")
Back to Journals » OncoTargets and Therapy » Volume 13
TMED3 Promotes Proliferation and Migration in Breast Cancer Cells by Activating Wnt/β-Catenin Signaling
Received 21 February 2020
Accepted for publication 31 May 2020
Published 19 June 2020 Volume 2020:13 Pages 5819—5830
DOI https://doi.org/10.2147/OTT.S250766
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Xiumei Zhang,1,2 Yalan Luo,3 Qingchang Li1
1College of Basic Medical Sciences, China Medical University and Department of Pathology, The First Affiliated Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China; 2Department of Pathology, Dalian Municipal Central Hospital Affiliated of Dalian Medical University, Dalian, Liaoning, People’s Republic of China; 3Department of Abdominal Emergency Surgery, The First Affiliated Hospital of Dalian Medical University, Dalian, Liaoning, People’s Republic of China
Correspondence: Qingchang Li
College of Basic Medical Sciences, China Medical University and Department of Pathology, The First Affiliated Hospital of China Medical University, Shenyang, Liaoning, People’s Republic of China
Tel +862423261638
Email [email protected]
Purpose: Evidence describing TMED3 in the context of breast cancer is scarce, and the effect of TMED3 on Wnt/β-catenin signaling in breast cancer has not been reported. The objective of this study was to determine the potential physiological functions and molecular mechanisms of TMED3 in breast cancer.
Materials and Methods: Quantitative real-time PCR and Western blot analysis were used to analyze the expression of TMED3 mRNA and protein in 182 paraffin-embedded primary breast cancer tissues and 60 paired noncancerous tissues and 25 fresh primary breast cancer tissues and surrounding adjacent noncancerous tissues. Associations between TMED3 expression and clinicopathologic factors or overall survival were determined. The effects of overexpression or knockdown of TMED3 on proliferation, migration, invasion, and cell cycle progression in breast cancer cell lines were investigated with the Cell Counting Kit-8, clone formation assay, transwell assay, wound healing assay, and flow cytometry, respectively. Immunofluorescence and Western blot analysis were used to detect the expression of cell cycle, migration-related, and Wnt/β-catenin signaling proteins.
Results: The expression of TMED3 mRNA and protein were significantly increased in breast cancer tissues and cell lines compared to normal controls. TMED3 upregulation was significantly correlated with clinicopathologic characteristics and predicted poor prognosis in patients with breast cancer. TMED3 overexpression promoted proliferation, migration, invasion, and cell cycle progression compared to controls in breast cancer cell lines. TMED3 knockdown suppressed proliferation, migration, invasion, and cell cycle progression compared to controls in breast cancer cell lines. TMED3 promoted proliferation and migration of breast cancer cells by a mechanism that involved Wnt/β-catenin signaling.
Conclusion: TMED3 behaves as an oncogene that promotes the proliferation and migration of breast cancer cells by a mechanism that involved Wnt/β-catenin signaling. Strategies targeting TMED3 have potential therapeutic implications for patients with breast cancer.
Keywords: TMED3, breast cancer, proliferation, invasion, Wnt/β-catenin signaling
Introduction
Globally, breast cancer is the most frequently occurring cancer in women and is a major public health concern.1 Traditional approaches to treatment, such as surgery, radiotherapy and chemotherapy, have improved outcomes of patients with breast cancer; however, breast cancer progression and metastasis represent a huge clinical problem.2 Elucidating the molecular mechanisms of carcinogenesis in the breast is vital for the discovery of novel therapeutic targets.
Transmembrane emp24 domain containing (TMED) proteins constitute a highly conserved family of proteins that exist as monomers or dimers of various compositions.3,4 TMED proteins share similar domain architectures, including a GOLD (Golgi dynamics) domain, a coiled-coil domain, a membrane-spanning domain, and a short cytoplasmic tail with several highly conserved motifs.5 TMED proteins dimerize and associate with coatomer protein complexes to regulate transport of various cargo proteins, including glycosylphosphatidylinositol-anchored proteins, G-protein-coupled receptors, and Wnt ligands.6–9
TMED proteins are essential for normal development, but have been implicated in pancreatic disease, immune system disorders, and cancer.5,6 Specifically, TMED2 promotes proliferation, migration, and invasion of ovarian cancer cells by activating the IGF2/IGF1R/PI3K/Akt pathway,10 TMED9 is upregulated in breast cancer, colon cancer, ovarian cancer, gastrointestinal cancer, lung cancer and hepatocellular cancer stem cells,11 and TMED10 induces autophagy in papillary thyroid cancer cells by activating the AMPK/mTOR pathway.12
The role of TMED3 in cancer is controversial. TMED3 suppresses distant colon cancer metastases,7 but promotes tumor progression in liver cancer and breast cancer.13,14 The Wnt/β-catenin pathway is involved in breast cancer tumorigenesis, progression, proliferation, invasion, and metastasis.15–18 Evidence describing TMED3 in the context of breast cancer and the effect of TMED3 on Wnt/β-catenin signaling in breast cancer is scarce.
The objective of this study was to determine the potential physiological functions and molecular mechanisms of TMED3 in breast cancer. Findings showed that the expression of TMED3 was increased in breast cancer tissues and cell lines, which was associated with an unfavorable prognosis. TMED3 promoted the proliferation, migration, and invasion of breast cancer cells by a mechanism that involved Wnt/β-catenin signaling.
Materials and Methods
Tissue Specimens
This study included 182 paraffin-embedded primary breast cancer tissues and 60 paired noncancerous tissues that had been collected between January 2010 and October 2018. Tissues were randomly selected from the archives of the Institute of Pathology, the Dalian Municipal Central Hospital Affiliated of Dalian Medical University. In addition, fresh primary breast cancer tissues and surrounding adjacent noncancerous tissues were obtained from 25 patients with newly diagnosed breast cancer who underwent surgical resection. Fresh samples were snap-frozen and stored at −80°C until analysis. The use of patient material was approved by the Ethics Committee of the Dalian Municipal Central Hospital Affiliated of Dalian Medical University (YN2019-054-01). This study was conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent.
Tissue Microarray (TMA)
For TMA, histological sections were evaluated, and representative areas of tumor tissue that were free from necrosis or hemorrhage were pre-marked on paraffin-embedded donor blocks. Two 2.0mm cores were punched out from each tumor sample, and all cores were embedded in a separate paraffin block. The TMA blocks were sectioned at a thickness of 4μm.
