")
Back to Journals » Cancer Management and Research » Volume 10
TMEM17 promotes malignant progression of breast cancer via AKT/GSK3β signaling
Authors Zhao Y, Song K, Zhang Y, Xu H, Zhang X , Wang L , Fan C, Jiang G, Wang E
Received 20 March 2018
Accepted for publication 14 May 2018
Published 2 August 2018 Volume 2018:10 Pages 2419—2428
DOI https://doi.org/10.2147/CMAR.S168723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Harikrishna Nakshatri
Yue Zhao,1 Kuiyuan Song,1 Yong Zhang,2 Hongtao Xu,1 Xiupeng Zhang,1 Liang Wang,1 Chuifeng Fan,1 Guiyang Jiang,1 Enhua Wang1
1Department of Pathology, First Affiliated Hospital and College of Basic Medical Sciences, China Medical University, Shenyang, China; 2Departments of Pathology, Cancer Hospital of China Medical University, Liaoning Cancer Hospital and Institute, Shenyang, China
Purpose: Current knowledge of TMEM17, a recently identified protein of the transmembrane (TMEM) family, is limited, especially with respect to its expression and biological functions in malignant tumors. This study analyzed TMEM17 expression in invasive breast cancer tissue and breast cell lines and its relevance to clinicopathological factors, and investigated the mechanisms underlying the biological effects of TMEM17 on breast cancer cells.
Patients and methods: TMEM17 protein expression was determined in 20 freshly harvested specimens (tumor and paired normal tissues) by Western blotting. Immunohistochemical analysis was performed to determine the expression and subcellular localization of TMEM17 in samples from 167 patients (mean age, 49 years) diagnosed with invasive ductal carcinoma (38 with triple-negative breast cancer; 129 with non-triple-negative breast cancer) who underwent complete resection in the First Affiliated Hospital of China Medical University between 2011 and 2013. Furthermore, TMEM17 was knocked down by small interfering RNAs in breast cancer cell lines.
Results: TMEM17 was found to be significantly upregulated in breast cancer tissues compared to the corresponding normal breast tissues by Western blotting (p=0.015). Immunohistochemical analysis revealed that TMEM was significantly upregulated in invasive breast cancer cells compared to adjacent normal breast duct glandular epithelial cells (10.78% vs 76.05%, p<0.001), and its expression was closely related to the patient’s T-stage (p=0.022), advanced TNM stages (p=0.007), and lymph node metastasis (p=0.012). After TMEM17 knockdown or overexpression in breast cancer cell lines, TMEM17 upregulated p-AKT, p-GSK3β, active β-catenin, and Snail, and downstream target proteins c-myc and cyclin D1, and downregulated E-cadherin, resulting in increased cancer cell proliferation, invasion, and migration. These effects were reversed by the AKT inhibitor LY294002.
Conclusion: Our results indicate that TMEM17 is upregulated in breast cancer tissues and can promote malignant progression of breast cancer cells by activating the AKT/GSK3β signaling pathway.
Keywords: proliferation, invasion, migration, β-catenin, Snail
Introduction
Breast cancer is the most common cancer among women worldwide.1 Owing to significant advancements in tumor diagnostic and therapeutic strategies, especially endocrine therapy, breast cancer mortality rates have steadily declined since the 1970s.2 However, owing to metastasis and genetic heterogeneity (especially triple-negative breast cancer [TNBC]), breast cancer is still one of the leading causes of cancer-related mortality among women.3,4 New biomarkers are required for more accurate prognosis and development of new targeted drugs for breast cancer.
TMEM17, a recently identified member of the transmembrane (TMEM) protein family, is involved in numerous physiological processes, including plasma membrane ion channel formation, signal transduction, cellular chemotaxis, adhesion, apoptosis, and autophagy.5–10 Emerging evidence indicates that the TMEM protein family is significantly associated with malignant progression and chemotherapeutic resistance in various cancers; however, the features and localization of the members of the TMEM protein family display considerable differences.11–18 Therefore, the functions of different TMEM family members in different tumors warrant detailed investigation. TMEM17 is a 198-amino acid protein encoded by a gene on human chromosome 2 and was initially reported to be a cilium-associated protein.19 Recently, another group in our laboratory reported that TMEM17 was downregulated in non-small cell lung cancer (NSCLC) and could inhibit invasion and metastasis of lung cancer cells.20 However, the current knowledge regarding TMEM17 expression and biological function in malignant tumors is still limited.
The present study aimed to analyze TMEM17 expression in invasive breast cancer tissue and breast cell lines and its relevance to clinicopathological factors. Furthermore, we investigated the mechanism underlying the biological effects of TMEM17 on breast cancer cells. Our results may provide a theoretical and experimental basis for the potential targeting of TMEM17 in the diagnosis and treatment of breast cancer.
Patients and methods
Patients and specimens
This study was approved by the local institutional review board of the China Medical University. The patients whose tissue samples were used in this research provided written informed consent. Primary tumor specimens were obtained from 167 patients (mean age, 49 years [range, 35–87 years]) diagnosed with invasive ductal carcinoma (38 with TNBC and 129 with non-TNBC) who underwent complete resection in the First Affiliated Hospital of China Medical University between 2011 and 2013. Patients who underwent neoadjuvant radiotherapy and/or chemotherapy were excluded. All patients received standard chemotherapy after surgery. Lymph node metastases were detected in 79 patients. The p-TNM staging system of the International Union Against Cancer (seventh edition) was used to classify specimens as stages I (n=21), II (n=78), III (n=66), and IV (n=2). In addition, 20 freshly harvested specimens, including both tumor and paired normal tissues, were stored at −80°C immediately after resection, until use.
Immunohistochemistry
All tissue specimens were fixed in neutral formaldehyde, embedded in paraffin, and sectioned (thickness, 4 μm). The streptavidin–peroxidase immunohistochemical method was used to enhance staining intensity. Tissue sections were incubated at 4°C overnight with TMEM17 mouse monoclonal antibody (G-10, sc-514433; 1:50 dilution; Santa Cruz Biotechnology Inc, Dallas, TX, USA); phosphate-buffered saline was used as a blank control. Sections were then incubated with biotin-labeled secondary antibody (Ultrasensitive; MaiXin, Fuzhou, China) at 37°C for 30 minutes, followed by diaminobenzidine for color formation. Finally, samples were lightly counterstained with hematoxylin, dehydrated in alcohol, and mounted. Two investigators blinded to the clinical data semiquantitatively scored the slides by evaluating the staining intensity and percentage of stained cells in representative areas. The staining intensity was scored as 0 (no signal), 1 (weak), 2 (moderate), or 3 (high). The percentage of cells stained was scored as 1 (1%–25%), 2 (26%–50%), 3 (51%–75%), or 4 (76%–100%). A final score of 0–12 was obtained by multiplying the intensity and percentage scores. Tumors were considered TMEM17 positive if a score of ≥4 was obtained. Tumor samples with scores between 1 and 3 were categorized as showing weak expression, whereas those with scores of 0 were considered to display no expression; both weak expression and no expression were defined as negative expression.
