")
Back to Journals » International Journal of Nanomedicine » Volume 16
Topical Delivery of Rapamycin by Means of Microenvironment-Sensitive Core-Multi-Shell Nanocarriers: Assessment of Anti-Inflammatory Activity in an ex vivo Skin/T Cell Co-Culture Model
Authors Rancan F , Guo X , Rajes K , Sidiropoulou P , Zabihi F, Hoffmann L, Hadam S, Blume-Peytavi U, Rühl E, Haag R, Vogt A
Received 27 July 2021
Accepted for publication 9 September 2021
Published 22 October 2021 Volume 2021:16 Pages 7137—7151
DOI https://doi.org/10.2147/IJN.S330716
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Israel (Rudi) Rubinstein
Fiorenza Rancan,1 Xiao Guo,1 Keerthana Rajes,2 Polytimi Sidiropoulou,1 Fatemeh Zabihi,2 Luisa Hoffmann,1 Sabrina Hadam,1 Ulrike Blume-Peytavi,1 Eckart Rühl,3 Rainer Haag,2 Annika Vogt1
1Clinical Research Center for Hair and Skin Science, Department of Dermatology and Allergy, Charité–Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; 2Institute of Chemistry and Biochemistry, Freie Universität Berlin, Berlin, Germany; 3Physical Chemistry, Institute of Chemistry and Biochemistry, Freie Universität Berlin, Berlin, Germany
Correspondence: Fiorenza Rancan
Clinical Research Center for Hair and Skin Science, Department of Dermatology and Allergy, Charitéplatz 1, Berlin, 10117, Germany
Tel +49 30 450 518 347
Fax +49 30 450 518 952
Email [email protected]
Introduction: Rapamycin (Rapa) is an immunosuppressive macrolide that inhibits the mechanistic target of rapamycin (mTOR) activity. Thanks to its anti-proliferative effects towards different cell types, including keratinocytes and T cells, Rapa shows promise in the treatment of skin diseases characterized by cell hyperproliferation. However, Rapa skin penetration is limited due to its lipophilic nature (log P = 4.3) and high molecular weight (MW = 914 g/mol). In previous studies, new microenvironment-sensitive core multishell (CMS) nanocarriers capable of sensing the redox state of inflamed skin were developed as more efficient and selective vehicles for macrolide delivery to inflamed skin.
Methods: In this study, we tested such redox-sensitive CMS nanocarriers using an inflammatory skin model based on human skin explants co-cultured with Jurkat T cells. Serine protease (SP) was applied on skin surface to induce skin barrier impairment and oxidative stress, whereas phytohaemagglutinin (PHA), IL-17A, and IL-22 were used to activate Jurkat cells. Activation markers, such as CD45 and CD69, phosphorylated ribosomal protein S6 (pRP-S6), and IL-2 release were monitored in activated T cells, whereas pro-inflammatory cytokines were measured in skin extracts and culture medium.
Results: We found that alteration of skin barrier proteins corneodesmosin (CDSN), occludin (Occl), and zonula occludens-1 (ZO-1) as well as oxidation-induced decrease of free thiol groups occurred upon SP-treatment. All Rapa formulations exerted inhibitory effects on T cells after penetration across ex vivo skin. No effects on skin inflammatory markers were detected. The superiority of the oxidative-sensitive CMS nanocarriers over the other formulations was observed with regard to drug delivery as well as downregulation of IL-2 release.
Conclusion: Overall, our results demonstrate that nanocarriers addressing features of diseased skin are promising approaches to improve the topical delivery of macrolide drugs.
Keywords: redox-sensitive nanoparticles, sirolimus, psoriasis, stratum corneum barrier, dermatology, drug release
Graphical Abstract:
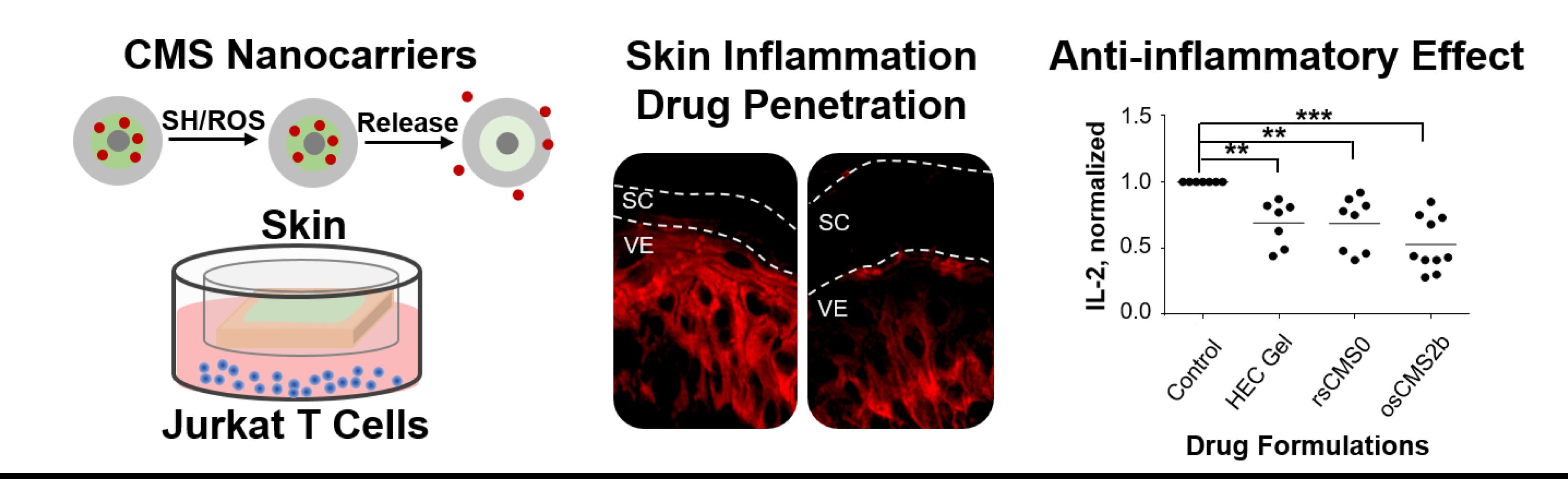
Introduction
Immunomodulatory drugs from the macrolide group, such as everolimus or tacrolimus, represent a valuable alternative to steroids and could complement the spectrum of anti-inflammatory drugs for the topical management of inflammatory skin conditions. Originally introduced for rejection prophylaxis in transplant recipients, rapamycin (Rapa), also known as sirolimus, additionally exerts anti-proliferative effects on keratinocytes, which makes it an interesting candidate for the topical treatment of hyperproliferative inflammatory skin disorders like psoriasis.1 As a potent inhibitor of the mechanistic target of rapamycin (mTOR), Rapa suppresses transcription factors regulating cell proliferation and differentiation2 and induces cell cycle arrest and apoptosis.3 A systematic review of the studies in which Rapa was used to treat dermatological conditions has shown evidence for the benefits in the treatment of certain inflammatory skin diseases such as lupus erythematosus, pemphigus vulgaris, or scleroderma.4 Less evidence was found for the use of Rapa to manage psoriasis. Nevertheless, one clinical trials has shown that, even is plaque thickness and erythema were not improved, topical application of Rapa could ameliorate psoriasis clinical score and reduce CD4+ cells as well as Ki67 expression in the epidermis.5 The reason for these limited outcomes may be due to Rapa high molecular weight and low skin permeability that largely hamper the use of this drug for topical therapies. Of note, in atopic dermatitis, a condition characterized by pronounced disruption of skin barrier integrity, topical tacrolimus can sufficiently penetrate across inflamed lesions to control mild to moderate flare-ups. In contrast, penetration processes in chronic hyperproliferative conditions such as psoriasis are more challenging, despite of the fact that keratinocyte hyper-proliferation, impaired differentiation, and the presence of immune infiltrates as well as pro-inflammatory mediators can all alter key barrier elements, including lipid composition, corneocyte architecture, and tight junction functionality.6–8
Many studies have provided convincing evidence to support the potential use of nanotechnology-based formulations for the topical delivery of immunosuppressants.9–11 Nanoparticles can improve the penetration of drugs by different mechanisms. Occlusion or skin hydration have been shown for solid lipid nanocarriers12 and nanogels,13 respectively. Particles with size of and above 200 nm can form depots in the hair follicle canals.14,15 Nanocrystals can increase the drug concentration gradient and, thus, favor the passive diffusion of the drug.16 Other nanocarriers, such as ethosomes, were shown to alter the skin barrier.17 Among the various approaches, core-multishell (CMS) nanocarriers possess highly promising characteristics for penetration enhancement. They are unimolecular carrier systems with a precise chemical composition, ie, a hydrophilic dendritic core molecule surrounded by covalently attached linear amphiphilic molecules that form an inner hydrophobic shell surrounded by an outer hydrophilic framework. Their low size and amphiphilic structure confer them high SC penetration ability.18 In addition, thanks to their intramolecular amphiphilic environment, they can transport a wide variety of cargos.19 In fact, previous studies have demonstrated their ability to effectively incorporate and deliver model drugs with high or low skin permeability such as dexamethasone20,21 and tacrolimus.22
In recent years, efforts have been focused on optimizing selective drug delivery by exploiting specific features of target tissues. For example, microenvironment-sensitive nanocarriers have been synthesized that release the loaded drug when triggered by changes in parameters like pH, temperature, or redox levels.23–26 In this regard, redox-sensitive nanocarriers represent a promising approach for drug delivery to skin. While healthy skin maintains a redox homeostasis and is rich in antioxidants, a state of oxidative stress is typically found in inflamed skin with an overproduction of reactive oxygen species (ROS) resulting in an oxidative environment.27–29
Thus, in this study, we addressed the question as to whether redox-sensitive CMS nanocarriers can improve the topical delivery of macrolides to inflamed skin thereby improving their anti-inflammatory effects. Following this concept, we tested the anti-inflammatory effects of reduction-sensitive (rsCMS) and oxidative-sensitive (osCMS) nanocarriers. The core of these CMS nanocarriers is a hyperbranched polyglycerol, whereas the inner shell is given by linear disulfide or thioether chains and the outer shell is formed by methoxy poly (ethylene glycol) groups. The rsCMS nanocarriers were prepared by introducing disulfide groups in the inner shell,30 while thioether groups were inserted to obtain the osCMS nanocarriers.31 The rsCMS nanocarriers have been shown to release the loaded cargo upon partial degradation following the reaction of free thiol groups (like glutathione) with the disulfide groups. As for the osCMS nanocarriers, the incorporated thioethers can be oxidized to sulfoxide by ROS. This results in a polarity alteration in the inner shell and consequent drug release.
