")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Toward antituberculosis drugs: in silico screening of synthetic compounds against Mycobacterium tuberculosis L,D-transpeptidase 2
Authors Billones J , Carrillo MC, Organo V, Macalino SJ, Sy JB, Emnacen I, Clavio NA, Concepcion G
Received 24 September 2015
Accepted for publication 31 December 2015
Published 11 March 2016 Volume 2016:10 Pages 1147—1157
DOI https://doi.org/10.2147/DDDT.S97043
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Junie B Billones,1,2 Maria Constancia O Carrillo,1 Voltaire G Organo,1 Stephani Joy Y Macalino,1 Jamie Bernadette A Sy,1 Inno A Emnacen,1 Nina Abigail B Clavio,1 Gisela P Concepcion3
1Office of the Vice President for Academic Affairs – Emerging Interdisciplinary Research Program: “Computer-aided Discovery of Compounds for the treatment of Tuberculosis in the Philippines,” Department of Physical Sciences and Mathematics, College of Arts and Sciences, 2Institute of Pharmaceutical Sciences, National Institutes of Health, University of the Philippines Manila, Manila, 3Marine Science Institute, University of the Philippines Diliman, Diliman, Quezon City, Philippines
Abstract: Mycobacterium tuberculosis (Mtb) the main causative agent of tuberculosis, is the main reason why this disease continues to be a global public health threat. It is therefore imperative to find a novel antitubercular drug target that is unique to the structural machinery or is essential to the growth and survival of the bacterium. One such target is the enzyme L,D-transpeptidase 2, also known as LdtMt2, a protein primarily responsible for the catalysis of 3→3 cross-linkages that make up the mycolyl–arabinogalactan–peptidoglycan complex of Mtb. In this study, structure-based pharmacophore screening, molecular docking, and in silico toxicity evaluations were employed in screening compounds from a database of synthetic compounds. Out of the 4.5 million database compounds, 18 structures were identified as high-scoring, high-binding hits with very satisfactory absorption, distribution, metabolism, excretion, and toxicity properties. Two out of the 18 compounds were further subjected to in vitro bioactivity assays, with one exhibiting a good inhibitory activity against the Mtb H37Ra strain.
Keywords: antituberculosis drug discovery, virtual screening, docking
Introduction
The cell wall of Mycobacterium tuberculosis (Mtb) is made up of a lipid membrane interspersed with cell wall proteins and complex layers of peptidoglycan, arabinogalactan, and mycolic acids known as the cell wall core or the mycolyl–arabinogalactan–peptidoglycan (mAGP) complex. While the part of the cell wall composed of lipids and proteins can be easily disrupted using solvents, the mAGP complex remains insoluble, making it indispensable for the survival of the pathogen and providing resistance to common antibiotics. Thus, the mAGP complex is a very attractive target for drug development against tuberculosis (TB).1 The peptidoglycan layer of mycobacteria, as well as other β-lactam resistant bacteria, is predominantly made of 3→3 linkages instead of the more common 4→3 cross-linkages. L,D-transpeptidases catalyze the formation of 3→3 cross-linkages, one type of which is the L,D-transpeptidase 2 (LdtMt2; MT2594, product of gene Rv2518c), a protein that was observed to be pertinent for Mtb’s virulence and growth during the chronic phase of the disease.2,3 While the combination of clavulanate and meropenem shows potential in treating patients with TB by inhibiting Mtb’s mycobacterial β-lactamase (BlaC) and LdtMt2, respectively, the short half-life of meropenem forestalls its use as an anti-TB agent.4 Furthermore, both imipenem, another carbapenem that has shown activity against LdtMt2, and meropenem, are compounds that are currently being tested for the treatment of a wide range of infections other than TB.5 Nonetheless, this may later lead to resistance, as the drug is not specific for TB.
TB, an infectious disease caused by Mtb, is one of the major health concerns worldwide, resulting in almost 1.5 million deaths in the year 2014.6 Despite the presence of current drug treatments for TB, including the use of isoniazid, rifampicin, ethambutol, pyrazinamide, and streptomycin,7 the incidence of TB remains high, especially in developing countries, such as the Philippines.6 The alarming rise of multi- and extensively drug-resistant TB patients has become a serious global health threat. The emergence of resistant strains is due to problems, such as the arrival of human immunodeficiency virus8 and poor patient compliance because of the extensive treatment regimen.9 Thus, new anti-TB drugs that can shorten the treatment regimen and/or target the resistant TB strains are urgently needed.
Depending on the target disease and treatment method, the whole process of drug discovery and development can take up to 15 years and cost millions of dollars before the drug can actually reach the market. Out of the millions of candidate drug compounds initially screened for a certain disease, only a few make it to clinical trials, and even after that, <10% of those compounds from clinical trials successfully get the final approval.10 For TB, the initial phase of drug discovery aims to identify “leads” with anti-TB activity and desirable physicochemical, pharmacokinetic, and toxicity properties through classic wet laboratory testing.11 However, this approach requires Biosafety Level 3 (BSL3) laboratory equipment and expert research skills in handling sensitive protocols used during drug screening.12 Also, with the continuous advancements in technology and the availability of the Mtb genome and three-dimensional (3D) structure of potential enzyme targets of TB, virtual target-based screening can be utilized. The use of computational screening methods can potentially reduce the cost, time, and effort needed for the initial screening of candidate drug compounds with pharmacological activity against TB.13
With the use of a powerful computer, a computational software, and structural data of a protein target and compound libraries, initial screening of millions of compounds can be performed in less time than what is used in the classic drug discovery and development process, helping to prioritize compound testing and minimizing randomization in the laboratory. In this work, >4 million synthetic compounds were screened based on a pharmacophore that satisfies the electronic and structural requirements of the drug target’s binding site. The high-scoring hits were subsequently docked to the target and were rank-ordered based on their binding energies. The high-affinity hits were further evaluated in silico for their potential pharmacokinetics and pharmacodynamics properties.