Cell culture
MCF-10A, MCF-7, T47D, MDA-MB-231, and MDA-MB-468 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in RPMI-1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal calf serum (Thermo Fisher Scientific), 100 IU/mL penicillin (Sigma-Aldrich Co, St Louis, MO, USA), and 100 μg/mL streptomycin (Sigma-Aldrich Co). Cells were cultured in sterilized culture dishes and subcultured every 2 days following trypsinization with 0.25% trypsin (Thermo Fisher Scientific).
Plasmid transfection and small interfering RNA (siRNA) treatment
Plasmids pCMV6-ddk-myc and pCMV6-ddk-myc-TMEM17 were purchased from OriGene (Rockville, MD, USA). TMEM17-siRNA (sc-94962) and NC-siRNA (sc-37007) were purchased from Santa Cruz Biotechnology Inc. Transfection was performed using Lipofectamine 3000 reagent (Thermo Fisher Scientific) in accordance with the manufacturer’s instructions.
Western blotting
Total protein was extracted using a lysis buffer (Thermo Fisher Scientific) and quantified through the Bradford method. In total, 50 μg of protein samples was separated through sodium dodecyl sulfate polyacrylamide gel electrophoresis (10% resolving gel) and electroblotted onto polyvinylidene fluoride membranes (Merck Millipore, Billerica, MA, USA). Membranes were incubated overnight at 4°C with the following primary antibodies: anti-TMEM17 (1:200; Santa Cruz Biotechnology Inc), anti-GAPDH (1:5000; Sigma-Aldrich Co), anti-Snail, anti-Myc-tag, anti-p-ERK, anti-ERK, anti-p-AKT, anti-AKT, anti-active β-catenin, anti-cyclin D1, anti-c-myc (1:500; Cell Signaling Technology, Danvers, MA, USA), anti-E-cadherin, and anti-β-catenin (1:500; BD Biosciences, San Jose, CA, USA). Membranes were washed and subsequently incubated with peroxidase-conjugated anti-mouse or anti-rabbit IgG (Santa Cruz Biotechnology Inc) at 37°C for 2 hours. Bound proteins were visualized using electrochemiluminescence (Thermo Fisher Scientific) and detected with a bio-imaging system (DNR Bio-Imaging Systems, Jerusalem, Israel).
MTT
Cells were plated in 96-well plates in medium containing 10% fetal bovine serum (FBS) at a density of ~3000 cells per well. For quantitation of cell viability, cultures were subjected to an MTT assay 5 days post-transfection. Briefly, 20 μL of 5 mg/mL MTT (thiazolyl blue) solution was added to each well and incubated for 4 hours at 37°C. Then, medium was aspirated from each well and the resultant MTT formazan was solubilized in 150 μL of DMSO. The results were obtained spectrophotometrically with a test wavelength of 570 nm.
Colony formation assay
The MCF-7 and MDA-MB-231 cells were transfected with siRNA targeting TMEM17 for 24 hours. Thereafter, cells were seeded into three 6-cm cell culture dishes (1000 per dish for MCF-7 and MDA-MB-231 cells) and incubated at 37°C. Twelve days later, plates were washed with PBS and stained with Giemsa. The number of colonies with >50 cells was counted.
Matrigel invasion
A cell invasion assay was performed using a 24-well transwell chamber with 8-μm pores (Costar Corp., Cambridge, MA, USA). The inserts were coated with 20 of μL Matrigel (1:3 dilution; BD Biosciences). At 24 hours after transfection, cells were trypsinized, and 3×105 cells in 100 μL of serum-free medium were transferred to the upper Matrigel chamber for 18 hours. Medium supplemented with 10% FBS was added to the lower chamber as a chemoattractant. After incubation, cells that passed through the filter were fixed with 4% paraformaldehyde and stained with hematoxylin. The invasive cells were microscopically counted in 10 randomly selected high-power fields.
Wound healing assay
In cultures with cell density <90%, at 48 hours after transfection, wounds were inflicted in confluent areas, using a 1000-μL pipette tip. Wound healing within the scrape line was observed at both 0 and 24 hours, and representative scrape lines for each cell line were photographed. Duplicate wells were examined for each condition, and each experiment was performed in triplicate. The optical wound distances were measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
SPSS version 22.0 for Windows (IBM Corporation, Armonk, NY, USA) was used for all statistical analyses. Pearson’s chi-square test was used to assess possible correlations between TMEM17 and clinicopathological factors. The Mann–Whitney U test was used for image analysis of Western blot results and the invasive assay results. p<0.05 was considered to indicate statistically significant differences.
Results
TMEM17 expression in breast cancer and its relevance to clinicopathological factors
We analyzed TMEM17 protein expression in 20 pairs of breast cancer tissue specimens through Western blotting. Normalized protein expression of TMEM17 (mean ± SEM =0.7086±0.06589, N=20) was significantly higher in breast cancer tissues than in the corresponding normal breast tissues (mean ± SEM =0.4760±0.06318, N=20, p=0.015, Figure 1A and B).