Immunohistochemistry Analysis
Immunostaining was performed using the EnVision kit (Maixin, Fuzhou, China), according to the manufacturer’s instructions. Sections were incubated with TMED3 primary antibody (1:50; Abcam, Cambridge, UK) at 4°C overnight, and subsequently with horseradish peroxidase (HRP) conjugated polymers for 30 minutes. Reactions were visualized with 3,3-diaminobenzidine tetrahydrochloride (DAB), and sections were counterstained with hematoxylin. Two investigators independently scored the immunostaining using 5 fields per slide, which were randomly selected and examined at 400 × magnification. Staining in the nucleus, cytoplasm, and membranes was considered positive. Immunostaining intensity was graded as: 0 (none), 1 (weak), 2 (moderate/strong). The percentage of positive tumor cells (intensity level =1 or 2) was classified as: 1 (1–25%), 2 (26– 50%), 3 (51–75%), 4 (76–100%). Results were scored by multiplying the intensity by the percentage of positive cells. TMED3 expression was categorized as negative/low expression (score <4) or overexpression (score ≥4).
Cell Culture and Transfection Procedure
Breast cancer cell lines MCF-7, T47D, MDA-MB-231 and human normal breast epithelial cells MCF-10A (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Hyclone, USA) with 10% fetal bovine serum (FBS) (Hyclone, USA), 100IU/mL penicillin, and 100ug/mL streptomycin in a humidified cell incubator at 37°C and 5% CO2.
TMED3 plasmid and siRNA sequences were provided by Genepharma Co., Ltd. (Shanghai, China). Three selected siRNA targeting TMED3 were used in this study, including siRNA duplex 1 (forward 5ʹ-CCCUGGAUUACCAGGUCAUTT-3ʹ, reverse 5ʹ-AUGACCUGGUAAUCCAGGGTT-3ʹ); siRNA duplex 2 (forward 5ʹ-GCUGAAGUCAAGGGCGUUUTT-3ʹ, reverse 5ʹ-AAACGCCCUUGACUUCAGCTT-3ʹ); and siRNA duplex 3 (forward 5ʹ-CCGAGUCUCUUACUGGUCUTT-3ʹ, reverse 5ʹ-AGACCAGUAAGAGACUCGGTT-3ʹ). siRNA duplex 2 produced a stronger knockdown compared to siRNA duplex1 or duplex3; therefore, duplex 2 was used in subsequent experiments. siRNA-β-catenin (Santa Cruz, CA, USA) and siRNA-Axin (Santa Cruz, CA, USA) were used to interfere with Wnt/β-catenin signaling. Cells were transfected using the Lipofectamine 3000 reagent (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions. Empty vectors served as the negative control. Cells were incubated in transfection media for 4 h, which was then replaced with standard media.
RNA Extraction and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted from frozen tissues or cell lines using the TRIzol reagent (Takara, Dalian, China), according to the manufacturer’s instructions. The primers used for amplifying TMED3 and the endogenous control β-actin were synthesized by Sangon Biotech (Shanghai, China). qRT-PCR was performed using the SYBR Green master mix kit (TianGen Biotech, Beijing, China), according to the manufacturer’s instructions. The fold change in the expression of target genes was calculated by the 2−ΔΔCT method.
Western Blot Analysis
Proteins were separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA), and incubated at 4°C overnight with antibodies against TMED3, Axin2, β-catenin, cyclin D1, cyclin E, CDK2, CDK4, CDK6, c-myc, Rock1, RhoA, RhoB, RhoC, MMP2, MMP7, MMP9 (1:1000; Proteintech, USA), or GAPDH (1:2000; Proteintech, USA). Membranes were incubated with HRP-conjugated anti-mouse or rabbit IgG antibody (1:1000, Affinity) at 37°C for 2h. Target protein bands were detected with enhanced chemiluminescence reagent (Thermo Fisher Scientific). A gel imaging system was used to analyze band density, which were compared against an internal control.
Immunofluorescence Analysis
Cells grown on coverslips were fixed with 4% paraformaldehyde in PBS on ice for 15 min. Coverslips were washed twice with PBS, permeabilized by incubation with 0.2% Triton X-100 (Sigma) for 10 min, washed twice with PBS, and incubated in 5% horse serum for 30 min. Coverslips were placed in a humidified chamber and incubated with Axin2 (1:200, Bioss Antibodies, China) and β-catenin (1:200, Proteintech, USA) antibodies at 4°C overnight. Subsequently, coverslips were washed twice with PBS, incubated with TRITC-conjugated anti-rabbit secondary antibody (1:100, Affinity) at 37°C for 30 min, washed with PBS, and incubated with 0.1% DAPI at 37°C for 30 min. Coverslips were mounted on a glass slide with 25 μL Slow Fade (Invitrogen). Slides were analyzed with an Olympus fluorescence microscope and images were evaluated using Axiovision software v4.6 (Carl Zeiss).
Cell Proliferation Assay
Cell proliferation was assessed using the Cell Counting Kit 8 (CCK-8; Beyotime), according to the manufacturer’s instructions. Cells were seeded into the wells of a microtitre plate. Each well contained 2000 cells, 100μL DMEM with 10% FBS, and 10μL CCK-8. The plate was cultured at 37°C for 2 hours. Absorbance of each well was measured at 450 nm, daily, for 5 days. The experiment was repeated 3 times.
Cell proliferation was further investigated using the colony formation assay. Transfected cells were seeded in 6-well plates (approx. 500 cells), incubated for 3 weeks, fixed with 4% paraformaldehyde for 15 min, washed with PBS, and stained with 0.2% crystal violet (Beyotime) for 10min. The number of colonies (>50 cells) was counted under a microscope. The experiment was performed in duplicate and repeated at least twice.
Flow Cytometry Assay
Cell cycles were analyzed by flow cytometry. Cells were harvested at 48h post- transfection by trypsinization, fixed in 500μL of 70% cold ethanol at 4°C overnight, stained with 100μL RNase A and 400μL propidium iodide (Beyotime) at 37°C for 30 min. 10,000 cells per sample were analyzed on a EPICS XL flow cytometer (Beckman Coulter, USA) with Modfit software.
Wound Healing Assay and Transwell Assay
For the wound healing assay, 5×105 cells were seeded in 6-well plates and incubated until they reached 100% confluence. The confluence plates were scratched using a sterile pipette tip, washed twice with PBS to remove the detached cells, and photographed under a microscope at 0 and 24h. Cell migration was measured by monitoring the width of the scratch over time.
The cell invasion assay was performed using a 24-well Transwell chamber (Corning, New York, USA) coated with Matrigel basement membrane matrix (BD Biosciences, Bedford, USA). Cells suspended in serum-free media were placed in the upper Matrigel chamber. Medium supplemented with 10% FBS was placed in the lower chamber. Cells that passed through the membrane after 24 hours were visualized with crystal violet staining.
Statistical Analysis
Statistical analysis was performed using SPSS version 17.0 for Windows. Data obtained from at least three independent experiments were used in all analyses. Correlations between TMED3 expression and clinicopathologic factors were analyzed with the χ2 test. Between group differences were compared with Student’s t-test. Overall survival was investigated using Kaplan–Meier survival analysis and the log‐rank test. P<0.05 was considered statistically significant.
