")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
Tirbanibulin for Actinic Keratosis: Insights into the Mechanism of Action
Authors Schlesinger T , Stockfleth E, Grada A , Berman B
Received 1 August 2022
Accepted for publication 19 October 2022
Published 16 November 2022 Volume 2022:15 Pages 2495—2506
DOI https://doi.org/10.2147/CCID.S374122
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Todd Schlesinger,1 Eggert Stockfleth,2 Ayman Grada,3 Brian Berman4
1Clinical Research Center of the Carolinas, Charleston, SC, USA; 2Department of Dermatology, Venereology and Allergology, Ruhr-University Bochum, Bochum, Germany; 3Department of Dermatology, Case Western Reserve University School of Medicine, Cleveland, OH, USA; 4Dr. Phillip Frost Department of Dermatology and Cutaneous Surgery, University of Miami Miller School of Medicine, Miami, FL, USA
Correspondence: Ayman Grada, Email [email protected]
Abstract: Actinic keratosis (AK) is a common pre-neoplastic skin lesion constituted by uncontrolled proliferation of atypical keratinocytes that may evolve to squamous cell carcinoma. With global prevalence increasing, AK is expected to be the most common carcinoma of the skin. Tirbanibulin is a reversible tubulin polymerization inhibitor with potent anti-proliferative and anti-tumoral effects. In-vivo and in-vitro studies have shown that tirbanibulin significantly inhibits cell proliferation, tumor growth and downregulates Src signaling with no overt toxicity. Early phase and Phase III trials have shown high lesion clearance, compliance, and few side effects of once daily tirbanibulin treatment. This review discusses tirbanibulin anti-cancer activity, focusing on tubulin polymerization and Src signaling inhibitory effects, highlighting relevant literature and novel preclinical results from the ATNXUS-KX01-001 study. Furthermore, we address the relevant findings obtained in recent clinical trials to evaluate the safety, efficacy, pharmacokinetics, clearance efficacy, and side effects of the 1% tirbanibulin ointment applied once daily. In summary, we highlight preclinical and clinical evidence on the use of tirbanibulin as an effective and safe treatment option for AK.
Keywords: actinic keratosis, tirbanibulin, microtubules, Src kinase inhibitor
Introduction
Actinic keratosis (AK) is a pre-cancerous skin lesion resulting from the proliferation of atypical keratinocytes in response to prolonged intermittent ultraviolet light exposure, predominantly on the face, balding scalp, and extremities. AK begins with DNA damage and mutation, followed by neoplastic transformation and proliferation.1 Once abnormal cell invasion involves the dermis structures, these lesions may progress to squamous cell carcinoma (SCC) and carry the potential to metastasize.2 Prolonged UV exposure induces multiple genetic and epigenetic changes, disrupting the function of key genes in keratinocytes driving AK to SCC progression.3 Oncogenes, especially Src family kinases (SFKs) can trigger the proliferation and progression to SCC4 and have been reported in hyperproliferative epidermal disorders and pre-malignant lesions such as AK.5 A recent large whole-exome sequencing study showed that genomic and copy number alterations already occur in AK before the transition to SCC6 and data from cohort studies suggest that approximately 60% of SCC arise from preexisting AK.7,8 Studies examining genomic alterations in AK and SCC identified NOTCH1 and TP53 mutations as early events in squamous cell carcinogenesis.9,10 Significant pathological gene expression changes have also been detected in SCC11,12 including alterations in key signaling pathways, such as TGF-β and mutations in ABI3BP and IMPA1 genes.6 Since a high mutational burden can drive AK to transform into invasive SCC (iSCC), all AK lesions should be carefully monitored and treated.13 Furthermore, the prevalence of AK is high and expected to increase significantly in the coming years.14,15 In the US, AK afflicts nearly 58 million people,16 costing over a billion dollars a year.17 Dermatologists diagnosed AK in more than 47 million visits over 10-years, and it was found to occur in 14% of patients.18 Geographic differences influence the prevalence of AK, as exposure to the sun represents the most considerable risk factor.19 Male gender, fair skin, and preexisting immune system disorders may increase the risk of developing AK.15,20,21 The primary treatment goals for AK are to decrease the risk of a patient developing invasive SCC, eradicate most clinical and subclinical AK, and extend the disease-free interval.22,23 Secondary aims include reducing side effects of AK therapies and improving the patient’s quality of life.2
Existing Therapeutic Options for Actinic Keratosis