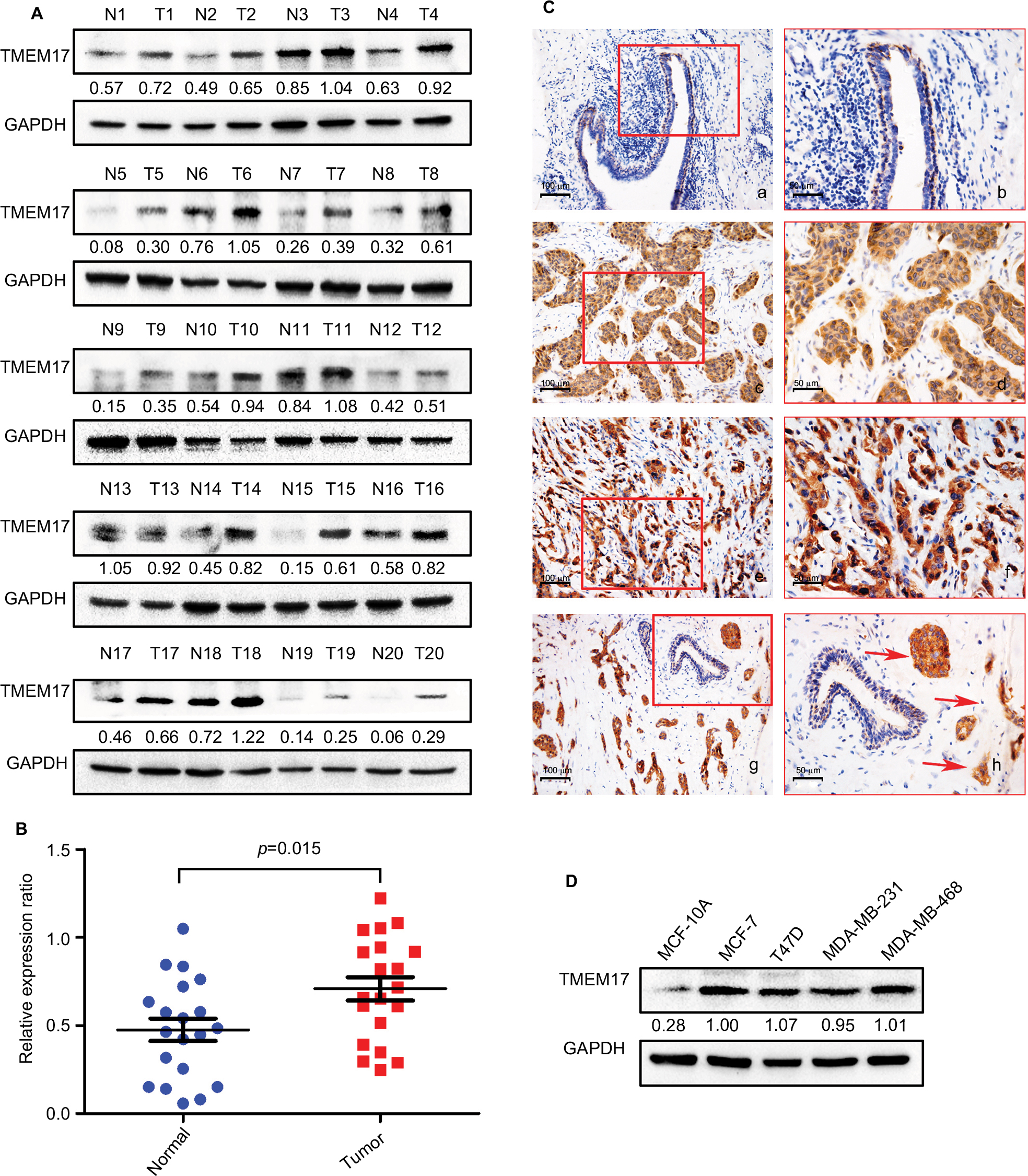 | Figure 1 TMEM17 expression in breast cancer tissues and cell lines. (A, B) Western blotting of 20 pairs of breast cancer tissue specimens showing that TMEM17 expression in breast cancer tissues was significantly higher than that in the corresponding normal breast tissues (p=0.015). (Ca, b) TMEM17 was negative in the normal breast duct glandular epithelium cells and weakly to moderately positive in the cytoplasm of normal breast duct myoepithelial cells. TMEM17 showed moderate (c, d) and strong (e, f) positive staining in the cytoplasm of cancer cells. (g, h) TMEM17 expression was significantly higher in breast cancer cells than in the adjacent normal breast duct glandular epithelium cells in the same field of view. Additionally, scattered nuclear staining was rarely visible in the cells. a, c, e, g 200×; b, d, f, h 400×. (D) TMEM17 expression was significantly higher in MCF-7, T47D, MDA-MB-231, and MDA-MB-468 breast cancer cells than in the normal human mammary epithelial cell line MCF-10A. |
We performed immunohistochemistry to analyze the expression and subcellular localization of TMEM17 in samples from 167 cases of breast cancer and adjacent noncancerous tissue specimens. TMEM17 expression was positive in the cytoplasm of normal breast duct myoepithelial cells (95.81%, 160/167), but negative or very weakly positive in the normal breast duct glandular epithelium (10.78%, 18/167, Figure 1Ca and b, Table 1). However, TMEM17 staining was significantly increased in the vast majority of invasive breast cancer, displaying moderate-to-strong positive staining in the cytoplasm of cancer cells (76.05%, 127/167, p<0.001, Figure 1Cc–f, Table 1). Scattered nuclear staining was rarely visible and not prominent. Figure 1Cg and h shows that TMEM17 expression was much higher in breast cancer cells than in adjacent normal breast duct glandular epithelial cells in the same field of view. Furthermore, TMEM17 positive expression was significantly correlated with the breast cancer patient’s T-stage (p=0.022), advanced TNM stages (p=0.007), and lymph node metastasis (p=0.012), but had no significant association with age (p=0.296) or estrogen receptor, progesterone receptor, or Her-2 status (p=0.698, Table 2).
