")
Back to Journals » Journal of Blood Medicine » Volume 9
To test or not: occurrence of sickle cell trait and assessment of the awareness toward its screening among patients attending Magale Health Center IV, Namisindwa District, Eastern Uganda
Authors Mandu K, Tusuubira SK , Mwambi B, Webbo F , Atuhairwe C, Taremwa IM
Received 14 June 2018
Accepted for publication 24 July 2018
Published 21 November 2018 Volume 2018:9 Pages 219—225
DOI https://doi.org/10.2147/JBM.S177203
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin Bluth
Keneth Mandu,1 Sharifu K Tusuubira,1,2 Bashir Mwambi,1 Fred Webbo,3 Christine Atuhairwe,4 Ivan Mugisha Taremwa1
1Institute of Allied Health Sciences, Clarke International University, Kampala, Uganda; 2Uganda Sickle Cell Rescue Foundation, Kampala, Uganda; 3Lancet Laboratories, Kampala, Uganda; 4Institute of Public Health and Management, Clarke International University, Kampala, Uganda
Purpose: To evaluate the occurrence of sickle cell trait (SCT), assess patient awareness and evaluate the performance of a sickle cell hemoglobin-S (dithionate-qualitative solubility) point-of-care test among patients seeking care at Magale Health Center IV, Namisindwa District, Eastern Uganda.
Materials and methods: We conducted a cross sectional study, in which we consecutively enrolled participants aged ≥18 years at Magale Health Center IV. Four milliliters of EDTA blood were collected by venipuncture and screened for SCT using solubility testing, and confirmed with hemoglobin (Hb) electrophoresis at Central Public Health Laboratory (CPHL), Kampala, Uganda. A structured questionnaire was used to assess participants’ awareness of SCT. Data were presented as proportion, and measurements of diagnostic test performance were calculated.
Results: We enrolled 242 participants, of these 58.7% (N = 142) were females. Their mean age was 26.4 years (range 18–49). Of the 242 participants, 11, who represent 4.5% (95% CI: 3.3–5.9), tested positive. The sensitivity, specificity, positive predictive value and negative predictive value of the rapid sickle cell test were 63.64%, 100%, 100% and 98.30%, respectively. There was knowledge gap regarding sickle cell awareness.
Conclusion: The occurrence of SCT was high, and the point-of-care test showed a high diagnostic reliability. The risk of SCT is associated with genetic predisposition as indicated by Hb electrophoresis. Community sensitization is key to avert the associated risk of Hb defects.
Keywords: sickle cell trait, point-of-care testing, Uganda
Introduction
Hemoglobin (Hb) defects are by far the most diagnosed genetic disease, with over 10,000 variants that involve a single amino acid substitution.1 A normal adult carries adult hemoglobin (HbA)1,2; however, mutations that cause substitution of glutamate for valine at position 6 of the β-globin chain result in Hb-S, a variant linked to life-long complications like sickle cell anemia.3–5 Globally, Hb disorders occur in about 7.87% of the population, representing 300 million individuals.6–8 The defects remain unacceptably high in sub-Saharan Africa where they affect 38% to 63% of the population.7 In Uganda, studies done in Sironko and Tororo districts found a prevalence of 17.5% and 19.5%, respectively.9–11
Universal screening and early intervention are thought to offer a protective advantage of sickle cell defects,12,13 however, diagnostic barriers due to high cost, sophisticated equipment and reliance on electricity limit the scope of Hb-S screening in resource-limited settings.8 This has necessitated tests that are inexpensive, reliable, rapid and fieldable.12,14–15 Cognizant to these, point-of-care testing (PoCT) has been explored, and is hoped to avert preventable morbidities and mortalities. There are varied PoCT assays and techniques, notable among these are: SICKLEDEX® (Streck, Omaha, NE, USA) that relies on the solubility differences in the HbA and sickle hemoglobin (HB-S) molecules,16 and the sickle cell hemoglobin-S (dithionate qualitative solubility) test (Bio Lab Diagnostics India Private Limited, Mumbai, Maharashtra, India) that is based on Hb solubility testing.17 To ascertain their diagnostic use, validation was conducted,12,15,18,19 and HB-S-PoCT was found to be practical in a resource-limited setting. For example, a paper-based validation test in a cohort of 159 newborns in Angola reported HB-S diagnostic sensitivity of 81.8% and specificity of 83.3%; while in the USA, a sensitivity of 94.2%, specificity of 97.7% and an overall diagnostic accuracy of 96.9% was reported.19
Although the invention of the sickle cell hemoglobin-S (dithionate-qualitative) test is pivotal to the diagnostic trend of sickle cell defect, its use remains at jeopardy due to scarcity of data on its validation in low resource settings. This coupled with limited awareness of Hb-S has led to immense sickle cell defects.9,10 This study established the occurrence of sickle cell trait (SCT), evaluated a point-of-care test and assessed sickle cell awareness among patients attending Magale Health Center IV, Namisindwa District in rural Eastern Uganda.