Results
Expression of TMED3 in Breast Cancer Is Associated with Clinicopathologic Factors
Analyses using the Oncomine database demonstrated that the expression of TMED3 mRNA was significantly increased in breast cancer tissues compared to normal breast tissues (Figure 1) (www.oncomine.org) (P<0.001). Consistent with these findings, expression of TMED3 protein and mRNA was significantly increased in fresh frozen breast cancer tissue samples compared to adjacent non-cancerous tissues (P<0.05) (Figure 2A and B).
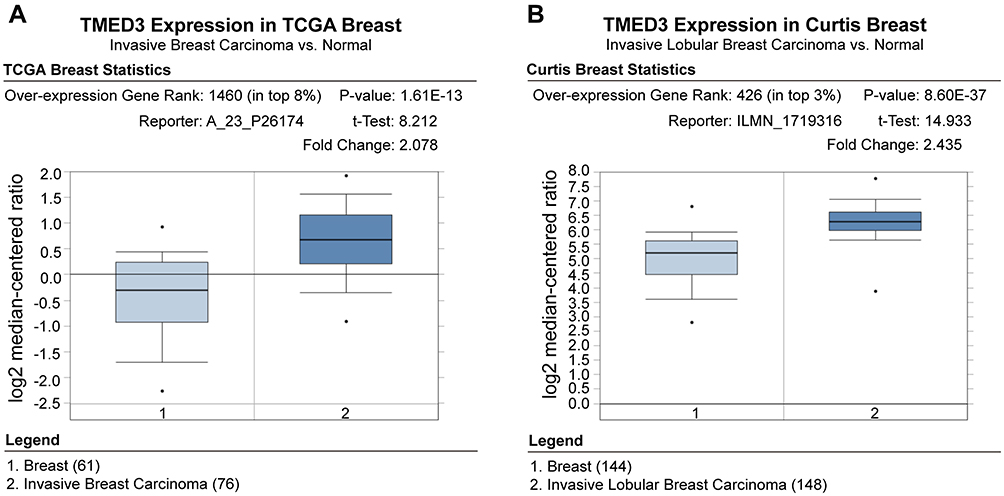 |
Figure 1 Box plots showing upregulation of TMED3 mRNA expression in breast cancer tissues relative to normal breast tissues; source: www.oncomine.org. Notes: (A) The Cancer Genome Atlas (TCGA) dataset: 76 breast cancer tissues and 61 normal breast tissues. (B) Curtis et al: 148 breast cancer tissues and 144 normal breast tissues. |
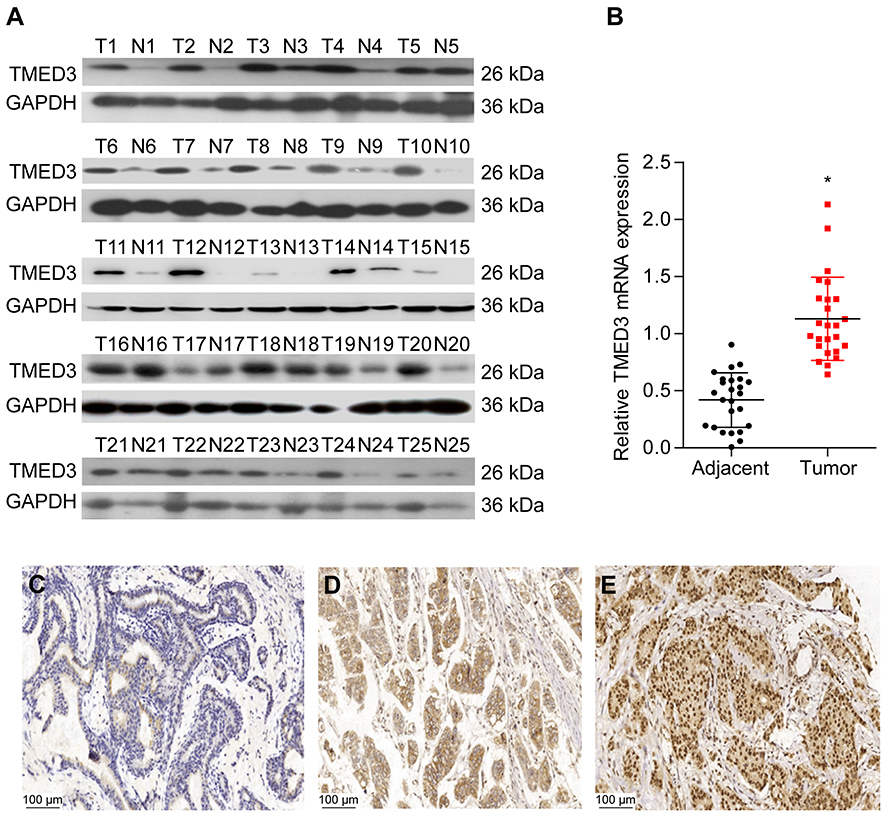 |
Figure 2 Expression of TMED3 protein and mRNA in breast cancer tissues. Notes: (A and B) Western blot and qPCR analysis showing the expression of TMED3 protein and mRNA in fresh breast cancer tissues and adjacent noncancerous tissues. (C) Immunohistochemistry showing negative or weak staining for TMED3 protein in noncancerous breast tissues (200×). (D) Immunohistochemistry showing moderate cytoplasmic and membranous staining for TMED3 protein in breast cancer tissues (200×). (E) Immunohistochemistry showing strong cytoplasmic and nuclear staining forTMED3 protein in breast cancer tissues (200×). *P<0.05. |
The expression and location of TMED3 protein in paraffin-embedded breast cancer and paired adjacent noncancerous tissues was investigated with immunohistochemistry. Findings showed negative/weak staining for TMED3 in normal breast tissues (Figure 2C) and moderate to strong cytoplasmic, nuclear and membranous staining for TMED3 in 113 of 182 breast cancer tissues (Figure 2D and E).
Overexpression of TMED3 protein was significantly associated with negative staining for estrogen receptor (ER, P=0.028), progesterone receptor (PR, P=0.032), and human epidermal growth factor receptor-2 (Her-2, P=0.040), as well as the presence of nodal metastases (P=0.002). Expression of TMED3 was not influenced by patient age, tumor size, grade, TNM stage, triple-negative status, or Ki67 index (Table 1). Kaplan–Meier survival analysis revealed that overexpression of TMED3 protein was associated with shorter overall survival compared to negative/low TMED3 expression (Log rank test, P=0.012; Figure 3).