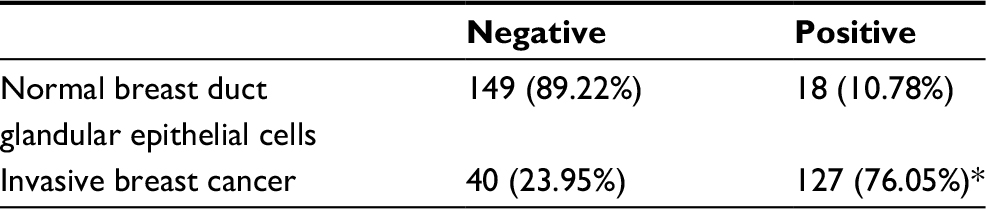 | Table 1 TMEM17 expression in adjacent normal breast tissue samples and invasive breast cancer specimens Note: *p<0.001, indicating statistical significance. |
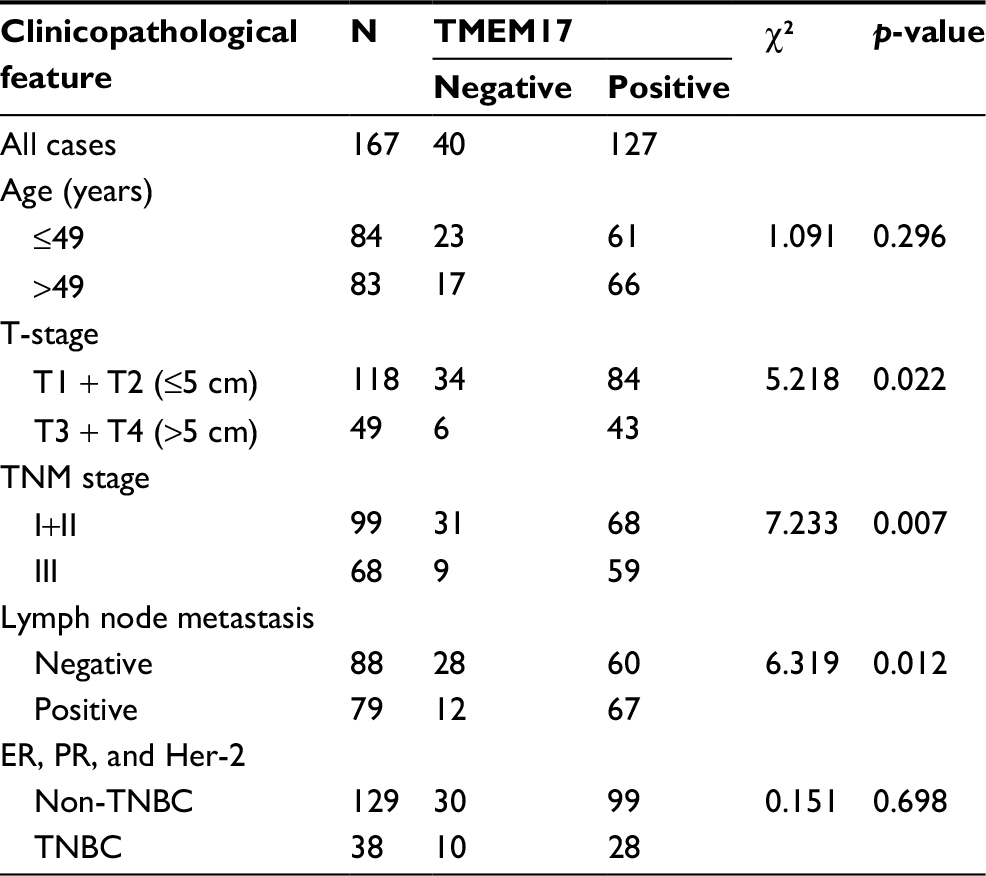 | Table 2 Summary of correlation of TMEM17 expression with clinicopathological characteristics of invasive breast cancer Abbreviations: TNBC, triple-negative breast cancer; ER, estrogen receptor; PR, progesterone receptor. |
TMEM17 expression was significantly higher in four breast cancer cell lines MCF-7, T47D, MDA-MB-231, and MDA-MB-468 than in the normal human mammary epithelial cell line MCF-10A (Figure 1D).
TMEM17 knockdown inhibited breast cancer cell proliferation, invasiveness, and migration
TMEM17 was knocked down by siRNAs in MCF-7 and MDA-MB-231 cells (Figure 2A). The MTT assay revealed that the siRNA targeting TMEM17 significantly inhibited the proliferation of MCF-7 (p<0.001 at the fifth day) and MDA-MB-231 (p<0.001 at the fifth day) cells (Figure 2B). Colony formation assays also revealed a significant reduction in the number of foci of MCF-7 (p=0.007) and MDA-MB-231 (p=0.011) cells after TMEM17 knockdown (Figure 2C). However, transwell analysis revealed that TMEM17 knockdown inhibited the invasion of MCF-7 (p=0.0025) and MDA-MB-231 (p=0.0037) cells (Figure 2D). Furthermore, a wound healing assay revealed that cell migration was also decreased by silencing of TMEM17 in MCF-7 (p=0.0363) and MDA-MB-231 (p=0.0044) cells (Figure 2E).
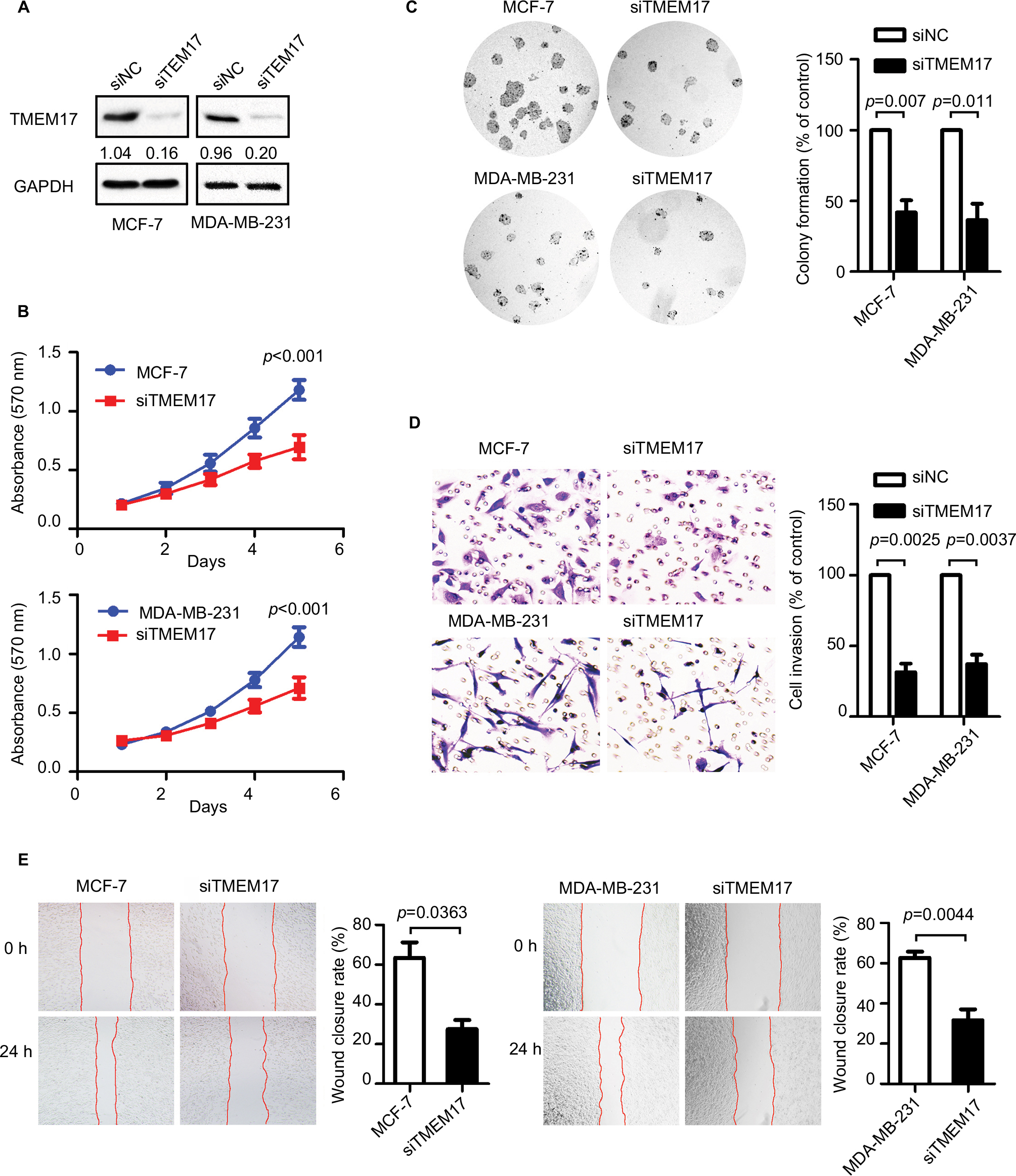 | Figure 2 TMEM17 knockdown inhibited proliferation, colony formation, invasion, and migration of MCF-7 and MDA-MB-231 cells. (A) Interference efficiency in MCF-7 and MDA-MB-231 cell lines after TMEM17 was knocked down by siRNA. (B) MTT assay shows that TMEM17 interference significantly inhibited proliferation in MCF-7 (the fifth day, p<0.001) and MDA-MB-231 (the fifth day, p<0.001) cells. (C) Colony formation assays revealed that TMEM17 knockdown significantly decreased the number of colonies of MCF-7 (p=0.007) and MDA-MB-231 (p=0.011) cells. (D) Transwell analysis revealed that TMEM17 knockdown inhibited the invasion of MCF-7 (p=0.0025) and MDA-MB-231 (p=0.0037). (E) Wound assay revealed that TMEM17 silencing also inhibited the migration of MCF-7 (p=0.0363) and MDA-MB-231 cells (p=0.0044). Abbreviations: siRNA, small interfering RNA; siNC, small interfering negative control. |