Materials and methods
Study design and setting
This was a cross sectional study that enrolled adults (>18 years) who were attending the out-patients department at Magale Health Center IV during the period of May to November, 2017. Magale Health Center IV is a private-for-profit health facility located in Namisindwa District, with a bed capacity of 86. The facility receives 178,902 patients annually and supervises 12 lower health care units in the Eastern region. It carries out sickle cell screening and sends positive samples for confirmation to Central Public Health Laboratory (CPHL). This study did not include participants with known sickle cell disease or reported sickling crisis in the previous 48hours, those who reportedly had blood transfusion in the previous 4 months and those who declined to consent.
Sample size determination and sampling procedure
Using Kish and Leslie formula20 for sample size determination, given as:
|
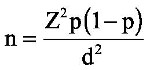
where n represents the desired sample size, Z is the statistical level at 95% CI, p is the estimated prevalence of sickle cell trait reported at 13.3%11 and d is the allowable error (at 5%), a total of 178 participants was calculated. We used a consecutive sampling technique in which participants were assessed for enrollment as they present to the health facility until the required sample size was attained.
Data collection
Data were obtained by laboratory analysis of blood for the HB-S as indicated in Figure 2. In addition, an interviewer administered a questionnaire to assess the awareness of SCT.
Sample collection and dried blood spot (DBS) preparation
Four milliliters of venous blood were collected into EDTA a vacutainer. Samples were kept at room temperature (22°C–27°C) and tested using rapid sickle cell test within 4 hours.
The preparation of a DBS was made by dispensing 100 µL of blood to a well labeled WhatmanTM 903 Protein Saver Card (GE Healthcare Ltd, Cardiff, UK), and stored in a humid free zip-locked bag with desiccant packets at −20°C until shipped to CPHL for analysis.
Laboratory sample analyses
Rapid sickle cell hemoglobin-S (dithionate qualitative solubility) test (Bio Lab Diagnostics India Private Limited)17 determines Hb containing variants in whole blood using visual inspection for turbidity due to difference in lysis of the insoluble HB-S and soluble form of HbA following a mixture of blood and a working solution in a test tube as shown in Figure 1.
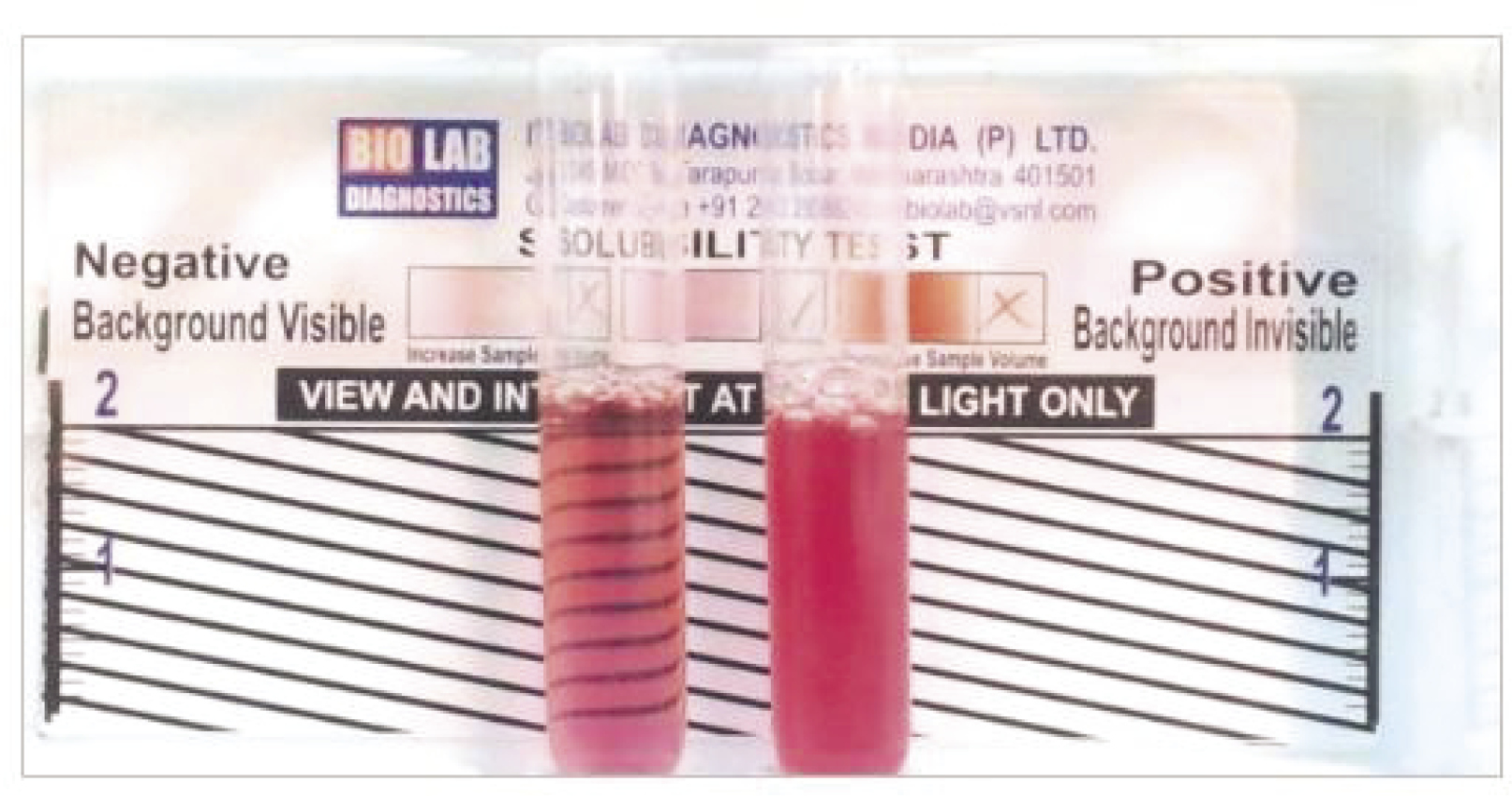 | Figure 1 Showing the reactivity pattern of the rapid sickle cell hemoglobin-S (dithionate qualitative solubility) test |
Constitution of the kit testing reagents