To test the biological efficacy of these innovative formulations, we developed an ex vivo inflammatory skin model based on human skin explants co-cultured with Jurkat T cells in a trans-well set-up. The skin surface was pre-treated with serine protease (SP) to induce an impairment of the SC barrier,32,33 activation of PAR2-mediated inflammatory cytokine release,34–37 and free radical formation.38 Furthermore, Jurkat cells were added to the system and activated with phytohaemagglutinin (PHA) and Th17 cytokines (IL-17A and IL-22) to reproduce the inflammatory environment typical for psoriasis.39 This skin-cell co-culture set-up allowed us to measure the selectivity of drug release by oxidative-sensitive CMS nanocarriers as well as the effects of the delivered drug towards inflammatory T cells.
Materials and Methods
Preparation and Characterization of Rapa-Loaded CMS Nanocarriers
In previous studies, different redox-sensitive nanocarriers with disulphide or thioether groups in the inner shell were prepared. In case of the oxidation-sensitive carriers, variations in the positions of the thioethers within the inner shell were investigated. These variations influenced the loading capacity, drug release, and skin penetration properties of the nanocarriers. The best results were reached for the CMS nanocarrier with two thioether groups in the δ- position to the carboxyl groups. The detailed synthesis of rsCMS and osCMS nanocarriers is reported in previous publications.30,31 Briefly, the CMS nanocarriers investigated in this study were prepared as follows: The rsCMS0 was synthesized by oxidating mercaptoundecanoic acid to disulfanediyldiundecanoic acid. This was then reacted with methoxy poly (ethylene glycol) amine through peptide coupling. The formed double shell was then coupled to hyperbranched polyglycerol amine using a peptide coupling reaction and forming the final carrier. The osCMS2b carrier was prepared by first synthesizing (pentanediylbis(sulfanediyl))dibutyric acid through a thiol-bromide coupling reaction of pentane dithiol and bromobutanoic acid. The diacid was then reacted with methoxy poly (ethylene glycol) amine and subsequently coupled to hyperbranched polyglycerol amine through peptide coupling. Stock solutions of the nanocarrier with concentration of 5 mg/mL were prepared in Milli-Q water.
In preliminary experiments, various methods for drug encapsulation were tested such as the film method (creating a film of the drug on a glass vial and stirring a carrier solution in it), the solvent method (adding a small amount of an organic solvent to increase the drug solubility in the carrier suspension and then remove it), or the horn sonication method. In case of the osCMS2b carrier, the film methods lead to lower drug encapsulation than horn sonication while the solvent method can lead to drug encapsulations of more than 10 wt% (data not shown). However, formulations with 10 wt% drug loading are known to be unstable as most of the drug will be located in the outer shell of the carrier leading to drug precipitation or carrier aggregation. For the rsCMS0 carrier, similar drug loading was reached with all methods. Effects of the different drug loading methods were studies using the model drugs Pheophorbide A and meso-tetra (m-hydroxyphenyl) porphyrin and are described in Rajes et al.30 In the present study, Rapa was loaded on the nanocarriers by horn sonication of the CMS carrier stock solution in the presence of 50 wt% Rapa. To remove non-encapsulated drug, the suspension was centrifuged at 570 × g and the supernatant kept. The drug loading was measured by HPLC as described previously30 and was approximately 5 wt% for both nanocarriers. For skin penetration experiments, the CMS nanocarriers were loaded with the fluorescent dye Atto Oxa12 NHS ester (AttoOxa, ATTO-TEC, Siegen, Germany). For the loading, 0.5 mL of the CMS suspension was stirred with 10 µL of a 3 mM AttoOxa solution overnight. Assuming that all molecules were encapsulated, a drug loading of 0.1 wt% was achieved. AttoOxa has a molecular weight of 835 g/mol and maxima of absorption and emission at 663 and 684 nm.
Skin Samples
Human skin was donated by healthy donors undergoing abdominal and breast cosmetic surgeries. The experimental protocol was in compliance with the guidelines of the Declaration of Helsinki. Approval for the study was obtained by the Ethics Committee of the Charité – Universitätsmedizin Berlin (approval EA1/135/06, January 2019). Written informed consent was signed by each donor. Skin from 15 donors was used. For skin barrier immunostaining and penetration experiments tissue from three donors was used, while tissue from other 12 donors was used for co-culture experiments. The excised skin was processed within 2 to 4 h after surgery. First, skin surface was examined to detect macroscopic damages or stretches. Only intact skin was used. Skin was carefully cleaned with sterile saline solution (NaCl, 0.9%), cut to pieces of approximately 1.5 × 1.5 (length, width) using a scalpel. Subcutaneous adipose tissue was removed with scissors.
SP-Treatment and Skin Culture Conditions
To partially remove SC lipids and facilitate SP penetration, 50 µL of a 1:1 mix of methanol: chloroform (Merck KGaA, Darmstadt, Germany) were dropped on a filter paper disc (SmartPractice Phoenix, AZ, USA) that was placed on the top of the skin sample. After 1 min, the filter paper discs were removed. Skin pieces were transferred on cell culture inserts (8 µm pore size membrane, BD Falcon™, Durham, NC, USA). The inserts were placed in six-well culture plates (BD Falcon™) and each well was filled with 2 mL RPMI-1640 medium (PAA, Heidelberg, Germany) supplemented with 10% fetal calf serum (FCS, PAA, Heidelberg, Germany), 100 I.E./mL penicillin, and 100 g/mL streptomycin (Sigma-Aldrich, Hamburg, Germany). Pig pancreas trypsin (Biochrom, Berlin, Germany) was applied on the top of the skin samples (20 µL of a 0.15 mg/mL solution, ie, 3 µg/cm2) leaving untreated margins of 0.5 cm to avoid the overflow of the applied solution. To skin without SP-treatment, 20 µL of 0.9% saline were applied topically. After 18 h incubation at 37 °C, 5% CO2, and 95% humidity, SP and saline were removed using a cotton swab.
Immunofluorescence Staining of Skin Cryosections
To investigate the effect of SP on skin barrier, corneodesmosomes and tight junction components, ie, corneodesmosin (CDSN), occludin (Occl) and zonula occludens-1 (ZO-1) were visualized by immunofluorescence staining. Skin from three different donors was treated with trypsin (+ SP) or with saline (−SP) as described above. After treatment, skin was frozen in liquid nitrogen and cut with a cryo-microtome (Frigocut 2800 N, Leica, Bensheim, Germany) to obtain 5 µm-thick sections. Tissue sections were fixed in acetone for 10 min at room temperature (r.t.) and incubated for 1 h at r.t. with DAKO® protein block (Dakocytomation, Carpinteria, USA). Polyclonal rabbit antibodies to human Occl (71–1500 Invitrogen, Carlsbad, CA, USA), ZO-1 (61–7300 Invitrogen) and CDSN/S protein (ab204235, Abcam, Cambridge, UK) were diluted at 1:100 in Dulbecco’s phosphate-buffered saline (PBS; Biochrom, Berlin, Germany) containing 5% FCS (PBS/5% FCS) and applied to the sections overnight (16 h) in a humidified chamber at 4 °C. After 3 washes in double-distilled water, sections were further incubated with the fluorescein-labelled secondary antibody (goat anti-rabbit IgG (H+L), FI-1000; Vector Laboratories, CA, USA) diluted 1:50 in PBS with 5% FCS. Sections were incubated in the dark for 45 min at r.t. in a humidified chamber. Finally, the slides were washed for 3 times for 5 min in double distilled water, air-dried for 2 h at r.t. and protected with a coverslip. Control sections using identical protocol but without antibodies were also prepared. Pictures of stained skin sections were taken using a CCD camera coupled to an LSM 700 confocal laser scanning microscope (Carl Zeiss Microscopy GmbH, Jena, Germany) using an excitation wavelength of 488 nm and detecting emission at wavelengths above 495 nm. Representative images for each barrier component and donor were taken using the same microscope and camera settings. The mean fluorescence intensity (MFI) of at least 15–20 areas per sample was evaluated using the ImageJ software, version 1.47 (National Institute of Health, Bethesda, MD, USA).