Materials and methods
All computational work was performed using Accelrys Discovery Studio 4.0 (DS 4.0) on a Windows 7 Home Edition with an Intel® Core™ i7-3770 3.40 GHz quad core processor, 4 GB RAM, and 64-bit operating system. Protein structures were downloaded from Research Collaboratory for Structural Bioinformatics protein databank, and imipenem and meropenem structures were taken from the National Center for Biotechnology Information website. Enamine real database containing compound structures was downloaded from the enamine website.14
Structure-based pharmacophore modeling
Preparation of 3D protein structure and library compounds
The 3D structure of LdtMt2 complexed with a peptidoglycan fragment (PDB ID: 3TUR) solved at 1.72 Å resolution2 was retrieved. The bound peptidoglycan fragment was removed, and the protein was prepared using the Prepare Protein protocol of DS 4.0 (BIOVIA, Tokyo, Japan) using the default parameters. The Prepare Protein protocol primes the protein for input into other protocols in DS 4.0 by inserting missing atoms in incomplete residues, optimizing side-chain conformation, modeling missing loop regions, removing alternate conformations, and protonating titratable residues at pH 7.4.15 The enamine compound database was downloaded and prepared using the Prepare Ligands protocol. The compounds in the enamine database were prepared using Prepare Ligands protocol.
Optimization of protein structure and root-mean-square deviation
Minimization protocol was used to optimize the protein structure for screening. The default algorithm parameter, Smart Minimizer, was used to minimize the structure by executing 1,000 steps of steepest descent using an RMS gradient acceptance of 3, followed by conjugate gradient minimization, which locates an unconstrained local minimum for the input structure.15,16 The root-mean-square deviation (RMSD) of the prepared protein structure was then calculated against the original protein file using the Superimpose Proteins tool. The protein structures were superimposed based on Cα pairs.
Generation of structure-based pharmacophore model
The binding site of LdtMt2 was identified based on literature’s description, that is, the site that contains the catalytic triad Cys354, His336, and Ser337.2,17,18 After identification of the binding site, a binding sphere was generated using the Binding Site tool in DS 4.0 with a radius of 10 Å. The Interaction Generation tool of DS 4.0 was used to generate a pharmacophore model that complements the chemical features (hydrophobic, H-donor, and H-acceptor) in the protein’s active site. The Edit and Cluster Pharmacophore tool was used to cluster the common pharmacophore properties down to <30 features.
Virtual screening of compounds
Preparation of 3D compound libraries
Approximately 4.5 million database compounds were screened in this work. The test compounds, as well as imipenem and meropenem, were prepared using the Prepare Ligands protocol with default parameters. The Prepare Ligands protocol primes the ligands for use in other protocols by removing duplicate structures, generating isomers and tautomers, generating 3D conformations, and other functions specified by the user.15
Database building
The Build 3D Database protocol was used to create compound databases for easier screening. The compound database was built based on Catalyst algorithms, which create compact, indexed compound databases used for pharmacophore screening.15
Pharmacophore-based screening
The generated structure-based pharmacophore model was employed to screen the compound databases using the Screen Library protocol, which uses the flexible search method of Catalyst. The Screen Library protocol enumerates numerous possible structures from an input query pharmacophore model and screens a compound 3D database. Screening was carried out twice, one for rigid fitting method and another for flexible fitting method. The rigid fitting method screens the compounds without modifying their input conformation, whereas in the flexible fitting method, each ligand conformation was slightly modified to better fit the pharmacophore model.15 The base fit value for rigid screening was arbitrarily set to 2.5, while the base fit value for flexible screening was set to 3.0. Any compound with a fit value <2.5 and <3.0 was not chosen for subsequent molecular docking and in silico toxicity screening, respectively.
Molecular docking
The hit compounds from the pharmacophore screening were docked to the prepared LdtMt2 active site using the CHARMm-based DOCKER (CDOCKER) docking protocol. CDOCKER is a grid-based molecular dynamics-simulated-annealing-based algorithm docking procedure that utilizes CHARMm force fields. It allows full ligand flexibility in the docking process by producing several ligand poses when the ligand is docked into the receptor’s binding site and by applying molecular dynamics-based simulated annealing and in situ minimization.19 Meropenem and imipenem, carbapenems known to inhibit LdtMt2,2,18 were also docked and compared to the original bound conformation to validate the docking method and compare binding affinities and active site interaction.
Calculation of binding energies
Calculate Binding Energies protocol of DS4.0 was used to calculate the binding affinity of all docked compounds. This protocol computes for the binding energy using the formula energy of binding = energy of complex − energy of ligand − energy of receptor.15 The calculated binding energies of meropenem and imipenem were used as the baseline comparison for the selection of compounds with the best binding affinity to LdtMt2.
In silico absorption, distribution, metabolism, excretion, and toxicity and toxicity prediction by komputer assisted technology screening
The compounds with the best binding affinity were further screened in silico for their pharmacokinetics and pharmacodynamics properties using the absorption, distribution, metabolism, excretion, and toxicity (ADMET) and toxicity prediction by komputer assisted technology (TOPKAT) protocols of DS 4.0. The solubility, absorption, plasma protein binding, CYP2D6 inhibition, and hepatotoxicity of each compound were evaluated by the use of ADMET.15 The candidate compounds were also subjected to TOPKAT calculations to determine the probability for carcinogenicity, mutagenicity, and other toxicity measures.
Resazurin-based microtiter plate assay
Bacterial strains and culture conditions
Mtb H37Ra (ATCC 25177) glycerol stocks, provided by the Marine Natural Products Laboratory at the University of the Philippines Marine Science Institute, were first thawed out and subcultured on Middlebrook 7H11 agar supplemented with 10% oleic acid–albumin–dextrose–catalase (Titan Media, Delhi, India). Plates were then incubated at 37°C for 3–4 weeks and then subcultured on Middlebrook 7H9 broth supplemented with 10% albumin–dextrose–catalase (Titan Media). Broth tubes were then incubated in a shaking incubator at 37°C, 150 rpm for another 3–4 weeks.
Compound preparation
Enamine compounds were all procured from Enamine Ltd (Kiev, Ukraine) and then solubilized at a stock concentration of 2 mg/mL in dimethyl sulfoxide (DMSO). Rifampicin (Sigma-Aldrich Co., St Louis, MO, USA), the assay positive control, was also solubilized in DMSO at a stock concentration of 1 mg/mL. All the compound stock solutions were stored at −20°C.