The Hb-S dithionate qualitative solubility kit comprises: reagent 1 (R1), composed of phosphate buffer; reagent 2 (R2), composed of 0.5 g sodium dithionate and reagent 3 (R3), composed of 0.5 g of white saponin. These were constituted to a working solution by transferring the contents of R2 and R3 to a bottle of R1, and were gently mixed for 15 minutes and well labeled.
Hb-S testing method
This involved addition of 2 mL of the working reagent to a labeled test tube, and 50 µL of blood. These were mixed well by gently shaking the test tube and incubated at room temperature (approximately 25°C) for 10 minutes. The test tube contents were visually examined and it was reported that the negative test was transparent, as seen in the background of the rapid sickle cell tube reader, while the background was not visible in the positive sample (HbAS or HbSS) given in Figure 1.
Two test tube readings were done independently with unaided readers, and in case of a discrepancy, a third reader was considered, and a result was concluded.
Confirmation was done using Hb electrophoresis. To ensure quality of the test results, experimental procedures were conducted according to the manufacturers’ instructions. Known HbAS and HbSS blood samples were used as positive controls.
Awareness assessment
We used an interviewer administered questionnaire to determine awareness of SCT among study participants. The questionnaire was developed based on literature from the previous studies,12,15,19–24 and pre-tested at St Benedict Medical Center in Mayuge District to check for clarity and comprehension. Following this, changes were made to suit the intended use. The questionnaire comprised: demographics of the participant, knowledge of the family history and transmission mode.
Statistical analysis
Data were recorded in hard copy and transferred to Microsoft® Office Excel, version 2007. Proportion of participants with SCT was analyzed as the number of those with the SCT divided by the total number of participants. Performance evaluation was based on measurement of: sensitivity = true positives (TP)/[TP + false negatives (FN)]; specificity = TN/[false positives (FP) + TN]; positive predictive value (PPV) = TP/(TP + FP) and negative predictive value (NPV) = TN/(TN + FN).
Ethical considerations
We received ethical approval from the research and ethics committee of Clarke International University, Kampala, Uganda (formerly, International Health Sciences University). Further, we obtained a signed informed consent from each of our participants.
Results
We approached 284 participants, of whom 242 were enrolled. The main reasons for exclusion were: being known HbSS, reported a sickling crisis in the previous 48 hours and history of blood transfusion in the previous 4 months. Participants’ mean age was 29.4 years (range 18–49), and they reported varied socio-demographic characteristics, as indicated in Table 1.