Staining of Skin Thiol Groups
Tissue sections were fixed in acetone for 10 min at r.t. and then stained with indocarbocyanine-maleimide (ICC-mal, 0.1 µg/mL, 10 min, r.t.), which was synthesized according to Reichert et al.40 Sections were washed three times with double distilled water and let dry for 30 min. Images were acquired with the LSM 700 microscope using the excitation wavelength of 550 nm and emission wavelengths above 570 nm. The MFI of at least 20 areas per sample was calculated using the ImageJ software.
Effects of SP on Skin Penetration of AttoOxa
Skin samples from three donors were incubated with trypsin (+SP) or saline (−SP) for 16 h and then were topically treated with AttoOxa-loaded osCMS2b and rsCMS0 nanocarriers as well as AttoOxa solution (DMSO/water 1:100). The nanocarrier suspensions as well as the dye solution were formulated in 2.5% hydroxyl ethyl cellulose (HEC) gel (final dye concentration of 5 µg/mL). A 40 µL volume of each formulation was applied on 1 cm2 of skin reaching a final applied dye amount of 0.2 µg/cm2. Samples were incubated for 24 h in cell culture inserts as described above. Non-penetrated material was removed from skin surface using a cotton swab and untreated borders were removed with a scalpel. The samples were frozen in liquid nitrogen, and stored at –20 °C until processing for cryosections. Images from at least 15 cryosections were acquired and analyzed with ImageJ as described above.
Co-Culture of Full-Thickness Human Skin and Jurkat Cells
To test the biological activity of Rapa-loaded CMS nanocarriers, an inflammatory ex vivo skin-cell co-culture model was used. Skin from 12 donors was used in these experiments. Jurkat cells (Clone E6-1, ATCC® TIB-152™) were added in the lower compartment of the trans-well set-up (1 × 106 cells/well in 2 mL supplemented RPMI 1640 medium) while skin samples pre-treated with trypsin (+ SP) or with saline only (−SP) were placed in the inserts as described above. Cells were stimulated with PHA-M (5 µg/mL, Roche Diagnostic, Mannheim, Germany), IL-22 (100 ng/mL, PromoCell, Heidelberg, Germany), and IL-17A (200 ng/mL, GenScript USA, Piscataway, NJ, USA). Rapa-loaded on rsCMS and osCMS nanocarriers (5 mg/mL, loading: 5% w/w) formulated in 2.5% HEC gel were applied on the skin surface (40 µL/cm2) reaching administered Rapa doses of about 10 µg/cm2. Rapa formulation in 70% ethanol and 2.5% HEC gel were also applied to reach a Rapa amount of approximately 10 µg/cm2. Control skin was treated with 40 µL of 2.5% HEC gel in water. After 24 h of incubation at 37 °C, 5% CO2, the non-penetrated formulation was removed using cotton swabs. Afterward, the central part of skin samples was collected using an 8 mm punch biopsy (Kai Europe, Solingen, Germany), shock-frozen in liquid nitrogen and stored at –20 °C for further processing. Jurkat cells were collected and centrifuged for 10 min at 380 × g. The supernatant was collected and stored at −20 °C previous cytokine analysis by enzyme-linked immunosorbent assay (ELISA). Cell pellets were re-suspended in 4% formaldehyde, incubated at r.t. for 10 min, and washed once with PBS. Cells were stored at 4 °C, previous immunohistochemical staining and flow cytometry analysis.
Preparation of Skin Extracts
Using a cryo-microtome (Frigocut 2800 N, Leica, Bensheim, Germany), horizontally sections of the frozen skin blocks were cut. For the epidermis (the upper 100 µm), 5 sections of 20 µm in thickness were cut, whereas for the upper dermis (the lower 900 µm) 18 sections of 50 µm thickness were prepared. Tissue samples were extracted by incubation of the sectioned tissue in 1 mL of cold extraction buffer (100 mM Tris-HCl; 150 mM NaCl; 1 mM EDTA; 1% Triton-X-100) in a Thermo mixer at 4 °C, 700 rpm for 90 min. Samples were then sonicated (70 Hz, 200 Weff) for 10 min at 4 °C, vortexed and centrifuged for 5 min at 380 × g and 4 °C. The supernatants were stored at –20 °C and used for cytokine analysis.
Enzyme-Linked Immunosorbent Assay (ELISA)
Twelve independent experiments with skin from different donors were performed. Human IL-2 and IL1-α were quantified using the R&D Systems ELISA kit (Minneapolis, MN, USA). Human IL-6 and IL-8 were quantified using the CytoSetTM kits (Invitrogen, Carlsbad, CA, USA). Each sample was run in duplicate, following the manufacturer instructions. The obtained amounts of IL1-α, IL-6, and IL-8 in tissue extracts were normalized to total protein content measured with the Pierce 660nm Protein Assay (Thermo Scientific Inc., Rockford, USA). Absorbance values were measured with the EnSpire Multimode plate reader (Perkin Elmer, Akron, OH, USA).
Immunofluorescence Staining of Jurkat Cell Markers and Flow Cytometry Analysis
Fixed Jurkat cells were incubated in 3% bovine serum albumin (BSA, Biomol, Hamburg, Germany) for 10 min, centrifuged (380× g, 10 min), and resuspended in 100 µL of 3% BSA. Cells were then stained for 20 min with 5 µL of FITC-labeled anti-CD45 (Biozol, München, Germany) and 5 µL of PE-labeled anti-CD69 antibodies (Biomol, Hamburg, Germany). After centrifugation and re-suspension with PBS, cells were centrifuged (380 × g, 10 min), re-suspended in 0.1% Tween-20 for 10 min at r. t. for permeabilization. After centrifugation (380 × g, 10 min), cells were re-suspended in 3% BSA and incubated for 1h at r.t. in the Thermo shaker (700 rpm). Cells were centrifuged (380 × g, 10 min), re-suspended in 100 µL of 3% BSA, and stained with 5 µL of APC-tagged antibody against the phosphorylated ribosomal protein S6 (pRP-S6) (Ser 235/236, Miltenyi, Auburn, CA, USA) for 20 min at r.t. in the Thermo mixer (700 rpm). Finally, cells were centrifuged (380× g, 10 min), re-suspended in PBS and stored at 4 °C until analysis by flow cytometry (FACS Calibur, BD, Germany). At least 50,000 total events were collected. The analysis of collected data was done with the FCS Express software version 3.1 (De Novo Software, USA). The percentages of positive cells were determined by setting a range gate (marker) using non-activated control cells as negative reference. Averages of positive cells from six independent experiments are reported.
Statistics
Means, standard deviations, standard errors, and statistics were calculated with Excel (Microsoft Corp., Redmond, WA, USA). One-way ANOVA followed by a Student’s t-test for comparison of two groups of data were used for statistics. Data were plotted using Excel or Prism GraphPad (GraphPad Software, CA, USA).
Results and Discussion
Effects of SP Treatment on Skin Barrier Proteins, Redox State, and CMS-Mediated Delivery of the High Molecular Weight Dye AttoOxa
SPs play an essential role in epidermal homeostasis, eg, SC desquamation, and are also involved in inflammatory processes.41,42 For this reason, trypsin, a member of the SP family, was employed in the ex vivo skin model to reproduce some of the key features of inflammatory skin.37 Here, to further characterize the effects of topically applied SP in the ex vivo skin model, we investigated the proteolytic effects of low concentrations (3 µg/cm2) of SP towards skin barrier proteins. CDSN is part of corneodesmosomes and its degradation by skin proteases is central in the process of physiological desquamation.43 Occl and ZO-1 are two major components of tight junctions, which act as physical barrier in the stratum granulosum and regulate the diffusion of molecules to the viable epidermis (VE).44 Representative pictures of immunolabelled sections show an intense positive staining for CDSN, Occl, and ZO-1 in control skin (−SP, Figure 1A–C), while a weaker staining is visible in trypsin-treated skin (+ SP, Figure 1A’–C’). The mean fluorescence intensity (MFI) values of the analyzed images confirmed these observations. For all three donors, the MFI of CDSN, Occl, and ZO-1 were significantly reduced in SP-treated skin with respect to untreated skin, indicating a degradation of barrier proteins following exposure to SP (Figure 1A”–C”).