Resazurin microtiter assay
Preparation of the assay inoculum was first done by adjusting the turbidity of the broth culture to match a McFarland No 1 standard (A625 nm ≈0.25). The adjusted culture was then diluted further to a 1:49 mixture of culture: M7H9 broth. This resulting inoculum was used for the assay proper within 30 minutes of preparation.
Stock solutions of enamine compounds and rifampicin were thawed and diluted to arrive at two concentrations: one at a high concentration of 2,500 μM, and the other at a low concentration of 20 μM. Since each test well has a final volume of 200 μL and only 2 μL of the compound is added per well, the final drug and rifampicin well concentrations were 25 μM and 0.2 μM. Each sample was tested in quadruplicates.
The resazurin assay proper was performed in 96-well, flat-bottomed microtiter plates. Four types of assay wells were prepared. First, sample wells consisted of 98 μL of Middlebrook 7H9 broth supplemented with 10% ADC, 100 μL of the H37Ra inoculum, and 2 μL of the prepared enamine compound. On the other hand, the rifampicin positive control wells were added with 98 μL of M7H9 broth with 10% ADC, 100 μL of the inoculum, and 2 μL of the prepared rifampicin standard. Sterility control wells were also prepared, which consisted of 2 μL of DMSO and 198 μL of M7H9 broth with 10% ADC. Moreover, growth control wells were prepared, which consisted of 98 μL of M7H9 broth with 10% ADC, 100 μL of H37Ra inoculum, and 2 μL of DMSO. Lastly, four media-only wells were also added with 200 μL of the M7H9 broth. Well plates were then incubated at 37°C, 150 rpm for 7 days in a shaking incubator. After 7 days, 20 μL of 0.02% resazurin (Sigma-Aldrich Co.) was added to all the wells and then incubated for an additional 24 hours. The fluorescence readings of all wells were then read at an excitation filter of 530 nm and an emission filter of 590 nm.
Ligand interaction analysis
Ligand interactions for both the originally bound and docked meropenem were analyzed with the aid of 2D protein–ligand interaction diagrams. Ligand interactions of the hit compounds were also analyzed to compare the interactions with that of the known inhibitor.
Results and discussion
Crystal structure of LdtMt2
Most bacteria rely on the action of D,D-transpeptidases in catalyzing the biosynthesis of the peptidoglycan layer of their cell walls, particularly in the formation of 4→3 cross-linkages (Figure 1B). A structurally unrelated transpeptidase also present in most bacteria, L,D-transpeptidase, only plays a minimal role in the organisms’ peptidoglycan synthesis.2,20 Presently, β-lactams are the mostly widely used class of antibacterial drugs that are designed to target 4→3 transpeptide linkages by acting as suicide substrates of D,DD-transpeptidases.21 However, in the case of the nonreplicating Mtb, 3→3 linkages (Figure 1A) predominate the transpeptide network of their peptidoglycan layer, which in turn are primarily catalyzed by L,D-transpeptidases.22,23
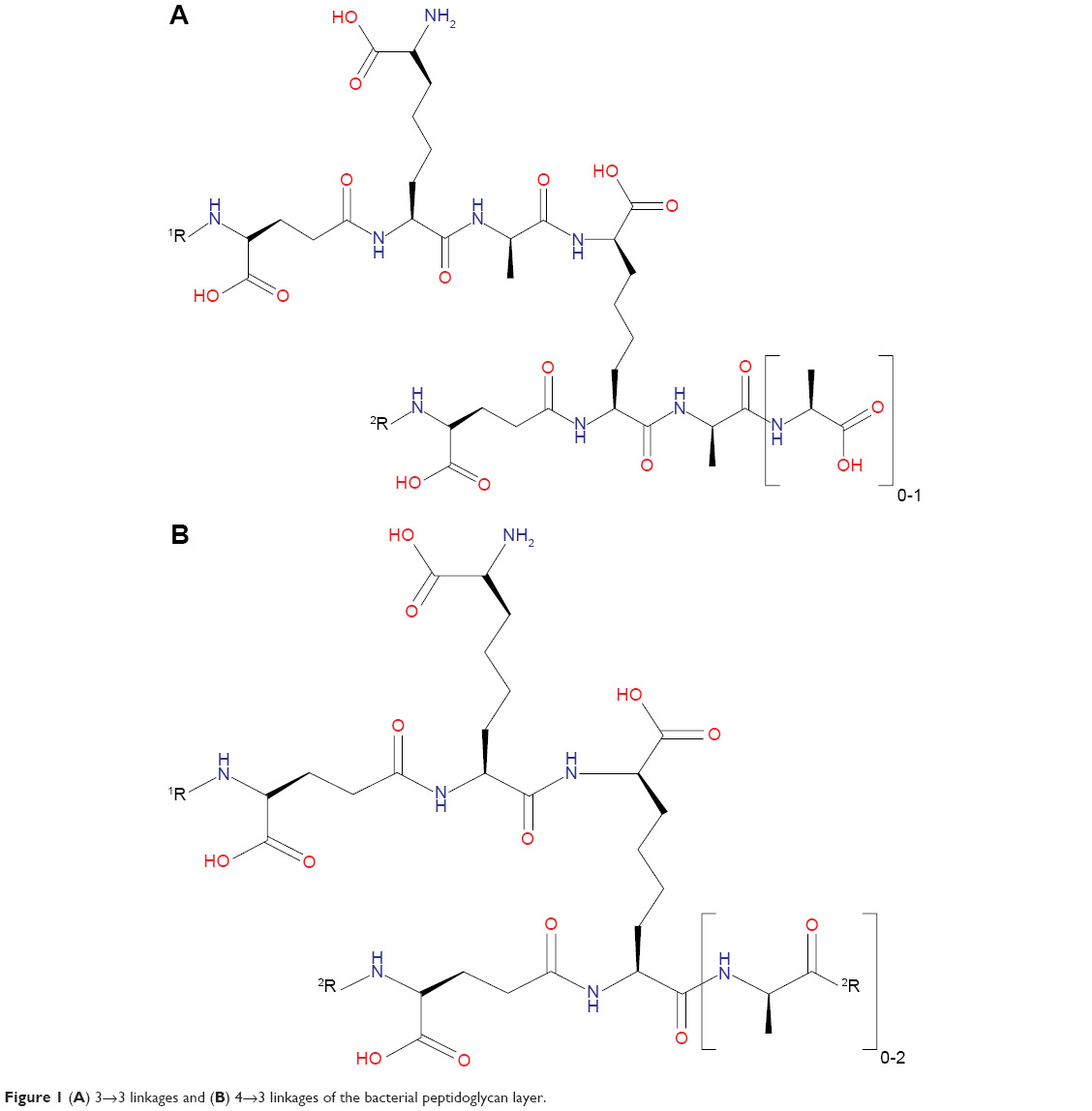 | Figure 1 (A) 3→3 linkages and (B) 4→3 linkages of the bacterial peptidoglycan layer. |
In 1974, Wietzerbin et al first discovered the existence of these 3→3 linkages in the peptidoglycan layer of mycobacteria.22 Nevertheless, their role and significance in the biochemistry pathway and physiology of mycobacteria remained unknown until recently, when a study conducted by Gupta et al3 showed that a specific LdtMt2 is essential for the maintenance of the virulence and drug resistance of Mtb strains. An inactivation of LdtMt2 resulted in an altered colony morphology, even though no difference in the lipid composition of the cell wall was detected.3 LdtMt2 is the primary L,D-transpeptidase in Mtb; LdtMt2 is expressed at least ten-fold higher than LdtMt1 in all phases of growth of the bacteria, which is evidenced by the fact that continuous LdtMt1 expression was not able to compensate the loss of LdtMt2 activity.3,18 Loss of LdtMt2 was also found to compromise the bacteria’s ability to acclimate during chronic infection and has displayed an increased susceptibility to β-lactams.3 Therefore, the protein LdtMt2 serves as an excellent target for novel drug discovery against the drug-resistant strains of Mtb.