Current treatments for AK are grouped into two main categories: lesion- or field-directed therapies.24,25 Lesion-directed therapies, including cryosurgery, curettage, or laser therapy, are directed at individual lesions. Cryosurgery is widely used; however, this procedure lacks standardization, causes pain, and is associated with a high recurrence rate of up to 96% within a year.22 Chemexfoliation using chemical peels, such as trichloroacetic acid, has demonstrated efficacy in clearing AKs and subclinical lesions.26 Although chemical AK removal also results in a low recurrence rate, it can cause chronic tissue damage and inflammatory changes to the skin.27 Field-directed therapies including photodynamic therapy (PDT), topical 5-fluorouracil (5-FU), diclofenac sodium, and imiquimod, are used for treating areas with multiple AKs grouped in one anatomical area, or for clinical evidence of field cancerization.24,25 Dirschka et al, divided field-directed therapies into cluster-directed therapies for a small field (under 25 cm2), and large field-directed therapies for a larger field (over 25 cm2), in their algorithm for AK treatment.22 PDT has proven high efficacy for treating AK, though this treatment cannot be self-applied and can result in significant side effects including severe pain.28 Topical 5-FU 5% is a DNA/RNA synthesis inhibitor that causes inhibition of cell proliferation and cell death, leading to inflammation and cell necrosis.29 5-FU 5% cream is self-applied twice daily for up to 4 weeks, with reported complete clearance rate ranging from 38% to 84% and a recurrence rate of 67% within a year.30,31 Diclofenac sodium gel is a nonsteroidal anti-inflammatory drug shown to be well tolerated despite limited efficacy to treat AK.32 Moreover, it requires a long treatment regimen of twice daily applications for 60 to 90 days.33 Imiquimod, 5% cream, is a toll-like receptor 7 agonist that can stimulate the innate immune system, inducing interferons and several cytokines.34 Complete clearance of AK lesion was reported with imiquimod, ranging from 20% to 45.1% when applied twice weekly for up to 16 weeks.35 In summary, while several effective therapies are available for AK, these are often associated with a high frequency of painful local skin reactions (LSRs) (irritation of the skin, erosions, ulcerations, edema, crusting, itching), irreversible skin changes (skin dyspigmentation, scarring) and occasionally with flu-like symptoms.36 Moreover, lengthy dosing regimens of topical therapies may lower patient compliance affecting adherence and efficacy of treatment.24,37 A recent systematic review suggests that topical therapies with shorter regimens are associated with improved patient-reported outcomes.38 Therefore, there is a need for developing well-tolerated and effective topical field therapies with shorter treatment duration.37
Microtubule-Targeting Agents
Currently, the pipeline for AK includes several drugs in different stages of clinical development.33 Among these, there are different anti-cancer agents targeting cell cycle regulation components, including the mitotic stage of tumor cell proliferation and the microtubules (MTs). MTs are composed of tubulin, a globular protein that exists as a heterodimer formed by α and β tubulin 50 kDa monomers that share 40% identity in amino acid homology.39 In dividing cells undergoing mitosis, the function of MTs is to rapidly form a mitotic spindle.40 As these structures are highly susceptible to modulation by anti-mitotic agents, they represent attractive chemotherapy anti-cancer drug targets. Most MT-targeting agents bind to β-tubulin in the αβ heterodimer and suppress MTs formation, causing a delay or blockade at the metaphase–anaphase transition during mitosis,39,41 disruption at the mitotic spindle and apoptosis.42,43 MT-targeting agents are usually classified into two main groups, stabilizers and depolymerizers.44 MTs stabilizers, such as paclitaxel and docetaxel, promote polymerization of tubulin and stabilize the polymer, halting depolymerization.45 In contrast, MT destabilizers such as vinblastine, colchicine, and nocodazole prevent MT polymerization.46,47 Many MT inhibitors have shown therapeutic efficacy in a wide range of malignancies,41,48,49 although their clinical efficacy is limited as both intrinsic and acquired drug resistance has been reported.50 In addition to drug resistance, the evaluation of certain tubulin-binding agents has been discontinued because of the onset of significant severe side effects, including peripheral neuropathy, bone marrow suppression, and severe weakness.51,52 Reduced neurotoxicity and chemoresistance represent critical objectives in the search of novel tubulin-targeting drugs.