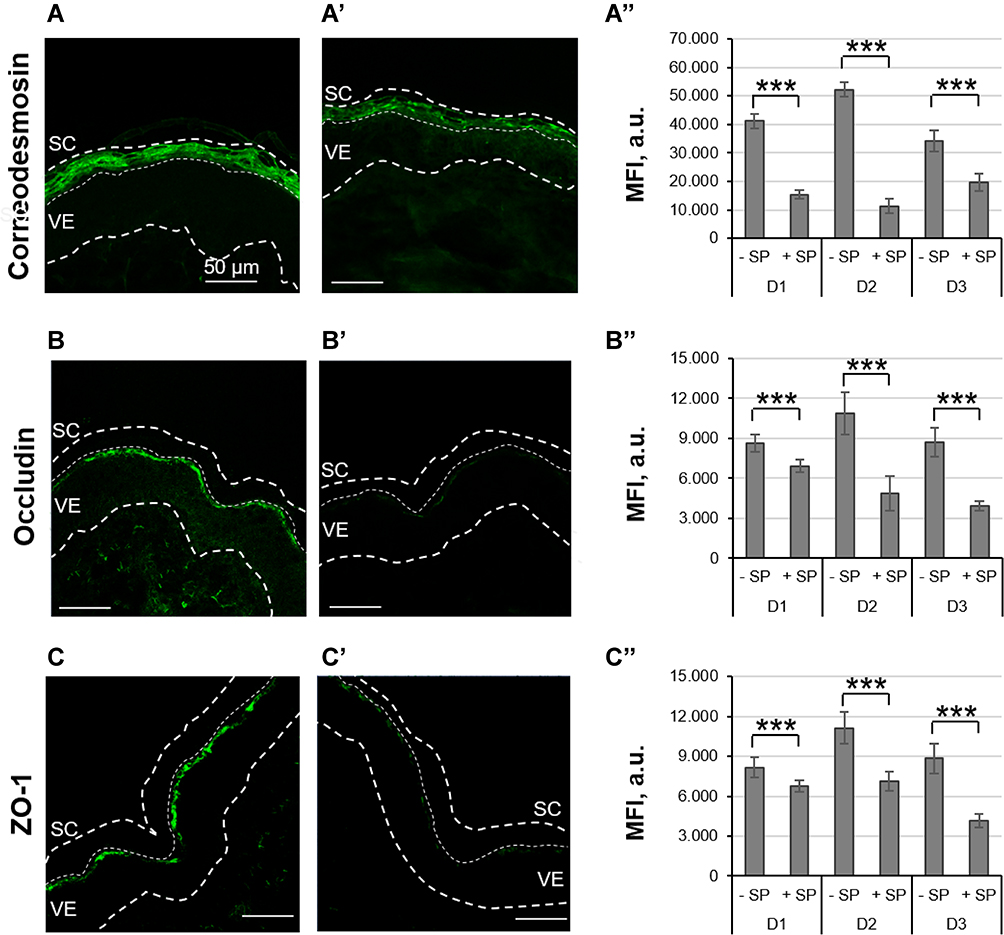 |
Figure 1 Effect of SP treatment on skin barrier proteins. Ex vivo skin was treated for 16 h with topically applied trypsin (+ SP), while control skin was treated with saline (−SP). Immunohistostaining of CDSN (A–A”), Occl (B–B”), and ZO-1 (C–C”) was performed on skin sections from three donors (D1-D3). Representative pictures of sections from saline-treated (A–C) and SP-treated (A’–C’) skin show lower fluorescence intensity in skin incubated with SP. Scale bars = 50 µm. The analysis of several sections from three different donors are summarized in (A”–C”). Statistically lower MFI values were found for sections of SP-pretreated skin (***p < 0.001). |
In previous in vitro studies with keratinocytes, we have shown that SP can induce the formation of reactive oxygen species.38 In this study, we investigated the effect of SP-treatment on skin redox state by labelling free thiol groups with ICC-mal (Figure 2A and B). Skin is rich in glutathione, which represents one of the main reductive species involved in the maintenance of the cutaneous redox state.45,46 In inflamed skin, several oxidation processes are activated, resulting in a decrease of glutathione and, as consequence, of free thiol groups. Thus, the detection of free thiol groups in skin sections was used to put in evidence the SP-mediated oxidative effects on ex vivo human skin (Figure 2A). In control skin, all VE layers (stratum basale, stratum spinosum, and stratum granulosum) were positive for thiol group staining, which is in accordance with the wide presence of glutathione in the epidermis. A less intensive but still measurable thiol group-related fluorescence was detected in the SC. In contrast, the thiol group staining was markedly reduced in the SC and in the stratum granulosum of skin treated with SP, most likely as a result of free radical-mediated oxidation. These results are in line with the findings of Pickard et al, who reported the presence of a thiol reach layer in the lower part of the SC and its depletion after skin exposure to 2,4-dinitrothiocyanobenzene.47 The semi-quantitative analysis of at least 30 images (Figure 2B) showed a significant reduction of thiol groups in both SC and VE of SP-treated skin and further evidenced the potential of SP to create an oxidative environment in the epidermis upper layers.
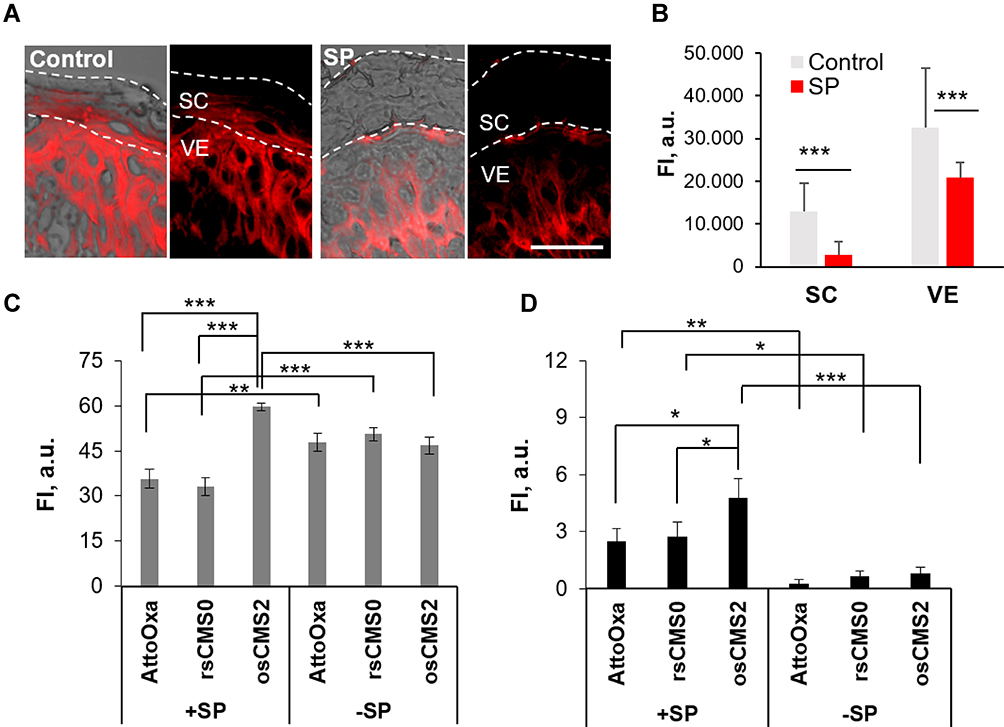 |
Figure 2 Effects of SP treatment on skin redox state and permeability. (A) Representative images of cryosections from skin samples after 18 h topical treatment with saline (−SP) or trypsin (+SP). Thiol groups were stained with ICC-maleimide. Scale bar = 25 µm. MFI analysis (B) was done in at least 30 different skin areas in SC and VE from different skin sections (n = 2). The effect of SP pre-treatment on skin permeability and CMS nanocarrier-mediated delivery of AttoOxa was tested by measuring the MFI in the SC (C) and VE (D) of skin samples incubated with or without SP (+SP, −SP) and treated with AttoOxa-loaded nanocarriers or free AttoOxa in 2.5% HEC gels (applied dye amount 0.2 µg/cm2) (n = 3). Statistical significance was found with *p < 0.05, **p < 0.01, and ***p < 0.001. |
Next, we used SP-treated skin to test the ability of reduction- and oxidative-sensitive CMS nanocarriers to deliver the lipophilic, high molecular weight dye AttoOxa (MW 835 g/mol). The fluorescence intensity of the penetrated dye was measured using ImageJ in pictures of at least 15 different skin sections per sample. The analysis of AttoOxa skin penetration (Figure 2C and D) revealed significant differences between untreated and SP-treated skin. For all tested formulations, a significant increase of dye fluorescence intensity was measured in the VE of trypsin-treated skin (+ SP) as compared to saline-treated skin (−SP). Considering the skin with intact barrier, no difference between the tested formulations were detected. In contrast, for SP-treated samples, only the oxidative-sensitive osCMS2b nanocarriers enabled a significantly higher dye penetration both in the SC (Figure 2C) and in the VE (Figure 2D).
These findings, along with the results about the proteolytic effects on skin barrier proteins, suggest that the proteolytic activity of topically applied SP improves skin permeability to topically applied drugs and confirm previous measurements using scanning transmission x-ray microscopy as label-free method to measure the penetration of drugs in skin.48 Perturbation of skin barrier permeability due to dysregulation of protease activity has been often reported in association with inflammatory skin disorders.42,49–52 It has been suggested that the increased permeability of the SC might be due to SP-mediated stimulation of the epidermal turnover, resulting in incomplete corneocyte maturation and, thus, in a defective skin barrier.52 Serine proteases and other proteolytic enzymes have already been used to enhance skin permeability to drugs.53,54 We have shown that the SP-treatment can enhance the penetration of dexamethasone in ex vivo human skin.37 The herein presented results show that SP can even increase the penetration of high MW molecules in the VE. Interestingly, in SP-treated skin the best penetration of AttoOxa in both SC and VE was achieved by the osCMS2b nanocarrier formulation. This is probably due to the ability of oxidative-sensitive CMS nanocarriers to sense the oxidative environment in SP-treated skin resulting in a triggered dye release with consequent high accumulation in the SC and the VE.