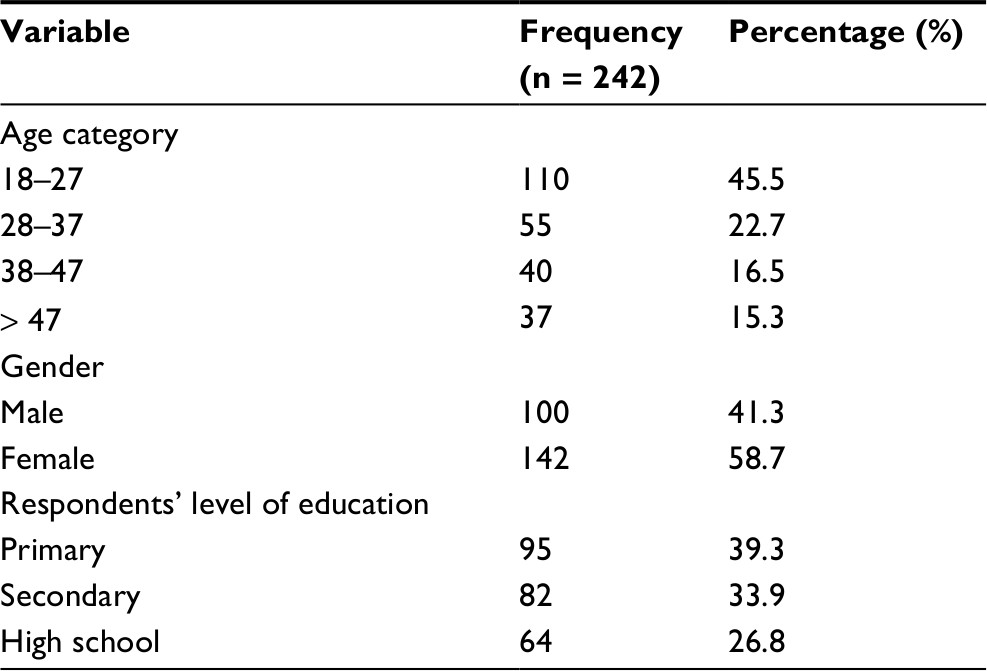 | Table 1 Showing socio-demographic characteristics of respondents |
Of the 242 participants, 11, who represent 4.5% (95% CI: 3.3–5.9), had SCT as reported by the Hb electrophoresis. The participant flow chart is presented in Figure 2.
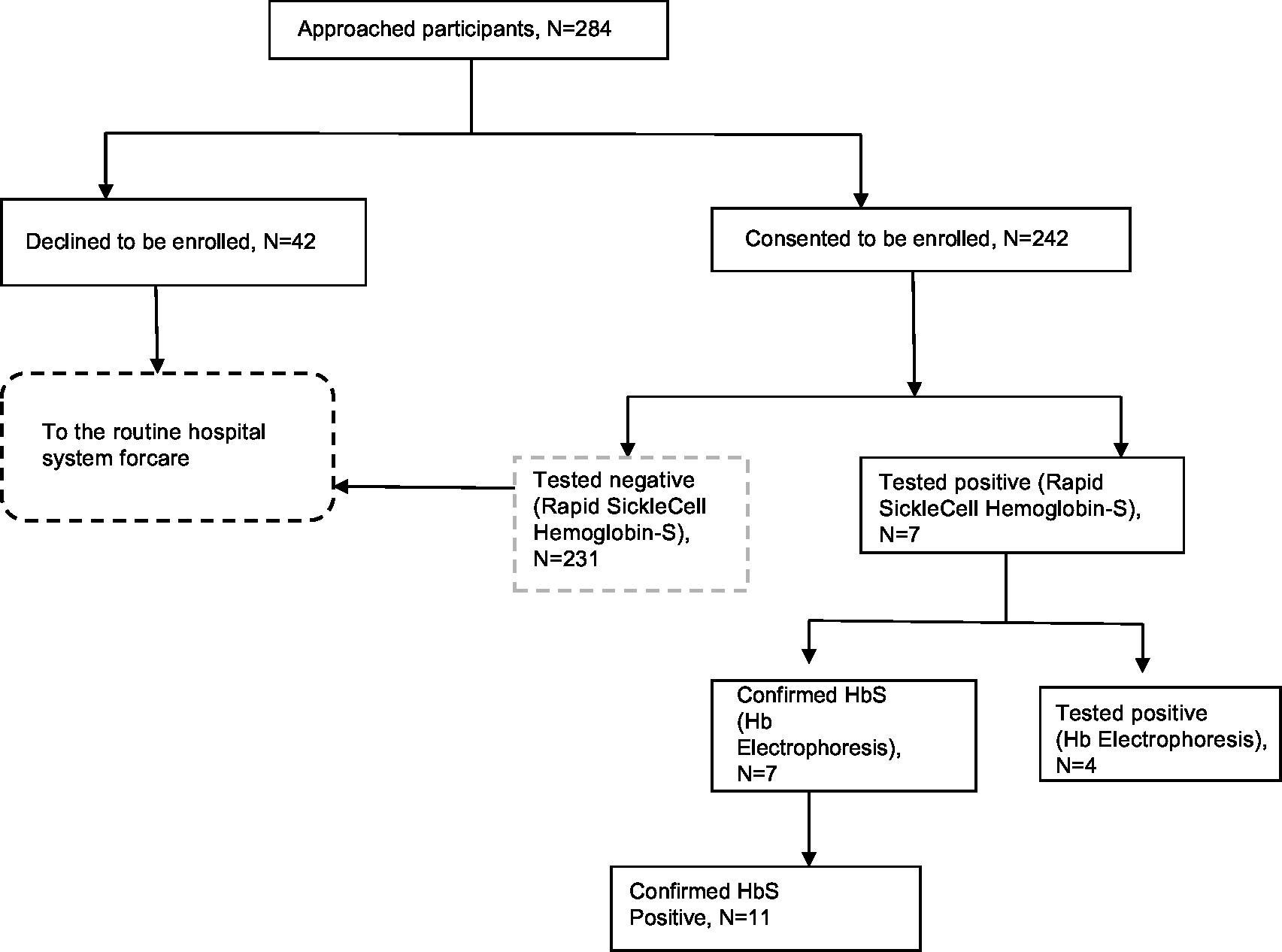 | Figure 2 Study participant flow chart and reactivity pattern of tests. Abbreviations: Hb, hemoglobin; HB-S, sickle hemoglobin. |