Tirbanibulin: Mechanism of Action
Tirbanibulin is a novel synthetic chemical entity that has shown potent anti-proliferative and anti-tumoral effects in-vitro and in-vivo by inducing cell cycle arrest and ultimately apoptotic cell death. These effects can be attributed to the ability of tirbanibulin to bind tubulin and inhibit polymerization.53 Cell-based experiments and crystal structure analysis of tubulin-tirbanibulin complex revealed that tirbanibulin reversibly binds to the colchicine-binding site on β-tubulin.54 However, Smolinski et al have shown that tirbanibulin binds to a novel site on α−β tubulin heterodimer.53 Additional data is needed to confirm these findings. The in-vitro ATNXUS-KX01-001 study identified α- and β-tubulins as tirbanibulin-binding sites through liquid chromatography with tandem mass spectrometry in colon cancer HT-29 cells. Photoaffinity labeling assays confirmed these data using purified tubulin and competitive binding assays with known tubulin-binding drugs (ie, colchicine, vincristine, docetaxel, guanosine diphosphate, and guanosine triphosphate). Importantly, tirbanibulin showed a dose-dependent inhibition of tubulin polymerization in an in-vitro immunofluorescence competitive binding assay with purified tubulin (Figure 1). Tubulin polymerization assay was started by incubation at 37°C in the absence or presence of test compounds (control, paclitaxel 10 µM, nocodazole 10 µM, tirbanibulin 10 µM, tirbanibulin 1 µM), and followed by absorption readings at 340 nm. The results have shown that tirbanibulin inhibition was comparable to that of nocodazole. Immunofluorescence staining demonstrated that tirbanibulin effectively disrupted the cellular MTs network via direct inhibition of tubulin in human peripheral blood mononuclear cells,53 prostate cancer PC3 cells, and immortalized keratinocyte CCD-1106 KERTr cells (ATNXUS-KX01-001 study) (Figure 2). Moreover, tirbanibulin induced complete cell cycle arrest at G2/M phase, detected by flow cytometry, of CCD-1106 KERTr cells incubated with tirbanibulin (50 nM) or control DMSO for 40 hours.55 Niu et al showed that compared to other MTs inhibitors, tirbanibulin binds