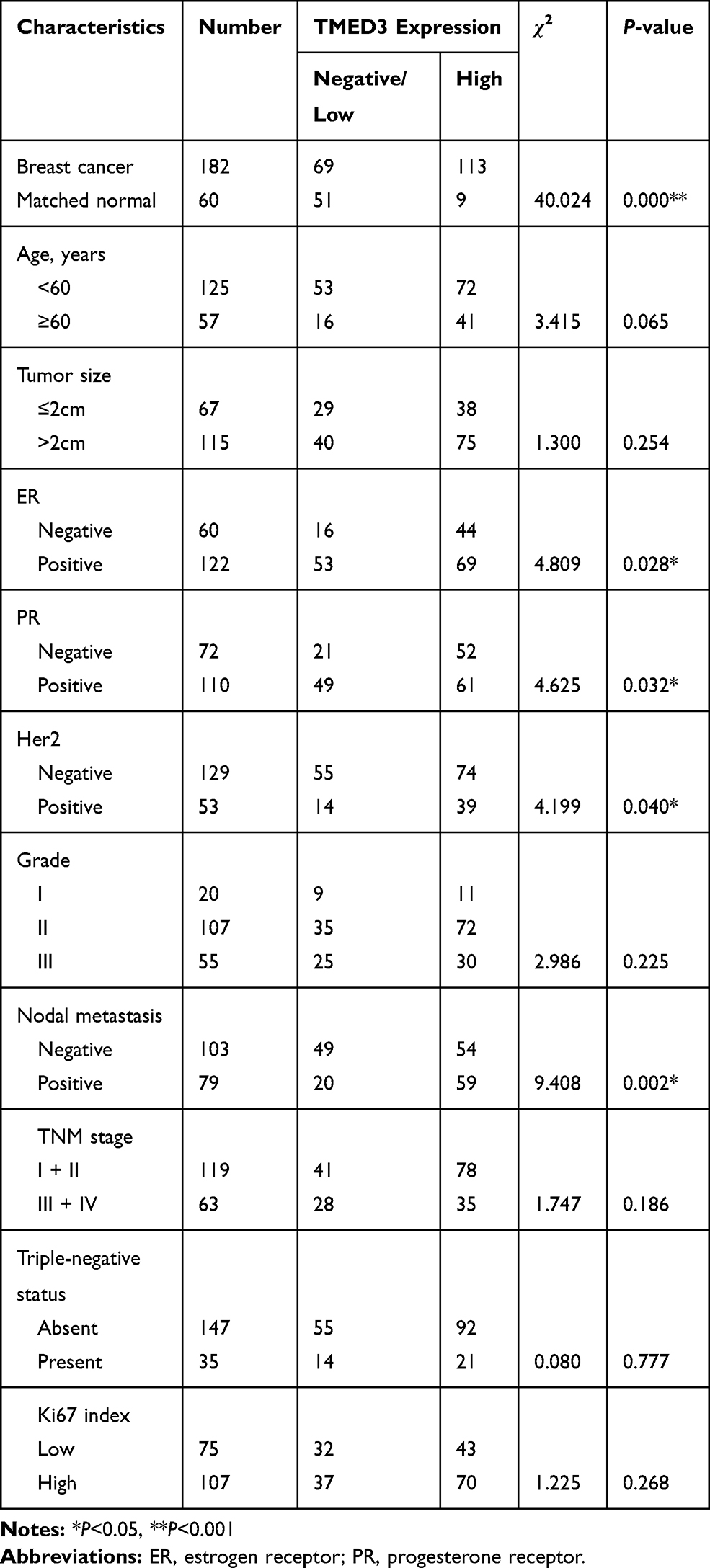 |
Table 1 Distribution of TMED3 Status in Breast Cancer According to Clinicopathologic Characteristics |
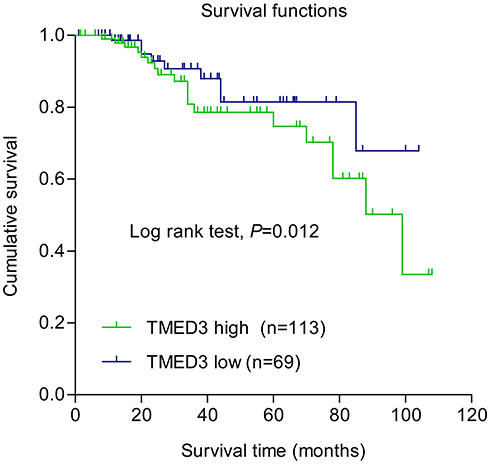 |
Figure 3 Kaplan–Meier survival analysis according to expression of TMED3 in patients with breast cancer (N=182). Note: Blue and green curves represent low and high TMED3 expression, respectively. |
TMED3 Was Upregulated in Breast Cancer Cells Lines
The expression of TMED3 mRNA and protein in a normal breast epithelial cell line (MCF-10A) and several breast cancer cell lines, including MCF-7, T47D and MDA-MB-231, were investigated with qPCR and Western blot analysis. Findings showed that TMED3 expression was higher in breast cancer cell lines (especially T47D and MDA-MB-231) compared to MCF-10A cells (Figure 4A) (P<0.05). The role of TMED3 in breast cancer cells was investigated in MCF-7 and T47D cells using gain-of-function and loss-of-function experiments. Transfection with TMED3 plasmid significantly increased the expression of TMED3 mRNA and protein compared to transfection with empty vector in MCF-7 cells (Figure 4B), and transfection with TMED3 siRNA significantly decreased the expression of TMED3 mRNA and protein compared to transfection with negative siRNA in T47D cells (P<0.05) (Figure 4C).