The diagnostic performance of the rapid sickle cell test against Hb electrophoresis as gold standard is presented in Table 2. From Table 2, the rapid test detected 7 cases of HbAS, while Hb electrophoresis reported 11 cases. The results have been summarized in Table 3 to aid the computation of the sensitivity, specificity, NPV and PPV.
Sensitivity=TP/(TP + FN)=7/(7+4)=7/11=63.64%
Specificity=TN/(FP + TN)=231/(0+231)=231/231=100.00%
PPV= TP/(TP + FP)=7/(7+0)=7/7=100.00%
NPV=TN/(TN + FN)=231/(231+4)=231/235=98.30%
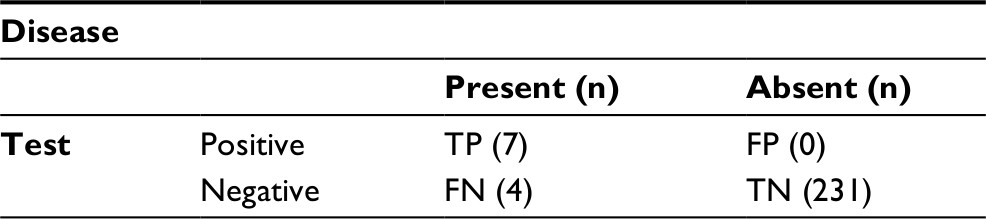 | Table 2 Performance evaluation of the solubility rapid screening test method against Hb electrophoresis (gold standard) Abbreviations: FN, false negative; FP, false positive; TN, true negative; TP, true positive; Hb, hemoglobin. |
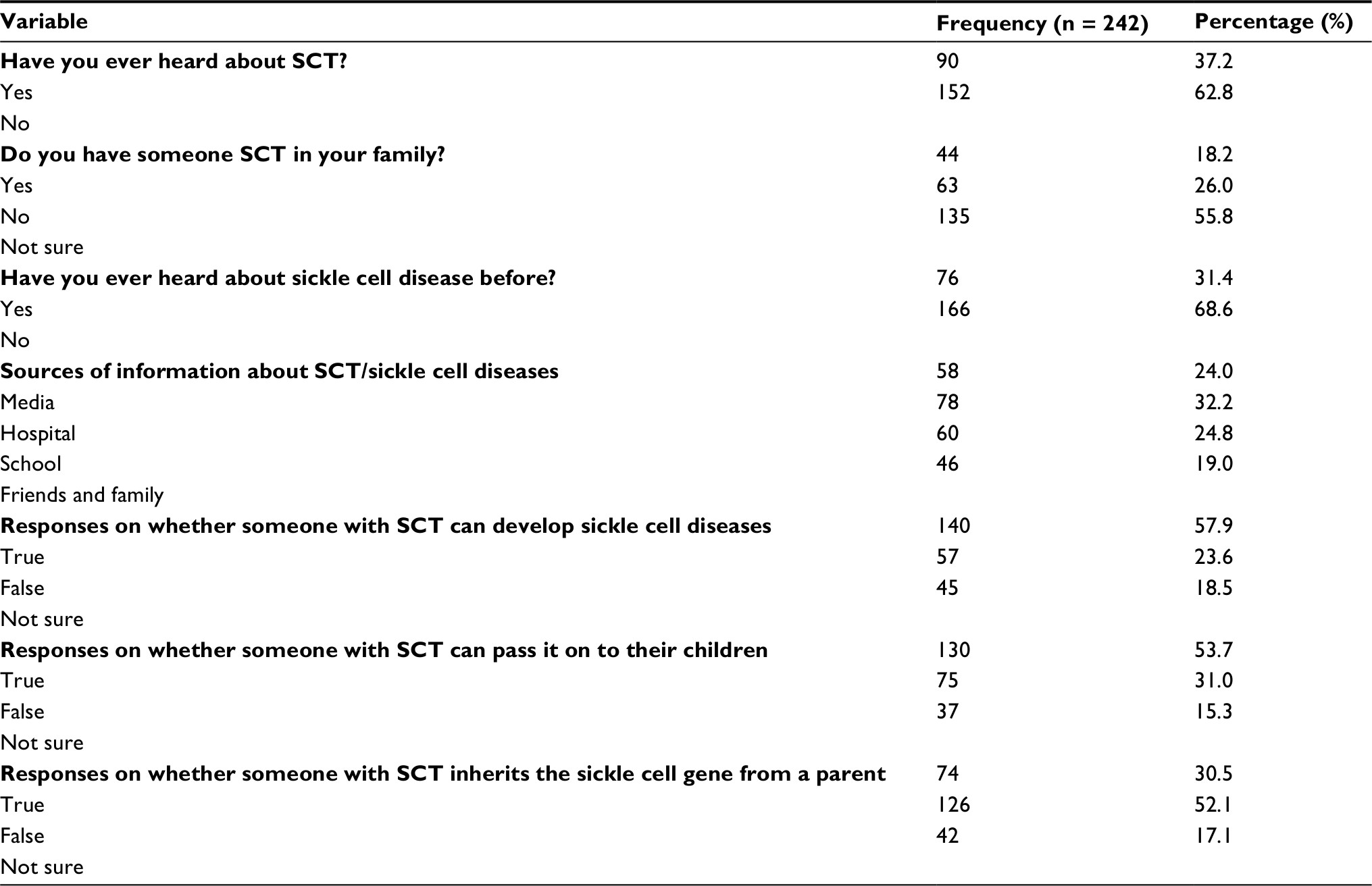 | Table 3 Participants’ responses regarding their awareness of the SCT Abbreviation: SCT, sickle cell trait. |
Participants’ response regarding the assessment of awareness of SCT indicated a knowledge gap, as presented in Table 3.