β-lactams were formerly thought to be ineffectual against Mtb due to the presence of the endogenous BlaC.24 However, a specific class of β-lactams called carbapenems were shown to have activity against L,D-transpeptidases from Enterococcus faecium (Ldtfm) and Mtb (LdtMt1), since they were poorly hydrolyzed by BlaC.2 In addition to this, clavulanic acid is found to be effective in inhibiting Mtb BlaC and can be used in combination with these carbapenems.24 Meropenem and imipenem, as known inhibitors of LdtMt2, were used as the reference in searching for lead-like compounds in this work.
The crystal structure of LdtMt2 with bound peptidoglycan fragment (PDB ID: 3TUR) was used in this study. The structural data of 3TUR contain two monomers of LdtMt2. However, these molecules are not equivalent to a physiological dimer, since it was determined that LdtMt2 behaves as a monomer in solution.2 A more recent crystal structure of LdtMt2 in complex with meropenem (PDB ID: 4GSU) that can be used to validate the docking results of meropenem became available.18 With the release of this new PDB data, the structural similarity of the protein file used in this study (ie, 3TUR) was calculated using 4GSU as a reference, yielding an RMSD value of 0.714 Å. This result indicates very tight structural resemblance between the two proteins. Subsequently, the 3D structure of the LdtMt2 protein with bound peptidoglycan fragments (Figure 2A) was subjected to Prepare Protein and Minimization protocols to clean and fix the protein and find the most stable protein conformation, respectively. The conformation of the 3D structure changed to some extent after optimization procedures were performed, albeit the deviation was minuscule (RMSD =0.66 Å; Figure 2B).
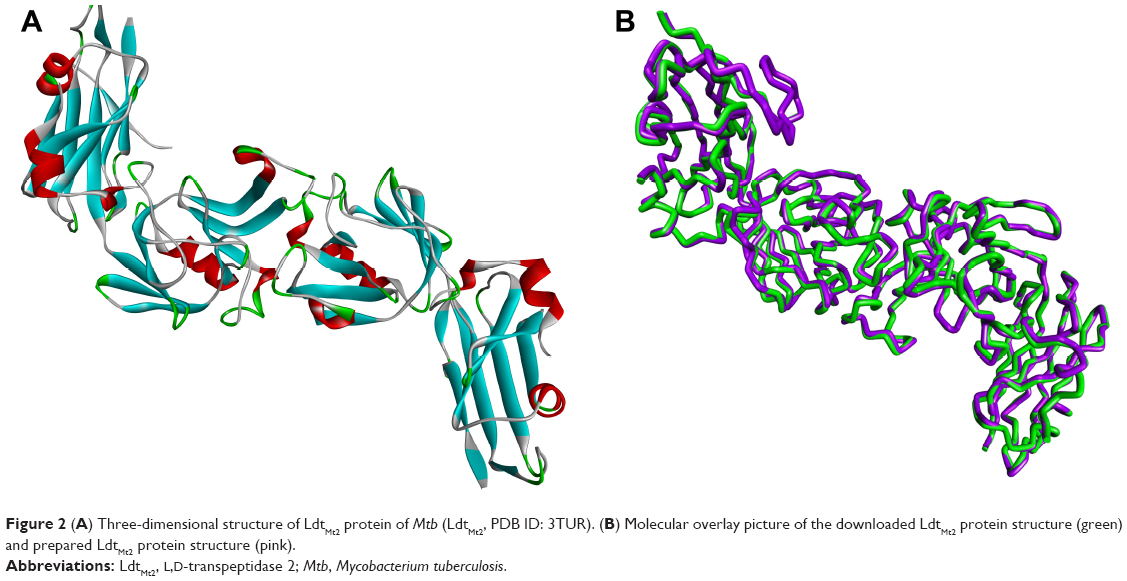 | Figure 2 (A) Three-dimensional structure of LdtMt2 protein of Mtb (LdtMt2, PDB ID: 3TUR). (B) Molecular overlay picture of the downloaded LdtMt2 protein structure (green) and prepared LdtMt2 protein structure (pink). |
Pharmacophore screening, molecular docking, in silico ADMET calculations, and resazurin microtiter assay
As in L,D-transpeptidases from Bacillus subtilis and E. faecium, the YkuD domain of LdtMt2 has a catalytic triad Cys354–His336–Ser337.2,17,18 These residues were used to identify the active site of LdtMt2, generate the binding sphere, and subsequently, the pharmacophore model. A pharmacophore is a set of steric and electronic characteristics utilized in screening of compounds to help prioritize compounds with potential pharmacological activity against the biological target structure to inhibit its activity.25 A structure-based pharmacophore model identifies areas in a target protein that are hydrophobic, hydrogen donor, or hydrogen acceptor. The generated pharmacophore model for LdtMt2 contains a total of 25 features: seven hydrophobic, ten donor, and eight acceptor. The model was employed to screen 4.5 million compounds from the enamine REAL database before docking to the prepared protein structure.