TMEM17 upregulated active β-catenin and Snail by promoting AKT and GSK3β phosphorylation
Because TMEM17 modulated ERK pathway activity in NSCLC cells,20 we first investigated the effect of TMEM17 on p-ERK and total ERK expression in breast cancer cells. However, after repeated verification, we confirmed that TMEM17 did not affect the expression of p-ERK or total ERK in MCF-7 and MDA-MB-231 cells. Interestingly, we found that TMEM17 upregulated p-AKT (Ser 473), p-GSK3β (Ser 9), active β-catenin, and Snail and their downstream target proteins; upregulated c-myc and cyclin D1; and downregulated E-cadherin expression in breast cancer cells (Figure 3). Furthermore, TMEM17-transfected MCF-7 cells were treated with the AKT inhibitor LY294002. LY294002 inhibited the TMEM17-induced increase in p-AKT (Ser 473), p-GSK3β (Ser 9), downregulated active β-catenin and Snail, and restored expression of c-myc, cyclin D1, and E-cadherin (Figure 4A). Correspondingly, MTT and transwell assays revealed that the increased cell proliferation and invasiveness caused by TMEM17 were also reversed by LY294002 (Figure 4B and C).
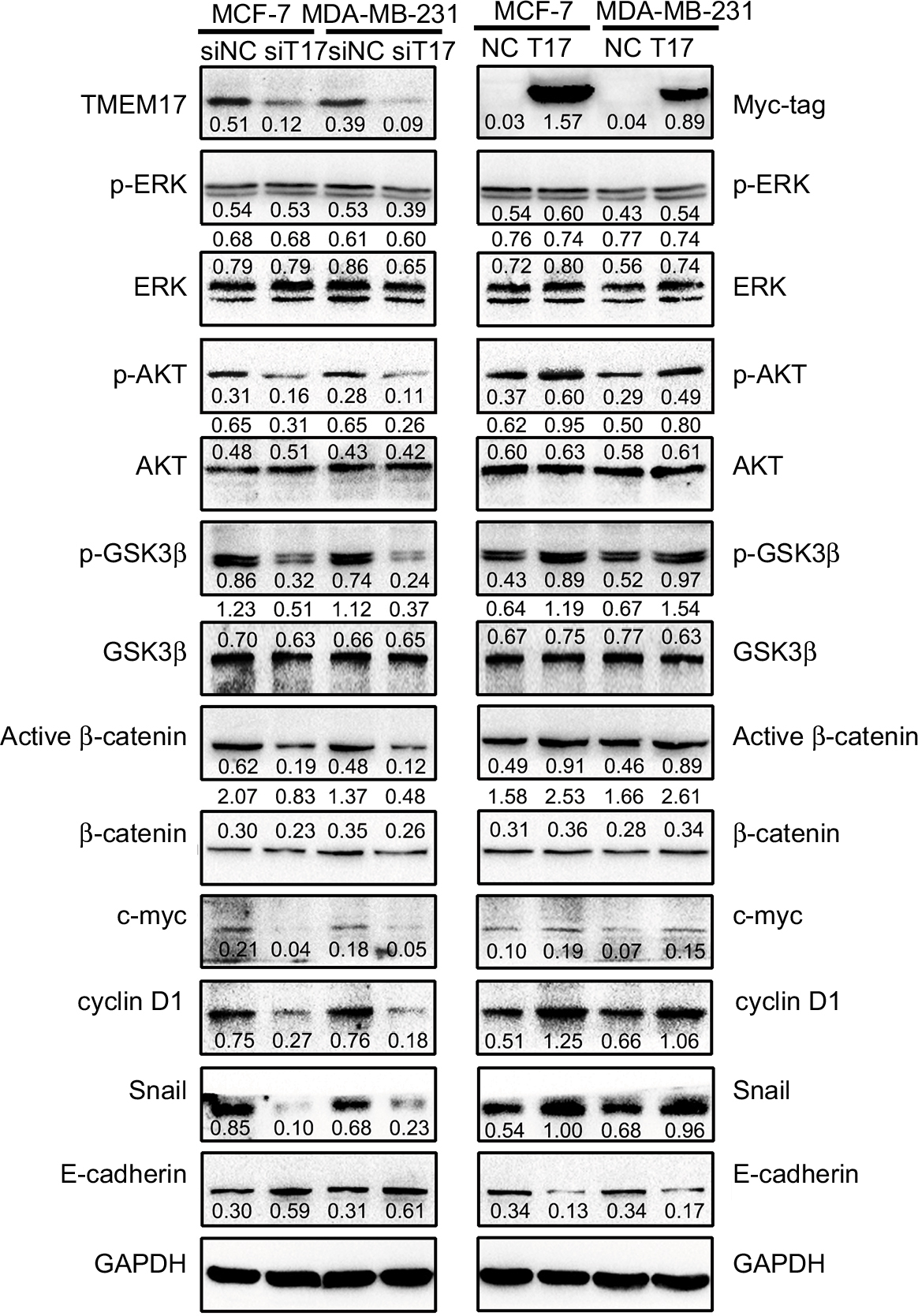 | Figure 3 TMEM17 upregulated active β-catenin and Snail by affecting p-AKT and p-GSK3β, but not p-ERK. TMEM17 could not affect the expression of p-ERK or total ERK in MCF-7 and MDA-MB-231 cells. However, TMEM17 could stably upregulate p-AKT (Ser 473), p-GSK3β (Ser 9), active β-catenin, and Snail; upregulate c-myc and cyclin D1; and downregulate E-cadherin expression. Abbreviations: siNC, small interfering negative control. NC, negative control. |
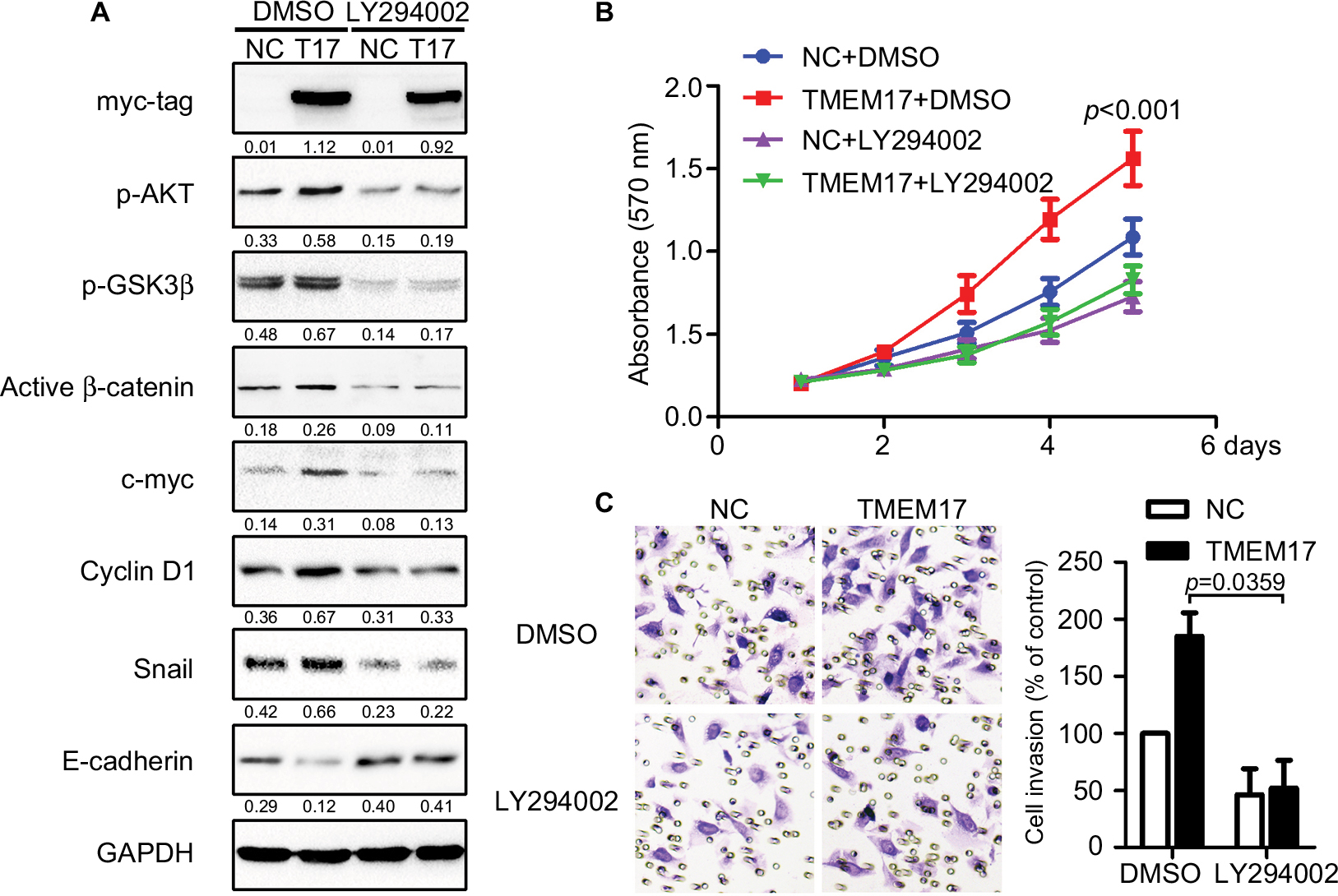 | Figure 4 TMEM17 promoted the proliferation and invasion of cancer cells via activation of AKT/GSK3β signaling. (A) The AKT inhibitor LY294002 (10 μM) could inhibit the TMEM17-induced increase with p-AKT (Ser 473) and p-GSK3β (Ser 9), and downregulate active β-catenin and Snail, and then restore the expression of c-myc, cyclin D1, and E-cadherin. (B) MTT assay revealed that increased cell proliferation caused by TMEM17 was reversed by LY294002 (the fifth day, p<0.001). (C) Transwell assay revealed that the increased invasiveness caused by TMEM17 was also reversed by LY294002 (p=0.002). |