with very high affinity and specificity to tubulin, exerting a robust anti-proliferative effect and halting cell division during the late interphase.54 Furthermore, tirbanibulin induced G2/M cell cycle arrest in HeLa cells and presented with no overt toxicity in-vitro, explained by tirbanibulin full reversible binding to tubulin.54 Tirbanibulin-dependent blunt arrest during G2/M phase triggered signals of programmed cell death by activating both intrinsic and extrinsic apoptotic pathways.55 Increased apoptosis was assessed in PC3-LN4 cells by annexin V and 7-amino-actinomycin D positive staining. The collapse of the mitochondrial membrane potential, a characteristic event of early-stage apoptosis, was also observed in tirbanibulin-treated PC3-LN4 cells.55 Moreover, tirbanibulin led to hyper-phosphorylation of Bcl-2, caspase 8 and 9 cleavages, activation of caspase 3 and subsequent poly(ADP-ribose) polymerase inhibitor (PARP) cleavage as demonstrated by immunoblot analysis.55
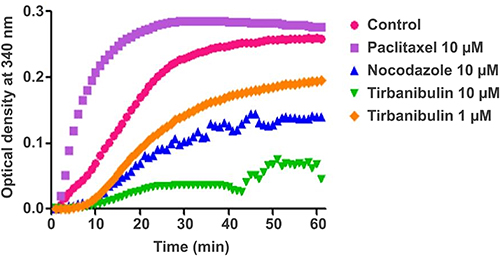 |
Figure 1 Tubulin polymerization inhibition by tirbanibulin and other known tubulin inhibitors (ATNXUS-KX01-001 study): effects of tirbanibulin on in-vitro tubulin polymerization. Tubulin polymerization was started by incubation at 37°C in the absence or presence of test compounds. The effect of tirbanibulin (10 μM and 1 μM), paclitaxel (10 μM), and nocodazole (10 μM) on tubulin polymerization was measured and plotted as changes in absorbance at 340 nm. |
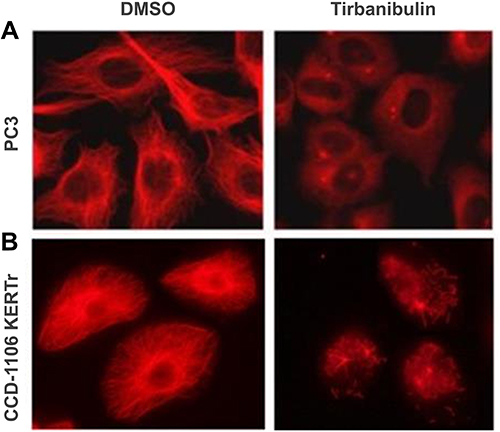 |
Figure 2 Disruption of microtubule architecture in PC3 and CCD-1106 KERTr cells by tirbanibulin (ATNXUS-KX01-001 study): representative figures of tubulin disruption in (A) PC3 and (B) CCD-1106 KERTr cells treated with tirbanibulin (100 and 200 nM, respectively) or control DMSO for 2 hours. Cells were then fixed, permeabilized, and stained with an antibody to tubulin. DMSO=dimethyl sulfoxide. |