The binding energies for meropenem was determined to be −112.83 kcal/mol. Hit compounds with high fit values (data not shown) were docked individually to the target protein using CDOCKER, and their affinity was measured by calculating their binding energies. Ninety-four compounds showed superior binding affinity than meropenem (data not shown).
Meanwhile, conventional drug discovery entails the need of wet laboratory analysis with the use of high-throughput screening, after which preclinical tests are performed to determine pharmacokinetic and toxicity properties of the compounds.12 Frequently, adverse findings are revealed at this late stage of drug discovery and development,26 wasting effort, time, and resources. In silico ADMET evaluation is an expedient step for predictive quantitative structure–property studies that can be applied in drug discovery.27 It reduces the need for expensive and expansive in vitro pharmacokinetics and toxicity screening.
ADMET and TOPKAT protocols of DS 4.0 were employed to furnish (absorption, distribution, metabolism, and excretion) and toxicity information for the hit compounds from the docking results. The ADMET protocol calculates for the probability of a query compound’s absorption, solubility, inhibition of CYP2D6, plasma protein binding, and hepatotoxicity. The TOPKAT module, on the other hand, computes for the probability of an input compound to be carcinogenic, mutagenic, ocular irritant, and other user-defined toxicity measures. In this study, TOPKAT was used to determine the probability of carcinogenicity, mutagenicity, developmental toxicity, and aerobic biodegradability.15 The 94 high-binding compounds were subjected to in silico ADMET screening, yielding 18 compounds that had the most satisfactory results as shown in Table 1.
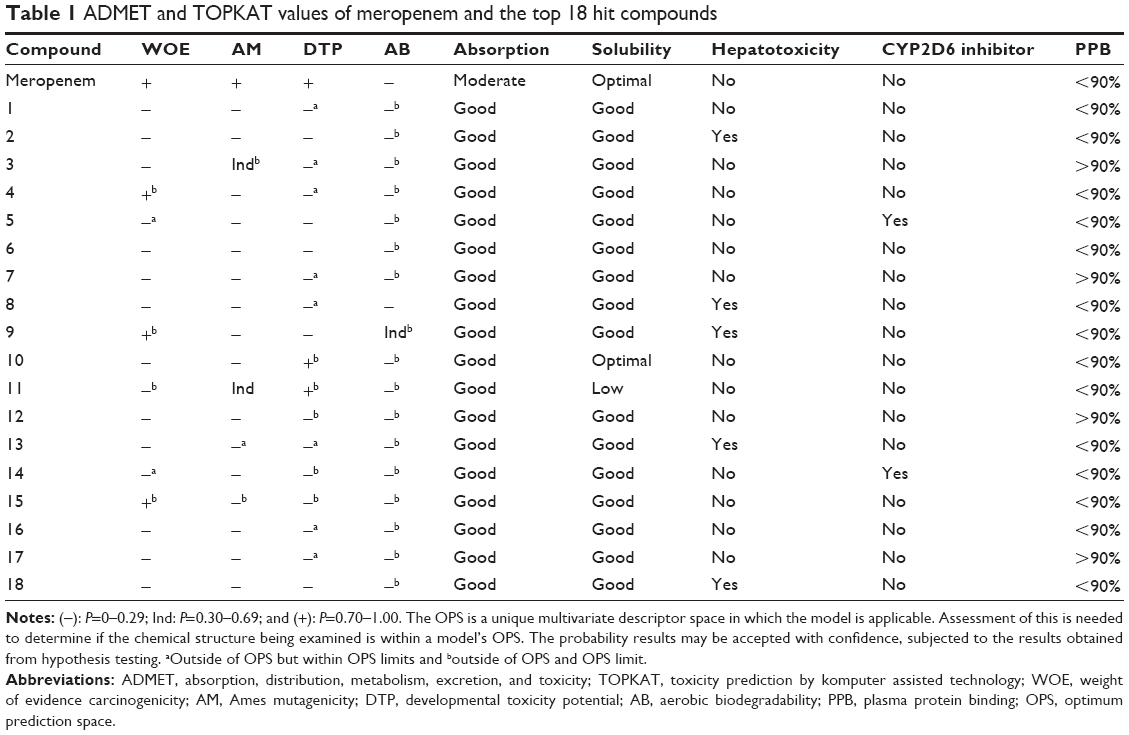 | Table 1 ADMET and TOPKAT values of meropenem and the top 18 hit compounds |
Among these 18 compounds, two were available for acquisition from Enamine Ltd. These two compounds, namely compound 1 and compound 2, consequently underwent in vitro antimycobacterial screening using the resazurin microtiter assay (REMA). However, due to limitations in biosafety facilities and resources, we were constrained to use only Mtb H37Ra, an avirulent strain of the mycobacterium. Results of the REMA screening showed that compound 2 demonstrated excellent activity against H37Ra with percentage growth inhibition of ~94% at a concentration of 25 μM and 89% at 0.2 μM (Figure 3). Moreover, this remarkable action of compound 2 was sustained for a period of at least 3 days (Figure 4).