Discussion
From this study, the rapid sickle cell test accurately identified 7 true positives out of 11 confirmed by the gold standard diagnosis, giving a sensitivity and specificity of 63.64% and 100%, respectively. Further, the PPV and NPV were 100% and 98.30%, respectively. The sensitivity of the rapid sickle cell (dithionate qualitative solubility test) is low compared to the 84% of a density-based test using aqueous multiphase systems done in Zambia,25 93% and 94% among children and post-partum women, respectively, in a study conducted in Angola,19 98.4% sensitivity using an immunoassay,14,26 and 99% using the Sickle SCAN™ test (BioMedomics, Durham, NC, USA).12 This assay has demonstrated a low sensitivity, which is attributed to interfering reactivity due to other Hb variants, particularly hemoglobin C (HbC)-Harlem and fetal hemoglobin (HbF).17 On the other hand, our study detected a 100% specificity, thus a high reliability toward confirming the SCT, which coupled with its diagnostic returns like being simple to use and rapid, makes it very advantageous in a limited resource set up. In addition, its simplicity makes it fieldable even where electricity is not available. This is thought to bridge the gap in the widely thought community outreaches to increase awareness of Hb-S status.8
Of the 242 participants, 231 (95.5%) tested negative, while 11 (4.5%) tested positive for SCT. This indicates a high burden of SCT, a finding comparable to a report from a review done in Africa.4 Further, our findings are in agreement with the 4.6% prevalence that was earlier reported in Southwestern Uganda.9 Four of the 11 participants who tested positive using the Hb electrophoresis were found to be negative when the Hb-solubility kit was used. This is attributable to the likely false negativity due to the high HbF variant interference.17
As the Hb electrophoresis indicated the HbAS variant, it is irrefutable that genetics had a role in the Hb presentation. The same finding has been reported in Eastern and Western Uganda,10 and in Ghana.27 Although complex, the HbAS variant offers a protective advantage to infections common in our set up, such as infection with Plasmodium species,6,28–30 and this risked 25% of the next generation to acquire HbSS if genetic counseling and testing is not emphasized.21,22 Further, findings from our study have indicated an information gap regarding the existence of the sickle cell defects, similar to what has been reported by other studies.7,9,10 Lack of awareness may portend the effort to lessen the risk of genetic cross-over that could multiply the trait incidence as well as the number of individuals who may inherit the sickle cell disease. It ought to be remembered that premarital counseling and screening for genetic disorders is key to their reduction and possible elimination of defects as a result of two partners with a carier state.4,8 Thus, in a set-up where genetic screening is neither indicated nor done, the career likelihood is rather amplified and may promulgate the genotypes and the associated defects. Our study ought to be interpreted in light of these limitations. First, the rapid sickle cell test does not identify other hemoglobinopathies which may present with varied forms of anemia. Second, though our study population consisted of adults in the child bearing age, we did not carry out partner-to-partner screening. Thus, this study cannot independently conclude the attributable carry over risk to the next generation.
Conclusion
Performance evaluation of the rapid sickle cell test indicated a high diagnostic reliability, which makes it suitable for field set-ups. Further, the study reports a high prevalence of SCT, and it is associated with genetic predisposition. Community sensitization is key to avert the associated risk of Hb defects.
Acknowledgments
The authors would like to acknowledge study participants and the management of Magale Health Center IV. We are grateful to the staff of Central Public Health Laboratories. We acknowledge the Uganda Sickle Cell Rescue Foundation who, with support from Hotel Africana, Roofings Group and AGT group, procured and donated the Sickle Cell screening kits to the study. This work did not receive any funding; however, we recognize the logistical support from Manafwa District Local Government.
Author contributions
KM, SKT, BM, FW, CA and IMT participated in study conception and design, data acquisition, analysis and interpretation, manuscript drafting and revising. CA, BM and IMT critically revised the manuscript. All authors read and approved the final manuscript. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Hatton MF. Molecular pathology of human haemoglobin. Nature. 2013;219(157):902–909. | ||
Perutz MF, Rossmann MG, Cullis MG, Muirhead H, Will G, North AC. Structure of haemoglobin: A three-dimensional Fourier synthesis at 5.5Å resolution, obtained by X-ray analysis. Nature. 2013;185(4711):416–422. | ||