Anti-Inflammatory Efficacy of Rapa Formulations
To test the anti-inflammatory efficacy of Rapa-loaded CMS nanocarrier formulations, we used human skin explants co-cultured with Jurkat cells in the presence of oxidative and pro-inflammatory stimuli (Figure 3A). It was postulated that the nanocarriers release the loaded drug after topical application on inflamed skin and that the drug, after crossing both epidermis and dermis, can act of the T cells in the lower compartment. After activation with PHA, Jurkat cells expression of activation markers (CD45, CD69) and the phosphorylation of the RP-S6 (mTOR pathway) were monitored (Figure 3B). PHA activates T cells by direct binding to CD45,55 which plays a key role in T cell receptor-mediated signal transduction.56 CD69 is the earliest inducible cell surface activation marker and is involved in T cell proliferation and differentiation. The role of CD69 in the inflammatory response is controversial with reports showing its regulatory effects57 and others demonstrating its overexpression in T cells from patients with chronic inflammatory diseases.58 The RP-S6 is a downstream protein of the mTOR pathway and is activated by phosphorylation. Thus, a decrease of pRP-S6 correlates with mTOR inhibition.59 For the FACS analysis, we followed the method used by Dieterlen et al60 and measured the percentage of marker-positive cells in Rapa-treated and untreated samples. The levels of CD45, CD69, and pRP-S6 increased after PHA stimulation (Figure 3B, black-filled vs grey-filled histograms). A shift to the left was recorded in most of Rapa treated samples (Figure 3B, open-coloured histograms), indicating that inhibition of both cell activation and RP-S6 protein phosphorylation had occurred. A trend is discernible that shows the highest Rapa-mediated mTOR inhibitory effects for the osCMS2b nanocarriers. These results show that topically applied formulations can deliver Rapa across the whole skin and exert inhibitory effects on Jurkat cells.
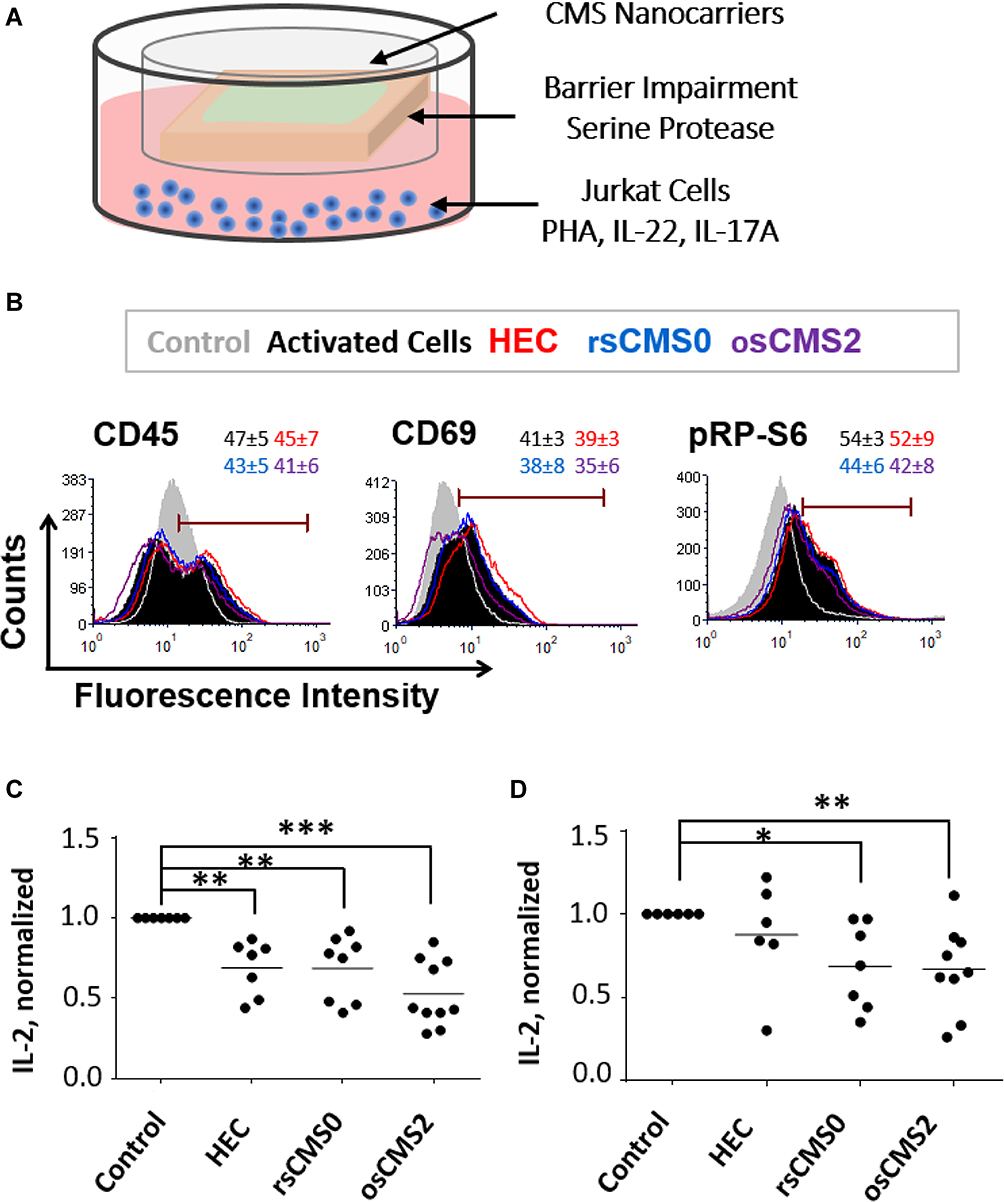 |
Figure 3 Anti-inflammatory effects of Rapa loaded on redox-sensitive CMS nanocarriers. (A) A trans-well set-up was used to co-culture ex vivo human skin and Jurkat T cells. Skin was pre-treated with SP to induce barrier-impairment and to create an oxidative environment. T cells were activated with PHA, IL-17A and IL-22. (B) Expression of activation markers (CD45, CD69) and phosphorylation of the ribosomal protein S6 (pRP-S6, mTOR pathway) by Jurkat cells co-cultured with skin treated with the different Rapa-formulations. Representative histograms of the flow cytometry analysis and average of positive cells within the range gate are shown (n = 3). (C and D) Released of the inflammatory cytokine IL-2 by Jurkat cells co-cultured with SP-treated (C) and SP-untreated (D) skin 24 h following topical application of Rapa formulations (applied Rapa doses: 10 µg/cm2). *p < 0.05; **p < 0.01; ***p < 0.001. |
As an inhibitor of the PI3K/Akt/mTOR pathway, Rapa can downregulate Th1/Th2 cytokine production in activated T cells.61 After activation by PHA, Jurkat cells release IL-2, which upon binding to its receptor, stimulates T cell proliferation and differentiation in an autocrine and paracrine mode via different intracellular signal pathways including the PI3K/Akt/mTOR one. In addition, Rapa was also shown to inhibit Jurkat cell proliferation by inducing G1 phase arrest.59 Thus, to test the validity of our inflammatory skin model, in preliminary experiments we tested the effect of Rapa on Jurkat cell proliferation and IL-2 release both in vitro (Supplemental Material, Figure S1) and in the ex vivo set-up (Supplemental Material Figure S2). We found in vitro inhibitory effects already at picomolar concentrations and a correlation between cells proliferation and amounts of IL-2 in culture medium. These results evidenced that IL-2 is a sensitive marker to detect Rapa effects on Jurkat cells and was therefore used to assess the efficacy of topically applied Rapa formulations in the co-culture set-up (Figure 3C and D). A clear reduction of IL-2 release was achieved in almost all samples and for all tested formulations. Whereas the values found for skin with intact barrier (Figure 3D) were more scattered, the inhibitory effect became evident in all SP-treated skin samples (Figure 3C). Interestingly, also with regard to IL-2 release, the results are in accordance with the observed enhanced permeability of skin treated with SP (Figure 2C and D). We also observed that Jurkat cells co-cultured with control SP-treated skin produced higher levels of IL-2 in comparison to cells co-cultured with control SP-untreated skin. This might be correlated to activation stimuli released by SP-treated ex vivo skin.37 The strongest inhibitory activity was measured for Rapa formulated in osCMS2b nanocarrier: For skin pre-treated with SP IL-2 mean values of 270, 190, 210, and 116 pg/mL were measured for control, HEC, rsCMS0, and osCMS2b formulations, respectively. For skin without SP pre-treatment, mean values of 195, 190, 169, and 93 pg/mL were measured for control, HEC, rsCMS0, and osCMS2b formulations, respectively. Among the SP-treated samples, highly significant differences (p < 0.001) were found when comparing the osCMS2b group with the control group, whereas lower p values were found when comparing the other two groups to the control group. When comparing the osCMS2b group with the nanocarrier-free Rapa group (HEC formulation), p values of 0.051 and 0.059 were found for the +SP and −SP samples, respectively. Higher p values were calculated when comparing the other treated groups with each other. Taken together, these findings indicate that topically applied Rapa formulation can inhibit IL-2 release by activated Jurkat cells. Of note, the strongest and most selective inhibitory effects were measured when Rapa was incorporated in osCMS2b nanocarriers.
