.molcel.2014.12.013
11. Moran-Jones K, Grindlay J, Jones M, Smith R, Norman JC. hnRNP A2 regulates alternative mRNA splicing of TP53INP2 to control invasive cell migration. Cancer Res. 2009;69(24):9219–9227. doi:10.1158/0008-5472.CAN-09-1852
12. Liu X, Wu Y, Zhou Z, et al. Celecoxib inhibits the epithelial-to-mesenchymal transition in bladder cancer via the miRNA-145/TGFBR2/Smad3 axis. Int J Mol Med. 2019;44(2):683–693. doi:10.3892/ijmm.2019.4241
13. Liu X, Xu X, Deng W, et al. CCL18 enhances migration, invasion and EMT by binding CCR8 in bladder cancer cells. Mol Med Rep. 2019;19(3):1678–1686. doi:10.3892/mmr.2018.9791
14. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45(W1):W98–102. doi:10.1093/nar/gkx247
15. Romero M, Sabaté-Pérez A, Francis VA, et al. TP53INP2 regulates adiposity by activating β-catenin through autophagy-dependent sequestration of GSK3β. Nat Cell Biol. 2018;20(4):443–454. doi:10.1038/s41556-018-0072-9
16. Zhou BP, Deng J, Xia W, et al. Dual regulation of Snail by GSK-3β-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6(10):931–940. doi:10.1038/ncb1173
17. Yook JI, Li XY, Ota I, et al. A Wnt-Axin2-GSK3β cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8(12):1398–1406. doi:10.1038/ncb1508
18. Taelman VF, Dobrowolski R, Plouhinec JL, et al. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143(7):1136–1148. doi:10.1016/j.cell.2010.11.034
19. Colella B, Faienza F, Di Bartolomeo S. EMT regulation by autophagy: a new perspective in glioblastoma biology. Cancers (Basel). 2019;11(3):1–21. doi:10.3390/cancers11030312
20. Mauvezin C, Sancho A, Ivanova S, Palacin M, Zorzano A. DOR undergoes nucleo-cytoplasmic shuttling, which involves passage through the nucleolus. FEBS Lett. 2012;586(19):3179–3186. doi:10.1016/j.febslet.2012.06.032
21. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi:10.1016/j.cell.2012.05.012
22. MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. doi:10.1016/j.devcel.2009.06.016
23. Valenta T, Hausmann G, Basler K. The many faces and functions of β-catenin. EMBO J. 2012;31:2714–2736.
24. Kimelman D, Xu W. β-Catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25(57):7482–7491. doi:10.1038/sj.onc.1210055
25. Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005;168(1):29–33. doi:10.1083/jcb.200409067
26. Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol. 2014;85(6):830–838. doi:10.1124/mol.114.091850
27. Catalano M, D’Alessandro G, Lepore F, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol. 2015;9(8):1612–1625. doi:10.1016/j.molonc.2015.04.016
28. Akalay I, Janji B, Hasmim M, et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from t-cell-mediated lysis. Cancer Res. 2013;73(8):2418–2427. doi:10.1158/0008-5472.CAN-12-2432
29. Hu Y, Li X, Xue W, et al. TP53INP2-related basal autophagy is involved in the growth and malignant progression in human liposarcoma cells. Biomed Pharmacother. 2017;88:562–568. doi:10.1016/j.biopha.2017.01.110
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.