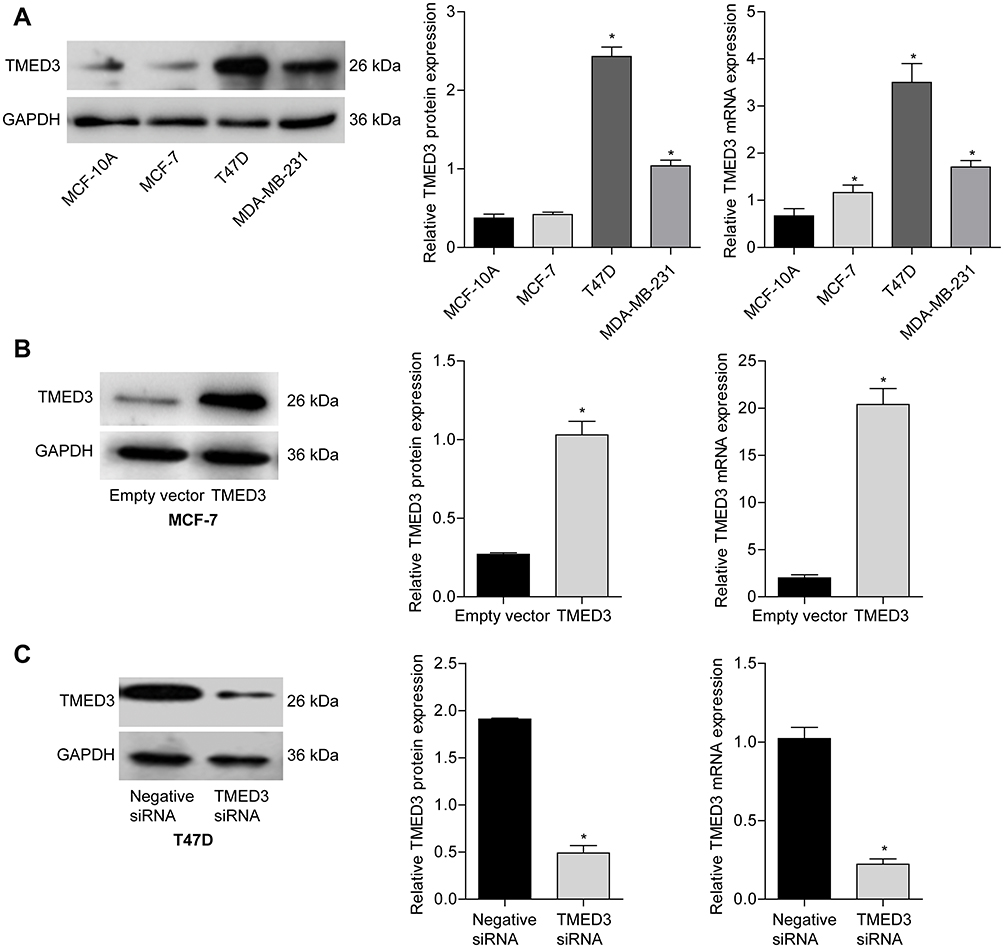 |
Figure 4 Expression of TMED3 mRNA and protein in a normal breast epithelial cell lines and several breast cancer cell lines. Notes: (A) qPCR and Western blot analysis showing the expression of TMED3 protein and mRNA in normal breast epithelial cells (MCF-10A) and three breast cancer cell lines (MCF-7, T47D, MDA-MB-231). (B) Transfection with TMED3 plasmid increased the expression of TMED3 mRNA and protein compared to empty vector in MCF-7 cells. (C) Transfection with TMED3 siRNA decreased the expression of TMED3 mRNA and protein compared to negative siRNA in T47D cells. *P<0.05. |
TMED3 Promoted Proliferation, Colony Formation and Cell Cycle Progression in Breast Cancer Cells
The role of TMED3 in breast cancer cell proliferation was investigated using the CCK-8 and colony formation assays. Findings showed that TMED3 overexpression significantly increased the proliferation of MCF-7 cells compared to control (P<0.05), while TMED3 knockdown significantly decreased the proliferation of T47D cells compared to control (P<0.05) (Figure 5A). TMED3 overexpression significantly enhanced colony formation in MCF-7 cells compared to control (P<0.05), while TMED3 knockdown significantly decreased colony formation in T47D cells compared to control (P<0.05) (Figure 5B). These data suggest that TMED3 promoted proliferation in breast cancer cell lines; therefore, the effect of TMED3 on cell cycle progression was explored. Findings showed that TMED3 overexpression increased the percentage of MCF-7 cells in S phase compared to control, while TMED3 knockdown significantly decreased the percentage of T47D cells in S phase compared to control (P<0.05) (Figure 5C). The effect of TMED3 expression on relevant cell cycle proteins was assessed using Western blot analysis. TMED3 overexpression significantly upregulated the expression of CDK2, CDK4, CDK6, cylinD1 and cyclin E proteins in MCF-7 cells compared to control (P<0.05), while knockdown of TMED3 significantly downregulated the expression of CDK2, CDK4, CDK6, cylinD1 and cyclin E proteins in T47D cells compared to control (P<0.05) (Figure 5D). These results indicate that TMED3 promoted cell proliferation and cell cycle progression in breast cancer cells by modulating the expression of relevant cell cycle proteins.
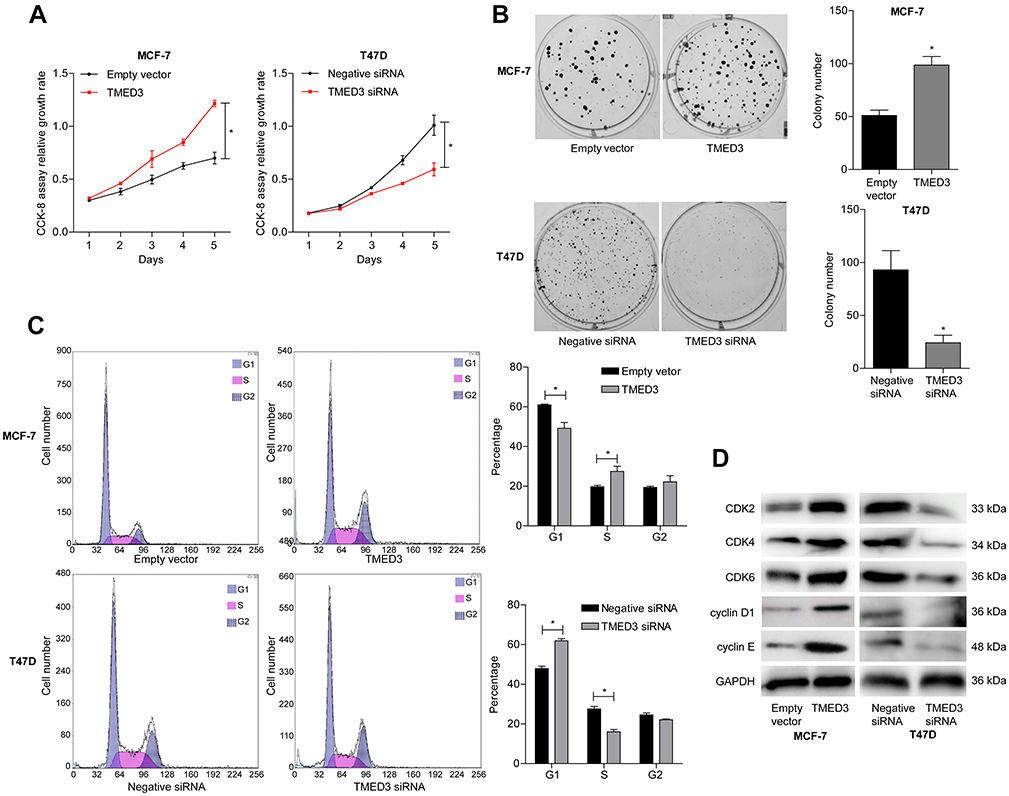 |
Figure 5 Effects of overexpression and knockdown of TMED3 on cell proliferation and cell cycle progression in breast cancer cells. Notes: (A) Proliferation in MCF-7 and T47D cells was detected using the CCK-8 assay. (B) The effect of TMED3 on MCF-7 and T47D cells proliferation was measured using the colony formation assay. (C) Flow cytometry showing overexpression of TMED3 induced cell cycle arrest in MCF-7 and T47D cells. (D) Western blot showing the levels of relevant cell cycle proteins in MCF-7 and T47D cells. *P<0.05. |
TMED3 Promoted Invasion and Migration in Breast Cancer Cells
The role of TMED3 in cell migration and invasion was investigated using the wound healing and transwell cell invasion assays. Findings showed TMED3 overexpression significantly increased migration and invasion of MCF-7 cells compared to control, while knockdown of TMED3 significantly decreased migration and invasion of T47D cells compared to control (both P<0.05) (Figure 6A and B). The effect of TMED3 expression on migration-related proteins was assessed using Western blot analysis. TMED3 overexpression significantly upregulated the expression of RhoA, RhoB, RHOC, ROCK1, MMP2, MMP7 and MMP9 proteins in MCF-7 cells compared to control (P<0.05), while knockdown of TMED3 significantly downregulated the expression of RHOA, RHOB, RHOC, ROCK1, MMP2, MMP7 and MMP9 proteins in T47D cells compared to control (P<0.05) (Figure 6C). These results indicate that TMED3 promoted invasion and migration in breast cancer cells by regulating the expression of migration-related proteins.