Discussion
TMEM proteins contain TMEM domains,9 and are predicted to localize at different cellular membranes, such as the plasma membrane, nuclear membrane, mitochondrial membrane, and membranes of the endoplasmic reticulum, lysosomes, and Golgi complex.6 It has been shown that TMEM proteins play important roles in various tumors, such as lung cancer,12 breast cancer,11,13 liver cancer,13 ovarian cancer,14 colorectal cancer,15 renal cell carcinoma,16 and lymphoma.17 However, there are marked differences in function and localization among TMEM proteins in various tumors.18 The present results show that TMEM17 is upregulated in the cytoplasm of breast cancer tissues, closely related with the T-stage of breast cancer patients, advanced TNM stages, and lymph node metastasis and could enhance breast cancer cell proliferation, invasiveness, and migration. This is completely different from the expression pattern and functions of TMEM17 in NSCLC reported by another group of our laboratory,20 which was initially contradictory. We repeated immunohistochemistry and Western blotting analyses using another TMEM17 polyclonal antibody (ab116550); however, we obtained the same results. Furthermore, data from The Human Protein Atlas (https://www.proteinatlas.org/) indicate that TMEM17 is upregulated in bronchial epithelial cells, downregulated in normal breast tissue, and markedly upregulated in breast cancer cells (data not shown), which is consistent with our results. Because our study specimens did not yield sufficient prognostic data, we performed a survival analysis of TMEM17 expression in breast cancer specimens, using the KM Plotter Online Tool (http://www.kmplot.com). The data thus obtained indicate that TMEM17 expression was significantly correlated with poor survival in breast cancer patients (relapse-free survival: n=1764, p=0.019; distant metastasis-free survival: n=664, p=0.0009, Figure S1). The aforementioned data support our results, and further confirm that the expression and function of TMEM17 are organization-specific in NSCLC and breast cancer.