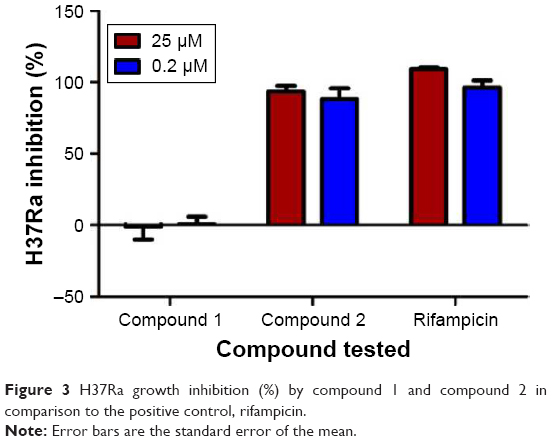 | Figure 3 H37Ra growth inhibition (%) by compound 1 and compound 2 in comparison to the positive control, rifampicin. |
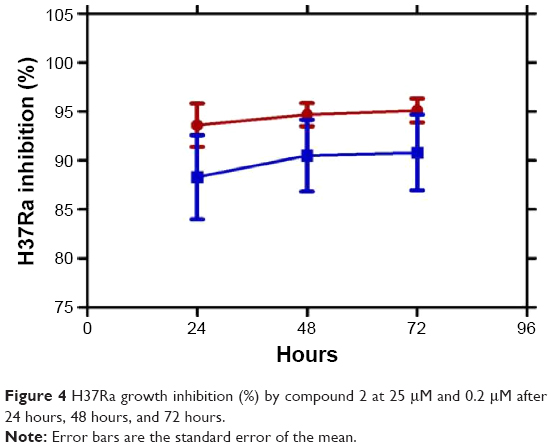 | Figure 4 H37Ra growth inhibition (%) by compound 2 at 25 μM and 0.2 μM after 24 hours, 48 hours, and 72 hours. |
Ligand interaction analysis
In silico docking of meropenem, a known inhibitor into the active site of LdtMt2 has shown characteristic protein–ligand interactions. A number of interactions with this complex was also observed in the Compound2-LdtMt2 complex (Table 2).
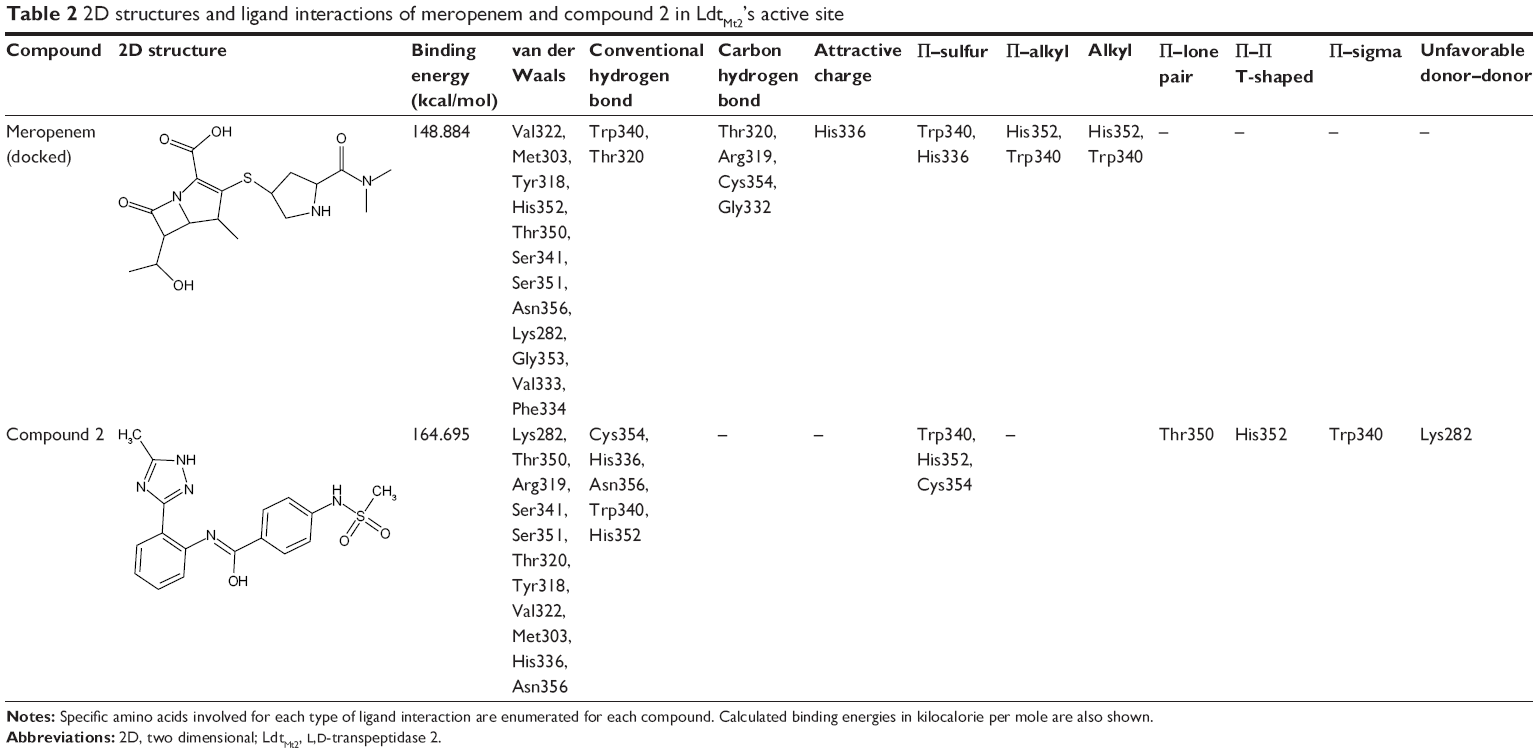 | Table 2 2D structures and ligand interactions of meropenem and compound 2 in LdtMt2’s active site |
Optimization of the protein model involved docking the meropenem ligand into the LdtMt2 binding site. The docked LdtMt2–meropenem complex, as shown in Figure 5, displayed hydrogen bonding with Cys354 (d1=3.02 Å, d2=3.74 Å) and Gly353 (d=3.38 Å). In LdtMt2, His352, Gly353, and Cys354 form an oxyanion cavity, wherein the carbonyl group of the opened meropenem β-lactam ring is hydrogen bound to the mentioned residues. The docking data of meropenem depicted H-bonds with His352 and Cys354, which were proposed as two of the three key catalytic residues in LdtMt2.17,18,28 The H-bond with Cys354 could be the representation of the thioester bond between meropenem and LdtMt2, since covalent bonding cannot be computed via Accelrys DS. In both originally bound and docked meropenem, a number of similar contacts were identified: van der Waals interaction with Val333; polar interaction with Gly353, Cys354, Tyr318, and His352; and side-chain H-bond with Cys354.