Lewis R. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature. 2006;180:326–328. | ||
Mulumba E, Wilson S. Comprehensive care in sickle cell disease: its impact on morbidity and mortality. Semin Hematol. 2015;28(3):220–226. | ||
Piel N, Nagel RL, Platt OS. Disorders of Hemoglobin. Cambridge: Cambridge University Press; 2010:494–526. | ||
Gamit D. Sickle cell trait and sudden death-bringing it home. J Natl Med Assoc. 2014;99(3):300–305. | ||
Jenerette L, Murdaugh D. New human haemoglobin variant from southern Arabia. Nature. 2008;219(159):1164–1166. | ||
World Health Organization. Hemoglobin Disorders; 2013. Available from: http://apps.who.int/iris/bitstream/10665/44665/1/9789241548274_eng.pdf. Accessed September 5, 2018. | ||
Ndeezi G, Kiyaga C, Hernandez AG, et al. Burden of sickle cell trait and disease in the Uganda Sickle Surveillance Study (US3): a cross-sectional study. Lancet Glob Health. 2016;4(3):e195–e200. | ||
Okwi AL, Byarugaba W, Ndugwa CM, Parkes A, Ocaido M, Tumwine JK. An up-date on the prevalence of sickle cell trait in Eastern and Western Uganda. BMC Blood Disord. 2010;10(1):5. | ||
Kiyaga C. Uganda Sickle Survey Results and Screening Program. 2014. Available from: http://www.hollyfoundationsicklecell.co.uk/wp-content/uploads/2015/06/The-Survey-in-Adobe-PDF-format.pdf. Accessed July 17, 2017. | ||
Kanter J, Telen MJ, Hoppe C, Roberts CL, Kim JS, Yang X. Validation of a novel point of care testing device for sickle cell disease. BMC Med. 2015;13(1):225. | ||
Kumar AA, Patton MR, Hennek JW, et al. Density-based separation in multiphase systems provides a simple method to identify sickle cell disease. Proc Natl Acad Sci. 2014;111(41):14864–14869. | ||
Bond M, Hunt B, Flynn B, Huhtinen P, Ware R, Richards-Kortum R. Towards a point-of-care strip test to diagnose sickle cell anemia. PLoS One. 2017;12(5):e0177732. | ||
Mcgann PT, Schaefer BA, Paniagua M, Howard TA, Ware RE. Characteristics of a rapid, point-of-care lateral flow immunoassay for the diagnosis of sickle cell disease. Am J Hematol. 2016;91(2):205–210. | ||
Insert SickleDex® Kit Streck, Omaha, NE, USA. Available from: http://labs-inc.org/pdf/22_3.pdf. Accessed July 10, 2018. | ||
Rapid Sickle Cell Hemoglobin-S (HbAS/HbSS) Dithionate Qualitative. Available from: http://biolabdiagnostics.com/image/catalog/pdf/Rapid%20Sickle%20Literature%20RSC%20R1+R2%209-13.pdf. Accessed September 5, 2018. | ||
Bond M, Hunt B, Flynn B, Huhtinen P, Ware R, Richards-Kortum R. Towards a point-of-care strip test to diagnose sickle cell anemia. Lam W, editor. PLoS One. 2017;12(5):e0177732. | ||
Nathaniel Z, Piety A, George S, et al. A Paper-Based Test for Screening Newborns for Sickle Cell Disease. Sci Rep. 2017;45488. | ||
Kish L. Survey Sampling. New York: John Wiley and Sons, Inc. Section 1.3, 13.1-13.6. Available from: https://biblioteca.ibge.gov.br/visualizacao/monografias/GEBIS%20-%20RJ/censocontinuo/Samples%20and%20Censuses.pdf. Accessed July 10, 2018. | ||
Harrison SE, Walcott CM, Warner TD. Knowledge and Awareness of Sickle Cell Trait Among Young African American Adults. West J Nurs Res. 2017;39(9):1222–1239. | ||
Obed SA, Oppong SA, Torto M, Aboagye S, Nuamah MA, Asah-Opoku K. Awareness of Sickle Cell Trait Status: A Cross-Sectional Survey of Antenatal Women in Ghana. Am J Trop Med Hyg. 2017;96(3):735–740. | ||
Creary S, Adan I, Stanek J, et al. Sickle cell trait knowledge and health literacy in caregivers who receive in-person sickle cell trait education. Mol Genet Genomic Med. 2017;5(6):692–699. | ||
Patil SS, Thikare AA, Wadhva SK, Narlawar UW, Shukla S, et al. Knowledge, attitude and practice regarding sickle cell disease in adult sufferers and carriers in a rural area. Int J Community Med Public Health. 2017;4(4):1075–1080. | ||
Kumar AA, Chunda-Liyoka C, Hennek JW, et al. Evaluation of a Density-Based Rapid Diagnostic Test for Sickle Cell Disease in a Clinical Setting in Zambia. PLoS One. 2014;9(12):e114540. | ||
Piety NZ, Yang X, Kanter J, Vignes SM, George A, Shevkoplyas SS. Validation of a Low-Cost Paper-Based Screening Test for Sickle Cell Anemia. PLoS One. 2016;11(1):e0144901. | ||
Brain J. Hereditary qualitative and quantitative erythrocyte defects in Ghana. An historical and geographical survey. Ghana Med J. 2014;7:118–119. | ||
Deore M, Cokic VP, Smith RD, et al. The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu Rev Biophys Biomol Struct. 2013;27:1–34. | ||
Vafa M, Troye-Blomberg M, Anchang J, Garcia A, Migot-Nabias F. Multiplicity of Plasmodium falciparum infection in asymptomatic children in Senegal: relation to transmission, age and erythrocyte variants. Malar J. 2008;7(1):17. | ||
Kwiatkowski DP. How malaria has affected the human genome and what human genetics can teach us about malaria. Am J Hum Genet. 2005;77(2):171–192. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.