Potential Irritative Effects of Topically Applied Rapa Formulations
Irritation effects have been observed by topical use of the macrolides like tacrolimus and sirolimus62,63 and the release of pro-inflammatory cytokines by skin cells has been reported in in vitro as well as ex vivo skin experiments.64,65 Therefore, we used the skin/T cell co-culture set-up to monitor eventual local irritation effects of the investigated Rapa formulations. At this purpose, IL-6, IL-8, and IL-1α were measured in skin extracts as well as in culture medium (Figure 4 and Figures S3–S4). These inflammatory mediators, which play a major role in skin inflammatory conditions like psoriasis,66,67 are also considered as markers for skin irritation.68 In cultured ex vivo human skin, these cytokines are expressed at low levels,69,70 but their release can be boosted by various stimuli like barrier disruption65,69 or stimulation of the protease-activated receptors 2 (PAR-2) signaling pathway.34–36 In the investigated samples, the expression of pro-inflammatory markers varied widely among the donors, possibly as a result of inter-individual variability but also of different baseline levels of inflammatory cytokines depending on anatomic region (breast vs abdomen), type and extent of surgery, as well as transportation time. Nevertheless, other than for tacrolimus,65 the tested Rapa formulations did not significantly increase or decrease the level of pro-inflammatory mediators with respect to control skin. Even if no significant differences were found, we observed reduced levels of IL-6 with respect to control in almost all donors of the group treated with SP and the Rapa-loaded osCMS2b nanocarriers (Figure 4A). Given that the penetration studies showed increased dye delivery by the osCMS2b nanocarriers (Figure 2D), the lower values of IL-6 might be due to possible Rapa inhibitory effects in keratinocytes. For instance, the effects of Rapa on IL-6 and IL-8 release by keratinocytes still remain controversial. While mTOR inhibition has been shown to increase the release of IL-6 and IL-8 by non-stimulated keratinocytes,71 in TNF-α stimulated keratinocytes the expression of IL-6 and IL-8 induced via the NFκB and mTOR pathway could be inhibited by Rapa.72,73 However, the skin inflammatory microenvironment is the result of a complex abnormal interplay between several pathways and mediators released by skin cells and infiltrating leukocytes. As for psoriasis, after induction of innate immunity processes, antimicrobial peptides and cytokines (eg, IL-1 and IL-8) released by keratinocytes stimulate dendritic cells that in turn release mediators like IL-23 and IL-12 and activate Th17 and Th22 cells in the early phase and Th1 cells in the late chronic phase.74,75 T cells, dendritic cells, and keratinocytes stimulate each other in a sort of self-amplifying loop, which results in the chronification of the inflammatory state. In the herein used inflammatory skin model, skin and the co-cultured Jurkat cells were exposed to different inflammatory stimuli to reproduce the typical features of psoriasis. These stimuli can directly or indirectly activate the mTOR pathway36,76,77 but also several other inflammatory mechanisms (eg, JAK-STAT and MAPK/ERK). Thus, the inhibition of the mTOR pathway was expected to have effects on T cell proliferation and cytokine release (eg, IL-2) but only limited inhibitory effects on pro-inflammatory cytokines released by keratinocytes.
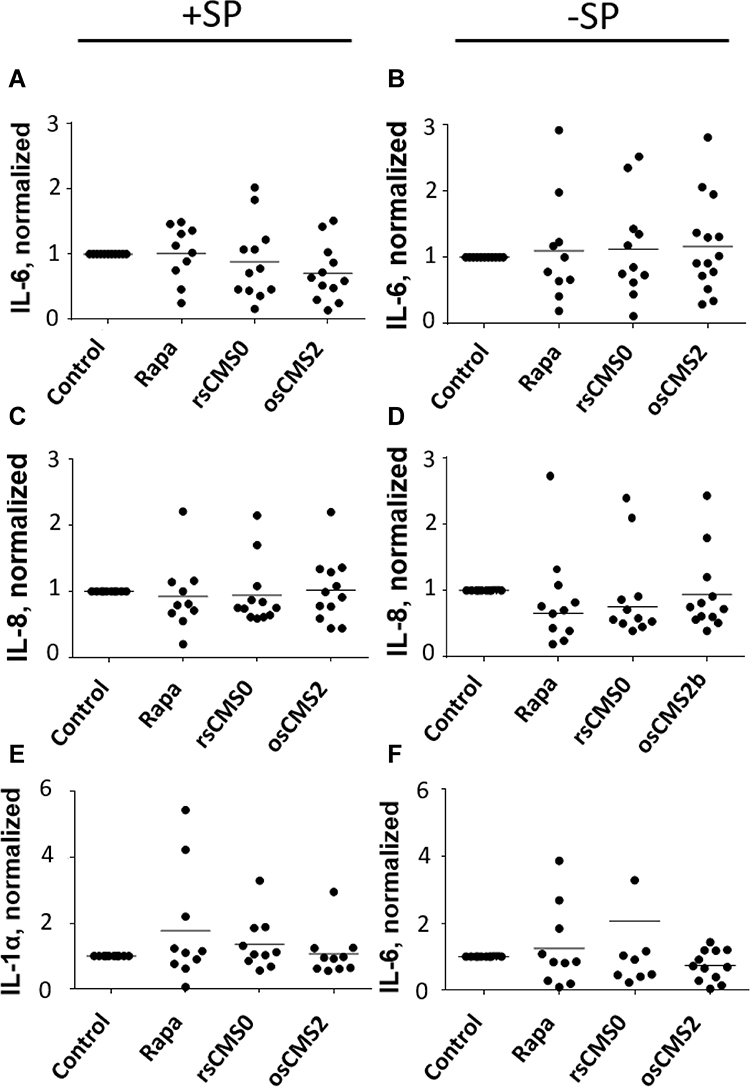 |
Figure 4 Normalized values of the inflammatory cytokine IL-6 (A and B), IL-8 (C and D), and IL-1α (E and F) extracted from the epidermis of skin pre-treated with trypsin (+ SP, A, C and E) or with saline (−SP, B, D and F) and incubated with the investigated Rapa formulations (24h, applied Rapa doses: 10 µg/cm2). |
Conclusion
In this study, we explored the possibility to take advantage of the oxidative microenvironment in inflamed skin and use it as a trigger for a more efficient and selective delivery of high MW drugs like sirolimus (Rapa). The insertion of sulfide groups in the frame of CMS nanocarriers resulted in formulations with selective drug delivery features. The use of an ex vivo skin model reproducing an oxidative and inflammatory environment typically found in a range of inflammatory skin disorders allowed us to demonstrate the superiority of oxidative-sensitive nanocarriers over reduction-sensitive nanocarrier or conventional formulations. The presented results indicate that the oxidation-sensitive nanocarrier osCMS2b can selectively deliver macrolide drugs to inflamed, barrier-disrupted skin creating a depot in the SC while favoring drug penetration to and across the skin. Clear inhibitory effects were found towards the co-cultured Jurkat T cell line. No significant effects on skin inflammatory mediators IL-1α, IL-6, and IL-8 were measured, which is indicative for a low irritation potential of both Rapa and the investigated nanocarriers. Rapa, as many other macrolide drugs, has a limited skin permeation, which is probably one of the reasons for the low clinical evidence after its topical application. Nevertheless, the reported results indicate that encapsulation in carrier systems and controlled release may improve drug efficacy while keeping the irritation potential at low levels. Further research work analyzing additional markers and using extended treatment protocols will further elucidate the potential of these oxidative-sensitive formulations as a new alternative option for the topical management of hyperproliferative inflammatory skin diseases such as psoriasis.
Abbreviations
AttoOxa, Atto Oxa12 NHS ester; BSA, bovine serum albumin; CDSN, corneodesmosin; CMS, core multishell; ELISA, enzyme-linked immunosorbent assay; Rapa, rapamycin; mTOR, mechanistic target of rapamycin; MW, molecular weight; FCS, fetal calf serum; HEC, hydroxyl ethyl cellulose; ICC-mal, indocarbocyanine-maleimide; MFI, mean fluorescence intensity; Occl, occluding; osCMS, oxidative-sensitive CMS; PBS, phosphate-buffered saline; PHA, phytohaemagglutinin; pRP-S6, phosphorylated ribosomal protein S6; ROS, reactive oxygen species; rsCMS, reduction-sensitive CMS; r.t., room temperature; SC, stratum corneum; SP, serine protease; ZO-1, zonula occludens-1.
Acknowledgments
The authors thank the Deutsche Forschungsgemeinschaft for funding the Project Vo 926/3-1. Polytimi Sidiropoulou thanks the (European Academy of Dermatology and Venereology) EADV for the Research Fellowship (RF-2019-43).
Disclosure
Dr Fiorenza Rancan reports grants from Deutsche Forschungsgemeinschaft, during the conduct of the study. Ms Keerthana Rajes reports grants from Verein Chemischer Industrie, grants from Deutsche Forschungsgemeinschaft, during the conduct of the study. Prof. Dr. Ulrike Blume-Peytavi reports grants from Pfizer, personal fees from Galderma, personal fees from Sanofi Regeneron, personal fees from Vichy, personal fees from Boots Healthcare, outside the submitted work; The authors report no other conflicts of interest in this work.