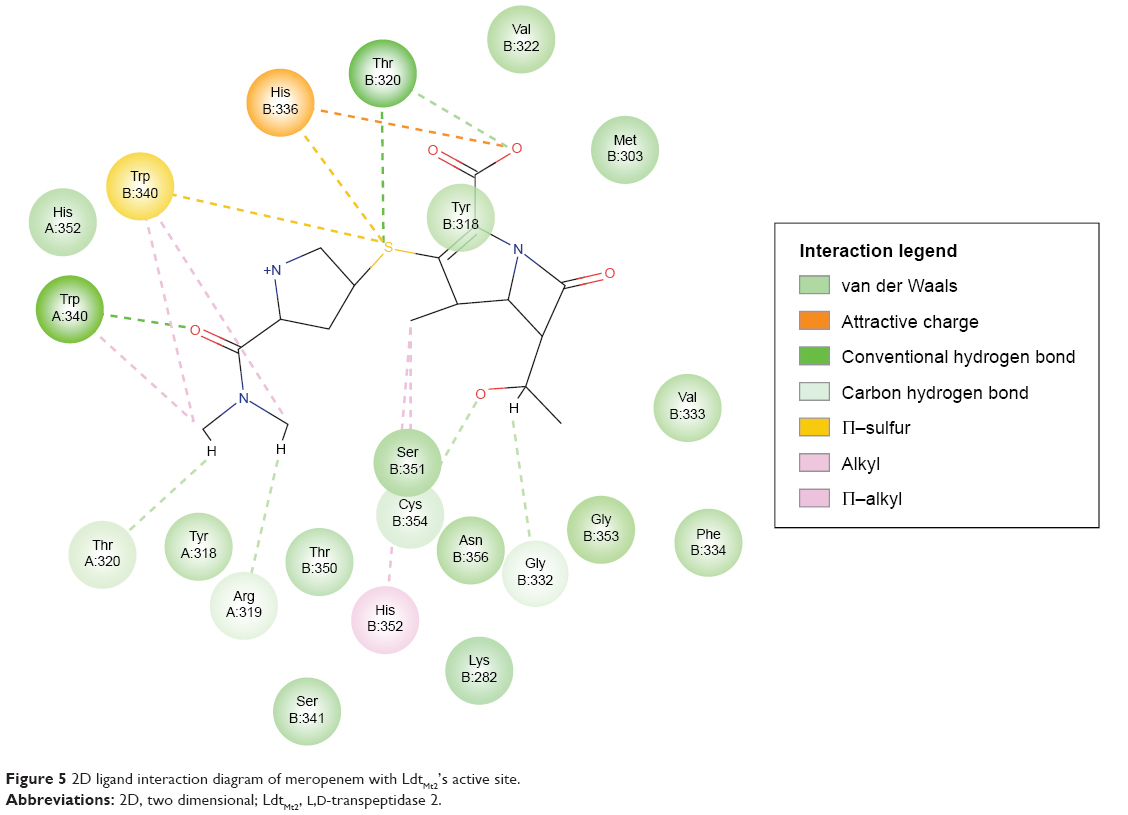 | Figure 5 2D ligand interaction diagram of meropenem with LdtMt2’s active site. |
Compound 2, which is the compound observed to be active in vitro against the TB strain, when docked within the LdtMt2 protein, was predicted to have pi interaction with the sulfur of Cys354, which could be parallel to the meropenem–Cys354 thioester bond.17,18 Of the catalytic triad, His336 and Cys354 were predicted to have H-bonding with meropenem, while other conventional H-bonds observed were with residues Trp340, His352, and Asn356. A Π–sulfur contact between meropenem and Trp340, a T-shaped Π–Π interaction with His352, and a Π–lone pair with Thr350 were observed. A number of van der Waals interactions were also predicted (Figure 6). A significant donor–donor clash was predicted between the meropenem pyrroline and Lys282. It is suggested that eliminating Lys282 or substituting the residue with another amino acid may further improve the stability of the complex.
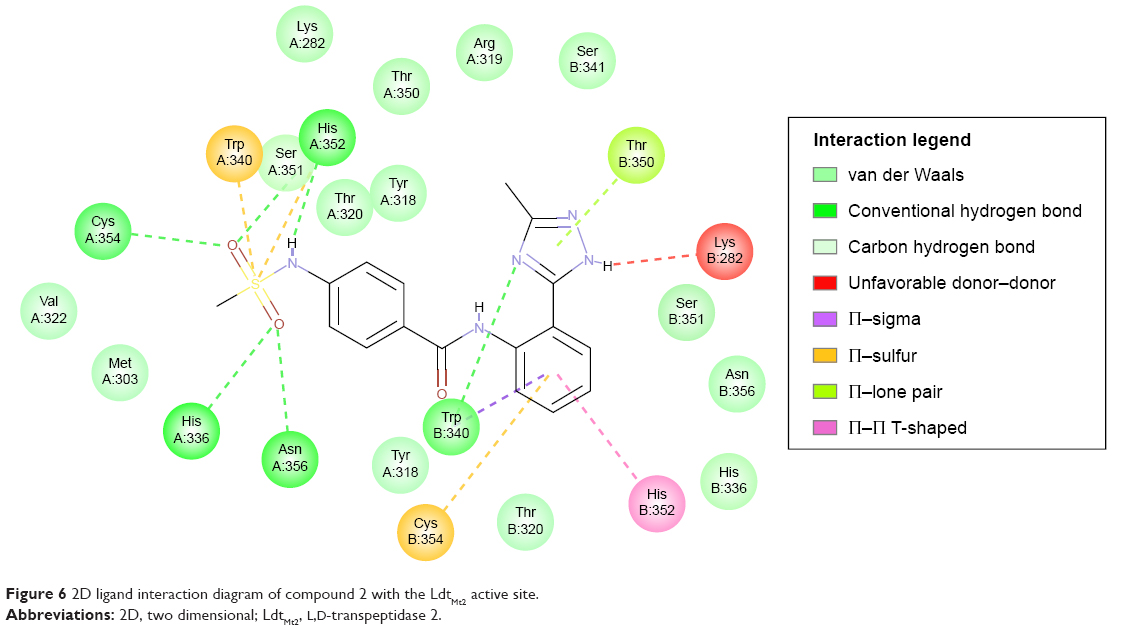 | Figure 6 2D ligand interaction diagram of compound 2 with the LdtMt2 active site. |
Conclusion
Pharmacophore-based virtual screening, molecular docking, as well as in silico ADMET evaluation of compounds from the enamine database were performed in order to identify a new class of potential antitubercular lead compounds. Out of the 4.5 million compounds screened, 18 compounds were found to have better binding energies than meropenem and with satisfactory in silico ADMET properties. Out of the two of the 18 compounds that were tested in vitro, compound 2 was found to have an excellent bioactivity against Mtb H37Ra. Consequently, this lead compound may lead to a novel class of anti-TB drugs in the future.