Correspondingly, after repeated verification and screening, we found that TMEM17 stably upregulated p-AKT, p-GSK3β, active β-catenin, c-myc, cyclin D1, and Snail and downregulated E-cadherin in breast cancer cells, but did not affect ERK signaling as in lung cancer. Both β-catenin and Snail were phosphorylated by GSK3β at Ser/Thr residues, and then subsequently degraded.21–23 TMEM17 activated AKT phosphorylation at Ser 473, which in turn phosphorylated GSK3β at Ser 9, thereby inhibiting its function,24,25 thus leading to the upregulation of active β-catenin and Snail. c-myc and cyclin D1 are classic downstream target proteins of active β-catenin, and play a prominent role in promoting cancer cell proliferation. However, Snail downregulated the important adhesion complex protein E-cadherin and enhanced the invasiveness and migration of cancer cells. Furthermore, an AKT inhibitor prevented GSK3β phosphorylation, subsequently reversing the upregulation of active β-catenin, c-myc, cyclin D1, and Snail and restoring the downregulation of E-cadherin that resulted from TMEM17 overexpression. The proliferation and invasiveness of breast cancer cells were also reversed upon AKT inhibitor treatment. Hence, our results reveal that TMEM17 promotes the malignant progression of breast cancer cells by activating the AKT/GSK3β signaling pathway. However, how TMEM17 affects p-AKT is still unclear, and the detailed mechanism should be elucidated in future studies.
Gupta et al reported that TMEM17 directly interacts with dishevelled-1 (DVL-1) and low-density lipoprotein-related protein 6,26 which are crucial factors in the canonical Wnt signaling pathway, and could also affect GSK3β phosphorylation and subsequently regulate active β-catenin and Snail.27 Moreover, the present results indicate that p-JNK, which has a complex relationship with DVL,28,29 can also be upregulated by TMEM17; however, the results were not stable (data not shown). These data imply that TMEM17 participates in more complex mechanisms in breast cancer; this will be further investigated in our future studies.
Conclusion
TMEM17 was downregulated in normal breast tissues and cell lines and significantly upregulated in metastatic breast cancer tissues and cell lines, as opposed to NSCLC. TMEM17 expression was significantly correlated with the T-stage of breast cancer patients, advanced TNM stages, and lymph node metastasis. TMEM17 upregulated active β-catenin and Snail via the AKT/GSK3β signaling pathway and enhanced proliferation, invasiveness, and migration of breast cancer cells. Additionally, our data indicate that there are no prominent differences in the expression and function of TMEM17 between TNBC and non-TNBC; hence, TMEM17 may be a potential diagnostic and therapeutic target for breast cancer, including TNBC.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No 81402369 to Guiyang Jiang, No 81572854 to Enhua Wang, No 81472599 to Chuifeng Fan) and the Natural Science Foundation of Liaoning Province (No 2015020499 to Yue Zhao). We would like to thank Editage (www.editage.cn) for English language editing.
Author contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Kohler BA, Sherman RL, Howlader N, et al. Annual report to the nation on the status of cancer, 1975–2011, featuring incidence of breast cancer subtypes by race/ethnicity, poverty, and state. J Natl Cancer Inst. 2015;107(6):djv048. | ||
Rakha EA. Pitfalls in outcome prediction of breast cancer. J Clin Pathol. 2013;66(6):458–464. | ||
Tremont A, Lu J, Cole JT. Endocrine therapy for early breast cancer: updated review. Ochsner J. 2017;17(4):405–411. | ||
Ishihara K, Suzuki J, Nagata S. Role of Ca(2+) in the stability and function of TMEM16F and 16K. Biochemistry. 2016;55(23):3180–3188. | ||
Wrzesinski T, Szelag M, Cieslikowski WA, et al. Expression of pre-selected TMEMs with predicted ER localization as potential classifiers of ccRCC tumors. BMC Cancer. 2015;15:518. | ||
Hayez A, Malaisse J, Roegiers E, et al. High TMEM45A expression is correlated to epidermal keratinization. Exp Dermatol. 2014;23(5):339–344. | ||