References
1. Leducq S, Giraudeau B, Tavernier E, Maruani A. Topical use of mammalian target of rapamycin inhibitors in dermatology: a systematic review with meta-analysis. J Am Acad Dermatol. 2019;80(3):735–742. doi:10.1016/j.jaad.2018.10.070
2. Zeng H, Chi H. mTOR signaling and transcriptional regulation in T lymphocytes. Transcription. 2014;5(2):e28263. doi:10.4161/trns.28263
3. Liu Y, Yang F, Zou S, Qu L. Rapamycin: a bacteria-derived immunosuppressant that has anti-atherosclerotic effects and its clinical application. Front Pharmacol. 2019;9:1520. doi:10.3389/fphar.2018.01520
4. Swarbrick AW, Frederiks AJ, Foster RS. Systematic review of sirolimus in dermatological conditions. Aust J Dermatol. 2021. doi:10.1111/ajd.13671
5. Ormerod A, Shah S, Copeland P, Omar G, Winfield A. Treatment of psoriasis with topical sirolimus: preclinical development and a randomized, double‐blind trial. Br J Dermatol. 2005;152(4):758–764. doi:10.1111/j.1365-2133.2005.06438.x
6. Kirschner N, Poetzl C, von den Driesch P, et al. Alteration of tight junction proteins is an early event in psoriasis: putative involvement of proinflammatory cytokines. Am J Pathol. 2009;175(3):1095–1106. doi:10.2353/ajpath.2009.080973
7. Yoshida Y, Morita K, Mizoguchi A, Ide C, Miyachi Y. Altered expression of occludin and tight junction formation in psoriasis. Arch Dermatol Res. 2001;293(5):239–244. doi:10.1007/s004030100221
8. Sano S. Psoriasis as a barrier disease. Dermatol Sin. 2015;33(2):64–69. doi:10.1016/j.dsi.2015.04.010
9. Pople PV, Singh KK. Development and evaluation of colloidal modified nanolipid carrier: application to topical delivery of tacrolimus. Eu J Pharmaceut Biopharmaceut. 2011;79(1):82–94. doi:10.1016/j.ejpb.2011.02.016
10. Gabriel D, Mugnier T, Courthion H, et al. Improved topical delivery of tacrolimus: a novel composite hydrogel formulation for the treatment of psoriasis. J Control Release. 2016;242:16–24. doi:10.1016/j.jconrel.2016.09.007
11. Damiani G, Pacifico A, Linder DM, et al. Nanodermatology‐based solutions for psoriasis: state‐of‐the art and future prospects. Dermatol Ther. 2019;32(6):e13113. doi:10.1111/dth.13113
12. Liu M, Wen J, Sharma M. Solid lipid nanoparticles for topical drug delivery: mechanisms, dosage form perspectives, and translational status. Curr Pharm Des. 2020;26(27):3203–3217. doi:10.2174/1381612826666200526145706
13. Giulbudagian M, Rancan F, Klossek A, et al. Correlation between the chemical composition of thermoresponsive nanogels and their interaction with the skin barrier. J Control Release. 2016;243:323–332. doi:10.1016/j.jconrel.2016.10.022
14. Patzelt A, Lademann J. Recent advances in follicular drug delivery of nanoparticles. Expert Opin Drug Deliv. 2020;17(1):49–60. doi:10.1080/17425247.2020.1700226
15. Rancan F, Afraz Z, Combadiere B, Blume-Peytavi U, Vogt A. Hair follicle targeting with nanoparticles. In: Nanotechnology in Dermatology. Springer; 2013:95–107.
16. Patel V, Sharma OP, Mehta T. Nanocrystal: a novel approach to overcome skin barriers for improved topical drug delivery. Expert Opin Drug Deliv. 2018;15(4):351–368. doi:10.1080/17425247.2018.1444025
17. Sala M, Diab R, Elaissari A, Fessi H. Lipid nanocarriers as skin drug delivery systems: properties, mechanisms of skin interactions and medical applications. Int J Pharm. 2018;535(1–2):1–17. doi:10.1016/j.ijpharm.2017.10.046
18. Du F, Hönzke S, Neumann F, et al. Development of biodegradable hyperbranched core-multishell nanocarriers for efficient topical drug delivery. J Control Release. 2016;242:42–49. doi:10.1016/j.jconrel.2016.06.048
19. Radowski MR, Shukla A, von Berlepsch H, et al. Supramolecular aggregates of dendritic multishell architectures as universal nanocarriers. Angew Chem Int Ed. 2007;46(8):1265–1269. doi:10.1002/anie.200603801
20. Yamamoto K, Klossek A, Flesch R, et al. Core-multishell nanocarriers: transport and release of dexamethasone probed by soft x-ray spectromicroscopy. J Control Release. 2016;242:64–70. doi:10.1016/j.jconrel.2016.08.028
21. Frombach J, Unbehauen M, Kurniasih IN, et al. Core-multishell nanocarriers enhance drug penetration and reach keratinocytes and antigen-presenting cells in intact human skin. J Control Release. 2019;299:138–148. doi:10.1016/j.jconrel.2019.02.028
22. Unbehauen ML, Fleige E, Paulus F, et al. Biodegradable core–multishell nanocarriers: influence of inner shell structure on the encapsulation behavior of Dexamethasone and Tacrolimus. Polymers. 2017;9(8):316. doi:10.3390/polym9080316
23. Wu Y, Zhang X, Li H, et al. A core/shell stabilized polysaccharide-based nanoparticle with intracellular environment-sensitive drug delivery for breast cancer therapy. J Mater Chem B. 2018;6(41):6646–6659. doi:10.1039/C8TB00633D
24. Yamazaki N, Sugimoto T, Fukushima M, et al. Dual-stimuli responsive liposomes using pH-and temperature-sensitive polymers for controlled transdermal delivery. Polym Chem. 2017;8(9):1507–1518. doi:10.1039/C6PY01754A
25. Qureshi D, Nayak SK, Maji S, Anis A, Kim D, Pal K. Environment sensitive hydrogels for drug delivery applications. Eur Polym J. 2019;53:109220.
26. Rancan F, Giulbudagian M, Jurisch J, Blume-Peytavi U, Calderon M, Vogt A. Drug delivery across intact and disrupted skin barrier: identification of cell populations interacting with penetrated thermoresponsive nanogels. Eur J Pharmaceut Biopharmaceut. 2017;116:4–11. doi:10.1016/j.ejpb.2016.11.017
27. Antille C, Sorg O, Lübbe J, Saurat J-H. Decreased oxidative state in non-lesional skin of atopic dermatitis. Dermatology. 2002;204(1):69–71. doi:10.1159/000051814
28. Briganti S, Picardo M. Antioxidant activity, lipid peroxidation and skin diseases. What’s new. J Eur Acad Dermatol Venereol. 2003;17(6):663–669. doi:10.1046/j.1468-3083.2003.00751.x
29. Cannavò SP, Riso G, Casciaro M, Di Salvo E, Gangemi S. Oxidative stress involvement in psoriasis: a systematic review. Free Radic Res. 2019;53(8):829–840. doi:10.1080/10715762.2019.1648800
30. Rajes K, Walker KA, Hadam S, et al. Redox-responsive nanocarrier for controlled release of drugs in inflammatory skin diseases. Pharmaceutics. 2021;13(1):37. doi:10.3390/pharmaceutics13010037
31. Rajes K, Walker KA, Hadam S, et al. Oxidation-sensitive core–multishell nanocarriers for the controlled delivery of hydrophobic drugs. ACS Biomater Sci Eng. 2021;7(6):2485–2495. doi:10.1021/acsbiomaterials.0c01771
32. Richard I. The genetic and molecular bases of monogenic disorders affecting proteolytic systems. J Med Genet. 2005;42(7):529–539. doi:10.1136/jmg.2004.028118
33. Ramachandran R, Hollenberg M. Proteinases and signalling: pathophysiological and therapeutic implications via PARs and more. Br J Pharmacol. 2008;153(S1):S263–S282.
34. Wakita H, Furukawa F, Takigawa M. Thrombin and trypsin induce granulocyte-macrophage colony-stimulating factor and interleukin-6 gene expression in cultured normal human keratinocytes. Proc Assoc Am Physicians. 1997;109(2):190–207.
35. Hou L, Kapas S, Cruchley A, et al. Immunolocalization of protease‐activated receptor‐2 in skin: receptor activation stimulates interleukin‐8 secretion by keratinocytes in vitro. Immunology. 1998;94(3):356–362. doi:10.1046/j.1365-2567.1998.00528.x
36. Caliendo G, Santagada V, Perissutti E, et al. Kallikrein protease activated receptor (PAR) axis: an attractive target for drug development. J Med Chem. 2012;55(15):6669–6686. doi:10.1021/jm300407t
37. Frombach J, Rancan F, Kübrich K, et al. Serine protease-mediated cutaneous inflammation: characterization of an ex vivo skin model for the assessment of dexamethasone-loaded core multishell-nanocarriers. Pharmaceutics. 2020;12(9):862.
38. Frombach J, Lohan SB, Lemm D, et al. Protease-mediated inflammation: an in vitro human keratinocyte-based screening tool for anti-inflammatory drug nanocarrier systems. Z Phys Chem. 2018;232(5–6):919–933. doi:10.1515/zpch-2017-1048
39. Smith SH, Peredo CE, Takeda Y, et al. Development of a topical treatment for psoriasis targeting RORγ: from bench to skin. PLoS One. 2016;11(2):e0147979.
40. Reichert S, Welker P, Calderón M, et al. Size‐dependent cellular uptake of dendritic polyglycerol. Small. 2011;7(6):820–829. doi:10.1002/smll.201002220
41. Suzuki Y, Nomura J, Koyama J, Horii I. The role of proteases in stratum corneum: involvement in stratum corneum desquamation. Arch Dermatol Res. 1994;286(5):249–253.