Severe LSRs determined by drugs used in the treatment of AK (eg, 5-FU) are caused by the release of pro-inflammatory cytokines, such as tumor necrosis factor- α (TNF-α) and interleukin-8 (IL-8).31,56 Interestingly, incubation with tirbanibulin for 24 hours induced only a small increase of IL-8 at the highest dose compared to the significant increase in TNF-alpha and IL-8 determined by 5-FU in CCD-1106 KERTr cells (Figure 3). Furthermore, tirbanibulin demonstrated a significant dose–response increase in IL-1α, a marker of cell death,57,58 compared to DMSO control and 5-FU. These results suggest that tirbanibulin induces a milder pro-inflammatory response when compared to 5-FU, potentially leading to milder LSRs. Moreover, tirbanibulin induced a more marked cell growth inhibition and cell death in rapidly dividing cells, maintained in normal complete media compared to cells with growth factor-reduced media.55 Additionally, incubation with tirbanibulin for 72 hours determined a potent anti-proliferative effect assessed by a cell viability assay, in a panel of tumor cell lines (Table 1). This test panel included renal cancer, lymphoma, melanoma, SCC, and gastric cancer-derived cell lines and multi-drug resistant cell lines suggesting that tirbanibulin selectively affects cells with a high proliferative profile. Moreover, tirbanibulin anti-tumor activity was shown in triple-negative breast cancer cell lines, determining cell growth and migration inhibition activity.59 Transformation of AK into iSCC occurs following the progressive migration in the epidermal layers of proliferating atypical keratinocytes, characterized by abnormal features, such as nuclear pleomorphism, hyperchromasia, increased mitotic rate, and hyperproliferation.60 Thus, tirbanibulin has the potential to target selectively those keratinocytes with an aberrant rate of cell growth. Finally, tirbanibulin may also induce tumor suppressor p53 expression,53 suggesting anti-proliferative effect by multiple mechanisms. It has been shown that p53 localizes on MTs and is transferred to the nucleus through the MTs in response to DNA damage.61 A previous study showed that treatment with MT-targeting agents determined the accumulation of p53 in the nucleus followed by the activation of downstream targets of p53, such as PARP cleavage and caspase-3 activation.62 This evidence could support a plausible role for tirbanibulin in p53 nuclear retention and potentiation of apoptotic cell death through the perturbation of MTs. Interestingly, two sites of mutations in the p53 gene, located on chromosome 17p132, were identified in AK and SCC.2