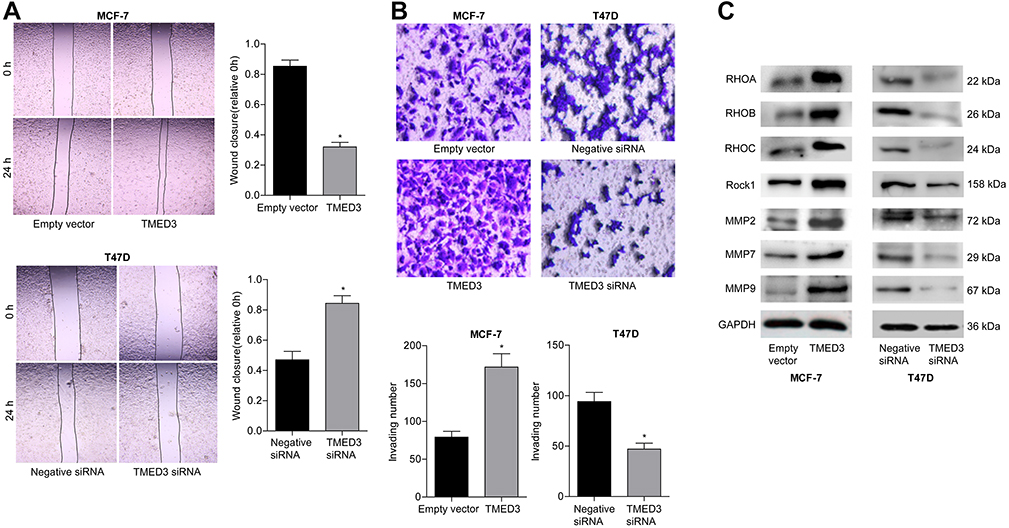 |
Figure 6 Effects of overexpression and knockdown of TMED3 on the migration and invasion of breast cancer cells. Notes: (A) Wound healing assay showing migration of MCF-7 and T47D cells (400×). (B) Transwell invasion assay showing the invasion of MCF-7 and T47D cells (400×). (C) Western blot analysis showing the expression of relevant migration-related proteins in MCF-7 and T47D cells. *P<0.05. |
TMED3 Activated Wnt/β-Catenin Signaling in Breast Cancer Cells
To explore the underlying molecular mechanism involved in TMED3-mediated promotion of proliferation, migration and invasion in breast cancer cells, immunofluorescence was used to investigate the expression of Axin2 and β-catenin proteins in MCF-7 cells. Axin2 and β-catenin are two key molecules involved in Wnt/β-catenin signaling. Findings showed that TMED3 overexpression resulted in substantial accumulation of β-catenin in the cytoplasm and nucleus and Axin2 in the cytoplasm of MCF-7 cells compared to control (P<0.05) (Figure 7A and B). Western blot analysis showed TMED3 overexpression significantly increased the expression of β-catenin, Axin2 cyclin D1, c-myc, MMP7 and TCF4 proteins in MCF-7 cells compared to control (P<0.05). Silencing β-catenin or Axin2 significantly reversed the effects of TMED3 on the expression of cyclin D1, c-myc, MMP7 and TCF4 proteins (both P<0.05) (Figure 7C and D). These results indicate that TMED3 activated Wnt/β-catenin signaling in breast cancer cells by regulating the expression of downstream target genes.
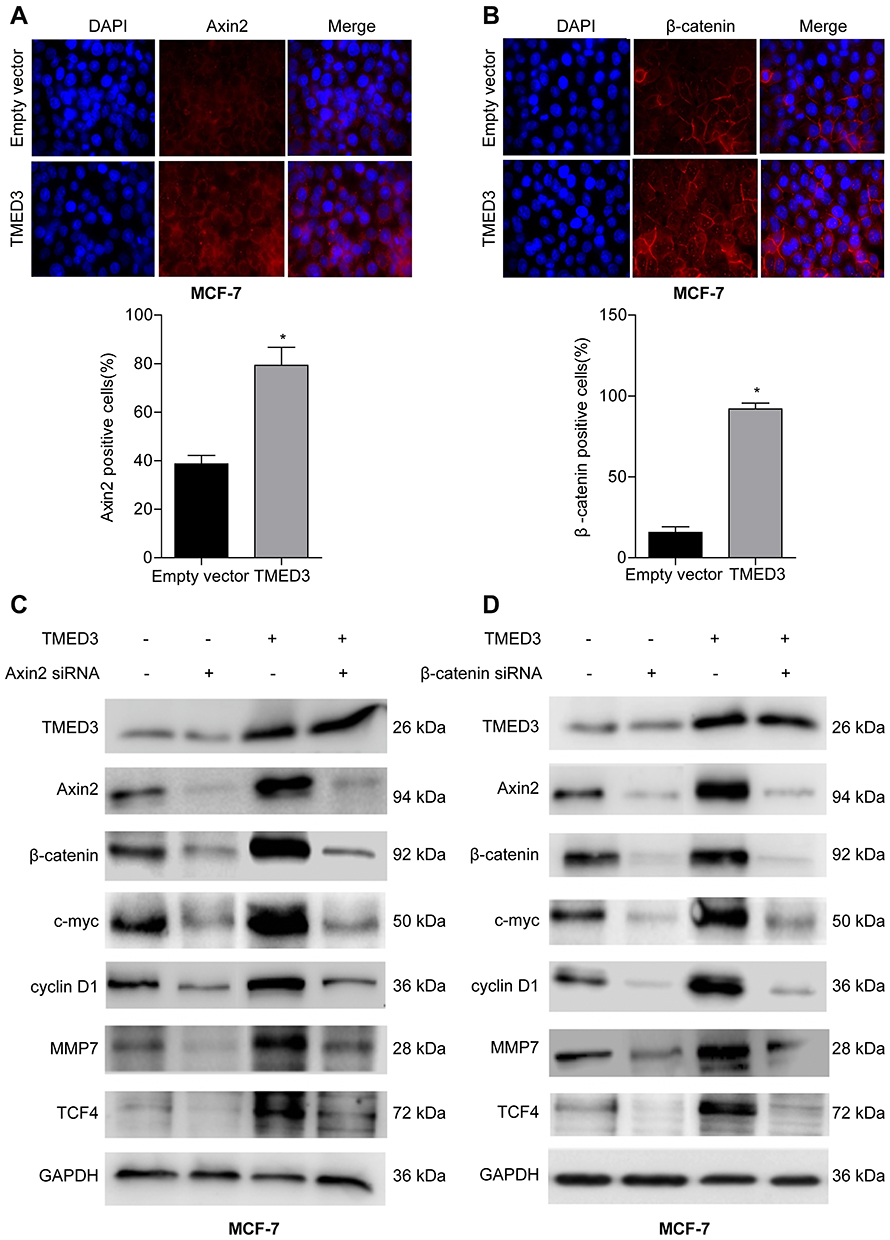 |
Figure 7 Effect of TMED3 on Wnt/β-catenin signaling in breast cancer cells. Notes: (A) Immunofluorescence showing overexpression of TMED3 increased expression of Axin2 protein in the cytoplasm of MCF-7 cells compared to control. (B) Immunofluorescence showing overexpression of TMED3 increased expression of β-catenin protein in the cytoplasm and nucleus of MCF-7 cells compared to control. (C and D) Western blot analysis showing the expression levels of Wnt/β-catenin signaling-related proteins after silencing Axin2 or β-catenin in MCF-7 cells ectopically overexpressing TMED3. *P<0.05. |
Discussion
The exact role of TMED3 in breast cancer remains to be elucidated. The present study explored the potential physiological functions and molecular mechanisms of TMED3 in breast cancer. Findings showed that TMED3 was overexpressed in breast cancer tissues and cell lines. TMED3 overexpression was significantly associated with clinicopathologic factors, including negative staining for ER, PR, and Her-2, as well as presence of nodal metastases. TMED3 overexpression was associated with poor prognosis in the population of patients with breast cancer included in this study. These data identify a prognostic role for TMED3 in breast cancer. The clinical significance of TMED3 has been investigated in other cancers. In accordance with our results, TMED3 overexpression was significantly correlated with poor prognosis in patients with hepatocellular carcinoma and clear cell renal cell carcinoma.14,19 Further, TMED3 was highly expressed in prostate cancer and metastatic prostate cancer, and high expression of TMED3 was associated with the expression of the androgen receptor and ERG oncogene.20
The present study revealed that TMED3 overexpression increased cell proliferation and cell cycle progression in breast cancer cell lines. Underlying mechanisms involved effects on cell cycle related proteins. TMED3 overexpression increased the percentage of breast cancer cells in S phase and the expression of cyclin D1, cyclin E, CDK2, CDK4 and CDK6 proteins. Cyclin D1 regulates the G1/S cell cycle checkpoint by phosphorylating retinoblastoma protein (pRB) complex, increasing the expression of cyclin E, and triggering uncontrolled cell growth and proliferation.21,22 Cyclin D1-CDK4/6 complexes and cyclin E-CDK2 complexes also control G1-S phase transition.23,24 To the authors’ knowledge, this is the first study to show that TMED3 induces G1/S phase arrest to promote breast cancer cell growth, playing a key role in the molecular mechanisms of carcinogenesis in the breast.