Acknowledgment
This work was funded by the UP System Emerging Interdisciplinary Research Program (OVPAA-EIDR 12-001-121102).
Disclosure
The authors report no conflicts of interest in this work.
References
Brennan P. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis. 2003;83(1–3):91–97. | ||
Erdemli SB, Gupta R, Bishai WR, Lamichhane G, Amzel LM, Bianchet MA. Targeting the cell wall of Mycobacterium tuberculosis: structure and mechanism of L,D-transpeptidase 2. Structure. 2012;20(12): 2103–2115. | ||
Gupta R, Lavollay M, Mainardi JL, Arthur M, Bishai WR, Lamichhane G. The Mycobacterium tuberculosis protein LdtMt2 is a nonclassicaltranspeptidase required for virulence and resistance to amoxicillin. Nat Med. 2010;16(4):466–469. | ||
Zumla A, Nahid P, Cole ST. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov. 2013;12(5):388–404. | ||
Dreetz M, Hamacher J, Eller J, et al. Serum bactericidal activities and comparative pharmacokinetics of meropenem and imipenem-cilastatin. Antimicrob Agents Chemother. 1996;40(1):105–109. | ||
WHO [webpage on the Internet]. World Health Organization Global Tuberculosis Report 2015. Available from: http://apps.who.int/iris/bitstream/10665/191102/1/9789241565059_eng.pdf?ua=1. Accessed November 6, 2015. | ||
Matviiuk T, Rodriguez F, Saffon N, et al. Design, chemical synthesis of 3-(9H-fluoren-9-yl)pyrrolidine-2,5-dione derivatives and biological activity against enoyl-ACP reductase (InhA) and Mycobacterium tuberculosis. Eur J Med Chem. 2013;70:37–48. | ||
Sharma SK, Mohan A. Multidrug-resistant tuberculosis. Indian J Med Res. 2004;120(4):354–376. | ||
Jain RF. Faulty prescription – an avoidable cause of MDR-TB. Indian J Tuberc. 1998;45:141–143. | ||
Showalter HDH, Denny WA. A roadmap for drug discovery and its translation to small molecule agents in clinical development for tuberculosis treatment. Tuberculosis (Edinb). 2008;88(suppl 1): S3–S17. | ||
Coxon GD, Cooper CB, Gillespie SH, McHugh TD. Strategies and challenges involved in the discovery of new chemical entities during early-stage tuberculosis drug discovery. J Infect Dis. 2012;205(suppl 2):S258–S264. | ||
Stanley SA, Grant SS, Kawate T, et al. Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem Biol. 2012;7(8):1377–1384. | ||
Lounnas V, Ritschel T, Kelder J, McGuire R, Bywater RP, Foloppe N. Current progress in structure-based rational drug design marks a new mindset in drug discovery. Comput Struct Biotechnol J. 2013;5:e201302011. | ||
Petrova T, Chuprina A, Parkesh R, Pushechnikov A. Structural enrichment of HTS compounds from available commercial libraries. Med Chem Commun. 2012;3:571–579. | ||
Accelrys Software Inc., Discovery Studio Modeling Environment, Release 4.0, San Diego: Accelrys Software Inc., 2007. | ||
Fletcher R. Function minimization by conjugate gradients. Comput J. 1964;7(2):149–154. | ||
Li WJ, Li DF, Hu YL, Zhang XE, Bi LJ, Wang DC. Crystal structure of L,D-transpeptidase LdtMt2 in complex with meropenem reveals the mechanism of carbapenem against Mycobacterium tuberculosis. Cell Res. 2013;23(5):728–731. | ||
Kim HS, Kim J, Im HN, et al. Structural basis for the inhibition of Mycobacterium tuberculosis L,D-transpeptidase by meropenem, a drug effective against extensively drug-resistant strains. Acta Crystallogr D Biol Crystallogr. 2013;69(pt 3):420–431. | ||
Wu G, Robertson DH, Brooks CL, Vieth M. Detailed analysis of grid-based molecular docking: a case study of CDOCKER-ACHARMm-based MD docking algorithm. J Comput Chem. 2003;24(13): 1549–1562. | ||
Triboulet S, Arthur M, Mainardi JL, et al. Inactivation kinetics of a new target of -lactam antibiotics. J Biol Chem. 2011;286(26): 22777–22784. | ||
Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32(2):234–258. | ||
Wietzerbin J, Das BC, Petit JF, Lederer E, Leyh-Bouille M, Ghuysen JM. Occurrence of D-alanyl-(D)-meso-diaminopimelic acid and meso-diaminopimelyl-meso-diaminopimelic acid interpeptide linkages in the peptidoglycan of Mycobacteria. Biochemistry. 1974;13(17):3471–3476. | ||
Lavollay M, Arthur M, Fourgeaud M, et al. The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by L,D-transpeptidation. J Bacteriol. 2008;190(12):4360–4366. | ||
England K, Boshoff HIM, Arora K, et al. Meropenem-Clavulanic acid shows activity against Mycobacterium tuberculosis in vivo. Antimicrob Agents Chemother. 2012;56(6):3384–3387. | ||
Wermuth CG, Ganellin CR, Lindberg P, Mitscher LA. Glossary of terms used in medicinal chemistry (IUPAC recommendations 1998). Pure Appl Chem. 1998;70(5):1129–1143. | ||
Van de Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov. 2003;2(3):192–204. | ||
Valerio LG Jr. In silico toxicology for the pharmaceutical sciences. Toxicol Appl Pharmacol. 2009;241(3):356–370. | ||
Lecoq L, Bougault C, Hugonnet JE, et al. Dynamics induced by β-lactam antibiotics in the active site of Bacillus subtilis L,D-transpeptidase. Structure. 2012;20(5):850–861. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.