42. Voegeli R, Rawlings A, Breternitz M, Doppler S, Schreier T, Fluhr J. Increased stratum corneum serine protease activity in acute eczematous atopic skin. Br J Dermatol. 2009;161(1):70–77. doi:10.1111/j.1365-2133.2009.09142.x
43. Simon M, Jonca N, Guerrin M, et al. Refined characterization of corneodesmosin proteolysis during terminal differentiation of human epidermis and its relationship to desquamation. J Biol Chem. 2001;276(23):20292–20299. doi:10.1074/jbc.M100201200
44. Yuki T, Haratake A, Koishikawa H, Morita K, Miyachi Y, Inoue S. Tight junction proteins in keratinocytes: localization and contribution to barrier function. Exp Dermatol. 2007;16(4):324–330. doi:10.1111/j.1600-0625.2006.00539.x
45. Ogura R, Knox JM, Griffin AC, Kusuhara M, Ogura J. The concentration of sulfhydryl and disulfide in human epidermis, hair and nail. J Invest Dermatol. 1962;38(2):69–75. doi:10.1038/jid.1962.16
46. Shindo Y, Witt E, Han D, Epstein W, Packer L. Enzymic and non-enzymic antioxidants in epidermis and dermis of human skin. J Invest Dermatol. 1994;102(1):122–124. doi:10.1111/1523-1747.ep12371744
47. Pickard C, Louafi F, McGuire C, et al. The cutaneous biochemical redox barrier: a component of the innate immune defenses against sensitization by highly reactive environmental xenobiotics. J Immunol. 2009;183(11):7576–7584. doi:10.4049/jimmunol.0901064
48. Germer G, Ohigashi T, Yuzawa H, et al. Improved skin permeability after topical treatment with serine protease: probing the penetration of rapamycin by scanning transmission X-ray microscopy. ACS Omega. 2021;6(18):12213–12222. doi:10.1021/acsomega.1c01058
49. Meyer-Hoffert U. Reddish, scaly, and itchy: how proteases and their inhibitors contribute to inflammatory skin diseases. Arch Immunol Ther Exp. 2009;57(5):345–354. doi:10.1007/s00005-009-0045-6
50. de Veer SJ, Furio L, Harris JM, Hovnanian A. Proteases and proteomics: cutting to the core of human skin pathologies. Proteomics Clin Appl. 2014;8(5–6):389–402. doi:10.1002/prca.201300081
51. Hachem J-P, Houben E, Crumrine D, et al. Serine protease signaling of epidermal permeability barrier homeostasis. J Invest Dermatol. 2006;126(9):2074–2086. doi:10.1038/sj.jid.5700351
52. Voegeli R, Rawlings A, Doppler S, Heiland J, Schreier T. Profiling of serine protease activities in human stratum corneum and detection of a stratum corneum tryptase‐like enzyme. Int J Cosmet Sci. 2007;29(3):191–200. doi:10.1111/j.1467-2494.2007.00386.x
53. Sim Y, Nam Y, Shin Y-H, et al. Proteolytic enzyme conjugated to SC-glucan as an enzymatic transdermal drug penetration enhancer. Pharmazie. 2003;58(4):252–256.
54. Nounou MI, Zaghloul TI, Ahmed NA, Eid AA, El-Khordagui LK. Skin permeability enhancement by Bacillus subtilis alkaline protease: application to transdermal drug delivery. Int J Pharm. 2017;529(1–2):423–432. doi:10.1016/j.ijpharm.2017.06.057
55. Monostori E, Hartyani Z, Ocsovszky I, et al. Effect of phytohaemagglutinin on CD45 in T cells. Immunol Lett. 1994;42(3):197–201. doi:10.1016/0165-2478(94)90086-8
56. Penninger JM, Irie-Sasaki J, Sasaki T, Oliveira-dos-Santos AJ. CD45: new jobs for an old acquaintance. Nat Immunol. 2001;2(5):389–396. doi:10.1038/87687
57. Cibrián D, Sánchez‐Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur J Immunol. 2017;47(6):946–953. doi:10.1002/eji.201646837
58. Cope AP. Studies of T-cell activation in chronic inflammation. Arthritis Res Ther. 2002;4(3):S197. doi:10.1186/ar557
59. Zhao Y, Zhou Q, Xu Y, Lai XY, Huang H. Antiproliferative effect of rapamycin on human T‐cell leukemia cell line Jurkat by cell cycle arrest and telomerase inhibition 1. Acta Pharmacol Sin. 2008;29(4):481–488. doi:10.1111/j.1745-7254.2008.00767.x
60. Dieterlen MT, Bittner HB, Klein S, et al. Assay validation of phosphorylated S6 ribosomal protein for a pharmacodynamic monitoring of mTOR‐inhibitors in peripheral human blood. Cytometry Part B Clin Cytom. 2012;82(3):151–157. doi:10.1002/cyto.b.21005
61. Herrero‐Sánchez MC, Rodríguez‐Serrano C, Almeida J, et al. Effect of mTORC 1/mTORC 2 inhibition on T cell function: potential role in graft‐versus‐host disease control. Br J Haematol. 2016;173(5):754–768. doi:10.1111/bjh.13984
62. Cinar S, Kartal D, Bayram A, et al. Topical sirolimus for the treatment of angiofibromas in tuberous sclerosis. Indian J Dermatol Venereol Leprol. 2017;83(1):27. doi:10.4103/0378-6323.190844
63. Bekersky I, Fitzsimmons W, Tanase A, Mahera RM, Hodosh E, Lawrence I. Nonclinical and early clinical development of tacrolimus ointment for the treatment of atopic dermatitis. J Am Acad Dermatol. 2001;44(1):S17–S27. doi:10.1067/mjd.2001.109816
64. Lan -C-CE, Yu H-S, Huang S-M, Wu C-S, Chen G-S. FK506 induces interleukin-6 secretion from UVB irradiated cultured human keratinocytes via p38 mitogen-activated protein kinase pathway: implication on mechanisms of tacrolimus-induced skin irritation. J Dermatol Sci. 2007;48(3):225–228. doi:10.1016/j.jdermsci.2007.08.003
65. Rancan F, Volkmann H, Giulbudagian M, et al. Dermal delivery of the high-molecular-weight drug tacrolimus by means of polyglycerol-based nanogels. Pharmaceutics. 2019;11(8):394. doi:10.3390/pharmaceutics11080394
66. Gillitzer R, Berger R, Mielke V, Müller C, Wolff K, Stingl G. Upper keratinocytes of psoriatic skin lesions express high levels of NAP-1/IL-8 mRNA in situ. J Invest Dermatol. 1991;97(1):73–79. doi:10.1111/1523-1747.ep12478128
67. Grossman RM, Krueger J, Yourish D, et al. Interleukin 6 is expressed in high levels in psoriatic skin and stimulates proliferation of cultured human keratinocytes. Proc Nat Acad Sci. 1989;86(16):6367–6371. doi:10.1073/pnas.86.16.6367
68. Perkins MA, Osterhues MA, Farage MA, Robinson MK. A noninvasive method to assess skin irritation and compromised skin conditions using simple tape adsorption of molecular markers of inflammation. Skin Res Technol. 2001;7(4):227–237. doi:10.1034/j.1600-0846.2001.70405.x
69. Rancan F, Afraz Z, Hadam S, et al. Topically applied virus-like particles containing HIV-1 Pr55gag protein reach skin antigen-presenting cells after mild skin barrier disruption. J Control Release. 2017;268:296–304. doi:10.1016/j.jconrel.2017.10.033
70. Döge N, Avetisyan A, Hadam S, et al. Assessment of skin barrier function and biochemical changes of ex vivo human skin in response to physical and chemical barrier disruption. Eur J Pharmaceut Biopharmaceut. 2017;116:138–148. doi:10.1016/j.ejpb.2016.12.012
71. DeTemple V, Satzger I, Walter A, Schaper K, Gutzmer R. Effects of mammalian target of rapamycin inhibitors on cytokine production and differentiation in keratinocytes. Exp Dermatol. 2016;25(10):775–782. doi:10.1111/exd.13079
72. Patel AB, Tsilioni I, Weng Z, Theoharides TC. TNF stimulates IL‐6, CXCL 8 and VEGF secretion from human keratinocytes via activation of mTOR, inhibited by tetramethoxyluteolin. Exp Dermatol. 2018;27(2):135–143. doi:10.1111/exd.13461
73. Young CN, Koepke JI, Terlecky LJ, Borkin MS, Boyd SL, Terlecky SR. Reactive oxygen species in tumor necrosis factor-α-activated primary human keratinocytes: implications for psoriasis and inflammatory skin disease. J Invest Dermatol. 2008;128(11):2606–2614. doi:10.1038/jid.2008.122
74. Albanesi C, Madonna S, Gisondi P, Girolomoni G. The interplay between keratinocytes and immune cells in the pathogenesis of psoriasis. Front Immunol. 2018;9:1549. doi:10.3389/fimmu.2018.01549
75. Billi AC, Gudjonsson JE, Voorhees JJ. Psoriasis: past, present, and future. J Invest Dermatol. 2019;139(11):e133. doi:10.1016/j.jid.2019.08.437
76. Buerger C, Shirsath N, Lang V, et al. Inflammation dependent mTORC1 signaling interferes with the switch from keratinocyte proliferation to differentiation. PLoS One. 2017;12(7):e0180853. doi:10.1371/journal.pone.0180853
77. Takei-Taniguchi R, Imai Y, Ishikawa C, et al. Interleukin-17- and protease-activated receptor 2-mediated production of CXCL1 and CXCL8 modulated by cyclosporine A, vitamin D3 and glucocorticoids in human keratinocytes. J Dermatol. 2012;39(7):625–631. doi:10.1111/j.1346-8138.2011.01462.x
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.