Increased migration and adhesion are important characteristics of cancer cells that are associated with metastasis and poor prognosis. The present study showed that silencing TMED3 significantly decreased the migration and invasion of breast cancer cells. Conversely, TMED3 overexpression significantly increased the migration and invasion of breast cancer cells, potentially explaining the association between TMED3 overexpression and worse overall survival in the patients with breast cancer in this study. Further, silencing TMED3 decreased the expression of migration-related proteins, while TMED3 overexpression significantly increased the expression of migration-related proteins. These results imply that TMED3 has migration- and invasion-promoting effects in breast cancer cells.
Evidence suggests that overactivation of Wnt/β-catenin signaling plays a key role in the development of breast cancer.25 Increased levels of β-catenin have been associated with metastasis and poor prognosis in patients with breast cancer.18,26 During Wnt/β-catenin signaling, β-catenin translocates from the cytoplasm into the nucleus where it associates with T-cell factor/lymphoid enhancer-binding factor (TCF/LEF) transcription factors and regulates downstream target genes, including those that affect cell proliferation, invasion and tumor progression, such as cyclin D1, c-myc and MMP7.27–31 Canonical Wnt signaling is mediated by β-catenin, whose degradation is controlled by a complex containing Axin2.32 In the present study, immunofluorescence staining revealed that TMED3 overexpression resulted in substantial accumulation of β-catenin in the cytoplasm and nucleus and Axin2 in the cytoplasm of MCF-7 cells. We speculate that the tumor-promoting effects of TMED3 are associated with activation of Wnt/β-catenin signaling. Accordingly, TMED3 overexpression increased expression of β-catenin and Axin2 and a complex series of downstream target genes of Wnt/β-catenin signaling, including cyclin D1, c-myc, MMP7 and TCF4. These effects were reversed by silencing β-catenin or Axin2. These results demonstrate that TMED3 promoted the proliferation and migration of breast cancer cells by activating Wnt/β-catenin signaling. Mechanistically, our results differ from previous reports that suggest TMED3 may be negatively modulated by miR-188-2p.13
Conclusions
In conclusion, the present study demonstrates that TMED3 is overexpressed in human breast cancer and serves as a predictor for poor prognosis. TMED3 behaves as an oncogene that promotes the proliferation and migration of breast cancer cells by activating Wnt/β-catenin signaling. Strategies targeting TMED3 have potential therapeutic implications for patients with breast cancer.
Abbreviation
TMED, transmembrane emp24 domain containing; TMA, tissue microarray; CCK-8, Cell Counting Kit 8; CDK, cyclin-dependent kinase; DMEM, Dulbecco’s Modified Eagle Medium; DAB, 3,3-diaminobenzidine tetrahydrochloride; mRNA, messenger ribonucleic acid; siRNA, small interference RNA; ECL, enhanced chemiluminescence; ER, estrogen receptor; PR, progesterone receptor; HRP, horseradish peroxidase; Her-2, human epidermal growth factor receptor-2; FBS, fetal bovine serum; MMP, matrix metalloproteinases; SPSS, statistical package for social science; SDS, sodium dodecyl sulfate; PAGE, polyacrylamide gel electrophoresis; RT-PCR, reverse transcription-polymerase chain reaction; TRITC, tetraethyl rhodamine isothiocyanate.