Ferrera L, Caputo A, Galietta LJ. TMEM16A protein: a new identity for Ca(2+)-dependent Cl(−) channels. Physiology. 2010;25(6):357–363. | ||
Dobashi S, Katagiri T, Hirota E, et al. Involvement of TMEM22 overexpression in the growth of renal cell carcinoma cells. Oncol Rep. 2009;21(2):305–312. | ||
Martin-Rendon E, Hale SJ, Ryan D, et al. Transcriptional profiling of human cord blood CD133+ and cultured bone marrow mesenchymal stem cells in response to hypoxia. Stem Cells. 2007;25(4):1003–1012. | ||
Yu X, Zhang X, Zhang Y, Jiang G, Mao X, Jin F. Cytosolic TMEM88 promotes triple-negative breast cancer by interacting with Dvl. Oncotarget. 2015;6(28):25034–25045. | ||
Zhang X, Yu X, Jiang G, et al. Cytosolic TMEM88 promotes invasion and metastasis in lung cancer cells by binding DVLS. Cancer Res. 2015;75(21):4527–4537. | ||
Flamant L, Roegiers E, Pierre M, et al. TMEM45A is essential for hypoxia-induced chemoresistance in breast and liver cancer cells. BMC Cancer. 2012;12:391. | ||
Guo J, Chen L, Luo N, Yang W, Qu X, Cheng Z. Inhibition of TMEM45A suppresses proliferation, induces cell cycle arrest and reduces cell invasion in human ovarian cancer cells. Oncol Rep. 2015;33(6):3124–3130. | ||
Hrasovec S, Hauptman N, Glavac D, Jelenc F, Ravnik-Glavac M. TMEM25 is a candidate biomarker methylated and down-regulated in colorectal cancer. Dis Markers. 2013;34(2):93–104. | ||
Abermil N, Guillaud-Bataille M, Burnichon N, et al. TMEM127 screening in a large cohort of patients with pheochromocytoma and/or paraganglioma. J Clin Endocrinol Metab. 2012;97(5):E805–E809. | ||
Cuajungco MP, Podevin W, Valluri VK, Bui Q, Nguyen VH, Taylor K. Abnormal accumulation of human transmembrane (TMEM)-176A and 176B proteins is associated with cancer pathology. Acta Histochem. 2012;114(7):705–712. | ||
Tran Q, Park J, Lee H, et al. TMEM39A and human diseases: a brief review. Toxicol Res. 2017;33(3):205–209. | ||
Li C, Jensen VL, Park K, et al. MKS5 and CEP290 dependent assembly pathway of the ciliary transition zone. PLoS Biol. 2016;14(3): e1002416. | ||
Zhang X, Zhang Y, Miao Y, Zhou H, Jiang G, Wang E. TMEM17 depresses invasion and metastasis in lung cancer cells via ERK signaling pathway. Oncotarget. 2017;8(41):70685–70694. | ||
Xu Y, Lee SH, Kim HS, et al. Role of CK1 in GSK3beta-mediated phosphorylation and degradation of snail. Oncogene. 2010;29(21):3124–3133. | ||
Zheng H, Li W, Wang Y, et al. Glycogen synthase kinase-3 beta regulates Snail and beta-catenin expression during Fas-induced epithelial–mesenchymal transition in gastrointestinal cancer. Eur J Cancer. 2013;49(12):2734–2746. | ||
van Kappel EC, Maurice MM. Molecular regulation and pharmacological targeting of the beta-catenin destruction complex. Br J Pharmacol. 2017;174(24):4575–4588. | ||
Freyberg Z, Ferrando SJ, Javitch JA. Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am J Psychiatry. 2010;167(4):388–396. | ||
Chen R, Yang Q, Lee JD. BMK1 kinase suppresses epithelial–mesenchymal transition through the Akt/GSK3beta signaling pathway. Cancer Res. 2012;72(6):1579–1587. | ||
Gupta GD, Coyaud E, Goncalves J, et al. A dynamic protein interaction landscape of the human centrosome–cilium interface. Cell. 2015;163(6):1484–1499. | ||
Brunt L, Scholpp S. The function of endocytosis in Wnt signaling. Cell Mol Life Sci. 2018;75(5):785–795. | ||
Lee HJ, Shi DL, Zheng JJ. Conformational change of Dishevelled plays a key regulatory role in the Wnt signaling pathways. Elife. 2015;4: e08142. | ||
Fukukawa C, Nagayama S, Tsunoda T, Toguchida J, Nakamura Y, Katagiri T. Activation of the non-canonical Dvl-Rac1-JNK pathway by frizzled homologue 10 in human synovial sarcoma. Oncogene. 2009;28(8):1110–1120. |
Supplementary material
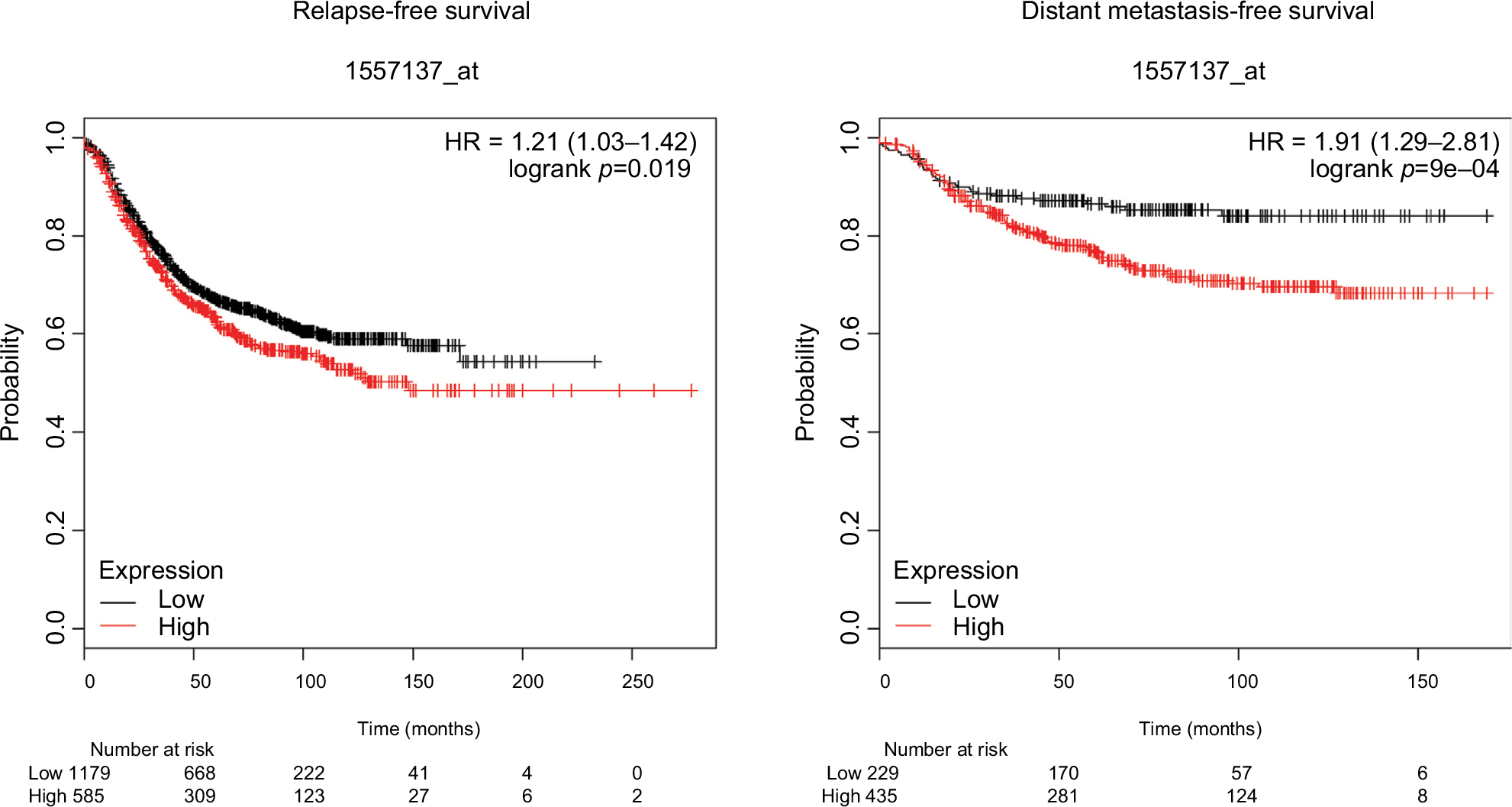 | Figure S1 TMEM17 expression is significantly correlated with the relapse-free survival and distant metastasis-free survival of breast cancer patients. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.