Data Sharing Statement
The datasets generated and analyzed during the present study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study was conducted after obtaining approval from the Ethics Committee of the Dalian Municipal Central Hospital Affiliated of Dalian Medical University and written informed consent from the patients (YN2019-054-01).
Author Contributions
All authors made substantial contributions to study conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tao Z, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast cancer: epidemiology and etiology. Cell Biochem Biophys. 2015;72(2):333–338. doi:10.1007/s12013-014-0459-6
2. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–289. doi:10.3322/caac.21349
3. Jenne N, Frey K, Brugger B, Wieland FT. Oligomeric state and stoichiometry of p24 proteins in the early secretory pathway. J Biol Chem. 2002;277(48):46504–46511. doi:10.1074/jbc.M206989200
4. Strating JR, Hafmans TG, Martens GJ. Functional diversity among p24 subfamily members. Biol Cell. 2009;101(4):207–219. doi:10.1042/BC20080075
5. Strating JR, Martens GJ. The p24 family and selective transport processes at the ER-Golgi interface. Biol Cell. 2009;101(9):495–509. doi:10.1042/BC20080233
6. Port F, Hausmann G, Basler K. A genome-wide RNA interference screen uncovers two p24 proteins as regulators of wingless secretion. EMBO Rep. 2011;12(11):1144–1152. doi:10.1038/embor.2011.165
7. Duquet A, Melotti A, Mishra S, et al. A novel genome-wide in vivo screen for metastatic suppressors in human colon cancer identifies the positive WNT-TCF pathway modulators TMED3 and SOX12. EMBO Mol Med. 2014;6(7):882–901. doi:10.15252/emmm.201303799
8. Buechling T, Chaudhary V, Spirohn K, Weiss M, Boutros M. p24 proteins are required for secretion of Wnt ligands. EMBO Rep. 2011;12(12):1265–1272. doi:10.1038/embor.2011.212
9. Li X, Wu Y, Shen C, Belenkaya TY, Ray L, Lin X. Drosophila p24 and Sec22 regulate wingless trafficking in the early secretory pathway. Biochem Biophys Res Commun. 2015;463(4):483–489. doi:10.1016/j.bbrc.2015.04.151
10. Shi-Peng G, Chun-Lin C, Huan W, et al. TMED2 promotes epithelial ovarian cancer growth. Oncotarget. 2017;8(55):94151–94165. doi:10.18632/oncotarget.21593
11. Seno A, Kasai T, Ikeda M, et al. Characterization of gene expression patterns among artificially developed cancer stem cells using spherical self-organizing map. Cancer Inform. 2016;15:163–178. doi:10.4137/CIN.S39839
12. Xu X, Gao H, Qin J, He L, Liu W. TMP21 modulates cell growth in papillary thyroid cancer cells by inducing autophagy through activation of the AMPK/mTOR pathway. Int J Clin Exp Pathol. 2015;8(9):10824–10831.
13. Pei J, Zhang J, Yang X, et al. TMED3 promotes cell proliferation and motility in breast cancer and is negatively modulated by miR-188-3p. Cancer Cell Int. 2019;19:75. doi:10.1186/s12935-019-0791-4
14. Zheng H, Yang Y, Han J, et al. TMED3 promotes hepatocellular carcinoma progression via IL-11/STAT3 signaling. Sci Rep. 2016;6:37070. doi:10.1038/srep37070
15. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi:10.1016/j.cell.2012.05.012
16. Nusse R, Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–999. doi:10.1016/j.cell.2017.05.016
17. King TD, Suto MJ, Li Y. The Wnt/beta-catenin signaling pathway: a potential therapeutic target in the treatment of triple negative breast cancer. J Cell Biochem. 2012;113(1):13–18. doi:10.1002/jcb.23350
18. Dey N, Barwick BG, Moreno CS, et al. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer. 2013;13:537. doi:10.1186/1471-2407-13-537
19. Ha M, Moon H, Choi D, et al. Prognostic role of TMED3 in clear cell renal cell carcinoma: a retrospective multi-cohort analysis. Front Genet. 2019;10:355. doi:10.3389/fgene.2019.00355
20. Vainio P, Mpindi JP, Kohonen P, et al. High-throughput transcriptomic and RNAi analysis identifies AIM1, ERGIC1, TMED3 and TPX2 as potential drug targets in prostate cancer. PLoS One. 2012;7(6):e39801. doi:10.1371/journal.pone.0039801
21. Roy PG, Thompson AM. Cyclin D1 and breast cancer. Breast. 2006;15(6):718–727. doi:10.1016/j.breast.2006.02.005
22. Loden M, Nielsen NH, Roos G, Emdin SO, Landberg G. Cyclin E dependent kinase activity in human breast cancer in relation to cyclin E, p27 and p21 expression and retinoblastoma protein phosphorylation. Oncogene. 1999;18(16):2557–2566. doi:10.1038/sj.onc.1202488
23. Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88(3):405–415. doi:10.1016/S0092-8674(00)81879-6
24. Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6(4):353–367. doi:10.1158/2159-8290.CD-15-0894
25. Wang Z, Li B, Zhou L, et al. Prodigiosin inhibits Wnt/beta-catenin signaling and exerts anticancer activity in breast cancer cells. Proc Natl Acad Sci U S A. 2016;113(46):13150–13155. doi:10.1073/pnas.1616336113
26. Wang J, Li M, Chen D, et al. Expression of C-myc and beta-catenin and their correlation in triple negative breast cancer. Minerva Med. 2017;108(6):513–517. doi:10.23736/S0026-4806.17.05213-2
27. Chen Y, Li Y, Xue J, et al. Wnt-induced deubiquitination FoxM1 ensures nucleus beta-catenin transactivation. EMBO J. 2016;35(6):668–684. doi:10.15252/embj.201592810
28. Shtutman M, Zhurinsky J, Simcha I, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96(10):5522–5527. doi:10.1073/pnas.96.10.5522
29. Yang X, Du X, Sun L, et al. SULT2B1b promotes epithelial-mesenchymal transition through activation of the beta-catenin/MMP7 pathway in hepatocytes. Biochem Biophys Res Commun. 2019;510(4):495–500. doi:10.1016/j.bbrc.2019.01.034
30. Xu J, Chen Y, Huo D, et al. Beta-catenin regulates c-Myc and CDKN1A expression in breast cancer cells. Mol Carcinog. 2016;55(5):431–439. doi:10.1002/mc.22292
31. Chen HJ, Hsu LS, Shia YT, Lin MW, Lin CM. The beta-catenin/TCF complex as a novel target of resveratrol in the Wnt/beta-catenin signaling pathway. Biochem Pharmacol. 2012;84(9):1143–1153. doi:10.1016/j.bcp.2012.08.011
32. Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22(4):1172–1183. doi:10.1128/MCB.22.4.1172-1183.2002
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.