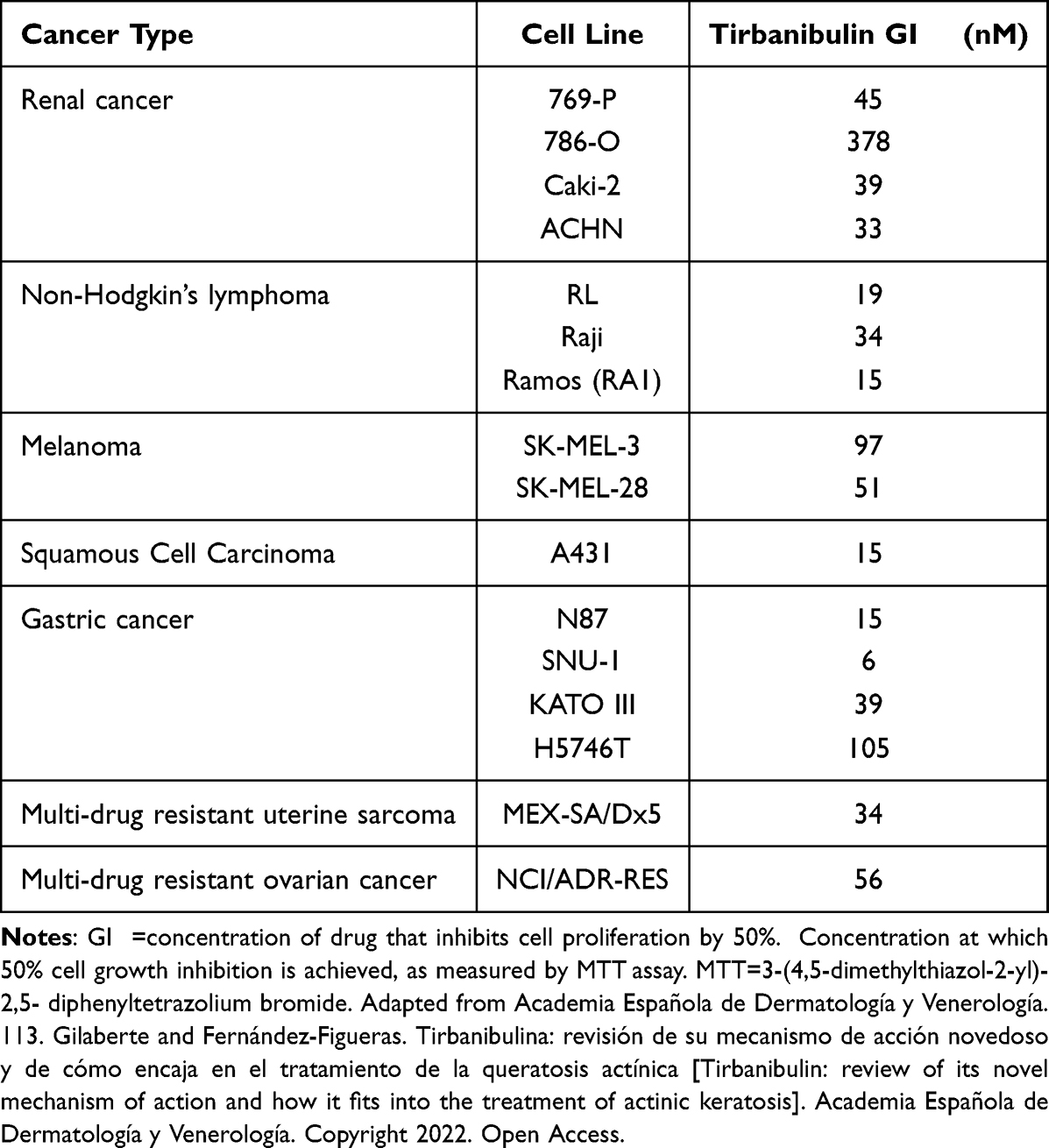 |
Table 1 In vitro Potency of Tirbanibulin in Various Cancer Cell Lines |
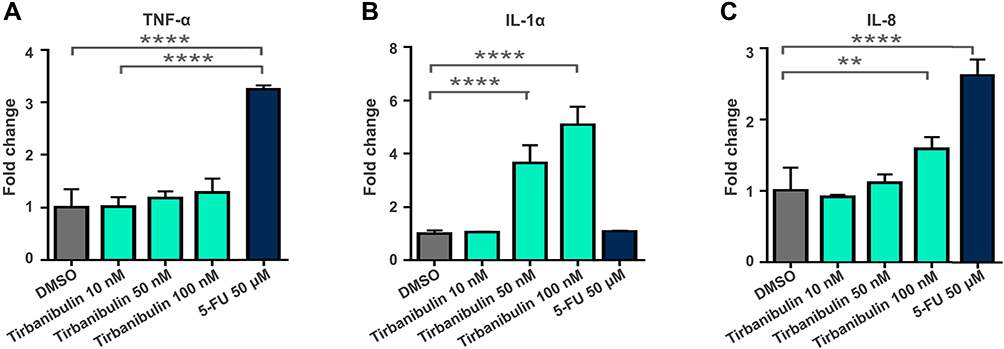 |
Figure 3 Tirbanibulin-induced pro-inflammatory response in-vitro (ATNXUS-KX01-001 study): Dose and time response of cytokine in CCD-1106 KERTr cells treated with tirbanibulin displayed concentrations for 24 h. Culture media was collected, and TNF-α (A), IL-1α (B) and IL8 (C) were measured with ELISA. **P=0.0055; ****P<0.0001. Abbreviations: 5-FU, 5-fluorouracil; DMSO, dimethyl sulfoxide; ELISA, enzyme-linked immunosorbent assay; IL, interleukin; TNF, tumor necrosis factor. |
Tirbanibulin anti-proliferative activity has been tested in-vivo in triple-negative breast cancer mouse xenograft models.59,63 Four weeks after treatment with tirbanibulin, mice showed significantly delayed tumor growth and reduced immunohistochemical staining levels of the tumor cell proliferation marker Ki-67, compared to vehicle control tissues. Moreover, tumor tissues from the tirbanibulin treatment group also displayed significantly increased numbers of apoptotic cells compared to the vehicle control sample, quantified by TUNEL assay.59 A summary of tirbanibulin potential mechanisms of action is depicted in Figure 4.
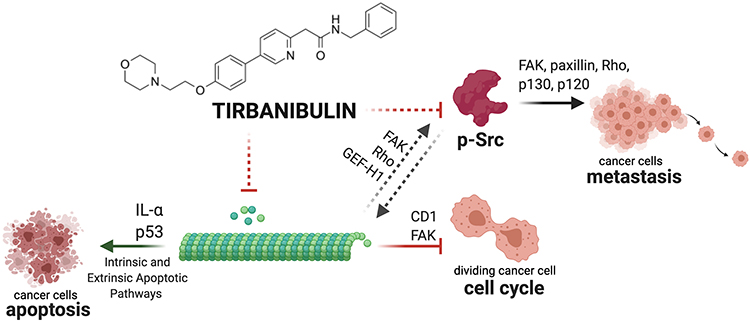 |
Figure 4 Graphical summary of tirbanibulin mechanism of action (ATNXUS-KX01-001 study): representative summary of tirbanibulin described and potential mechanisms of action. Tirbanibulin inhibits tubulin polymerization, activating intrinsic and extrinsic apoptotic pathways of cancer cells inducing IL-α and P53. The disruption of the cytoskeleton leads to the arrest of the cell cycle via multiple factors (eg, CD1, FAK) and might affect Src intracellular trafficking (FAK, Rho and GEF-H1) and Src-mediated signaling (FAK, paxillin, Rho, p130, p120) causing the inhibition of p-Src and metastasis. This figure was created with “BioRender.com”. Abbreviations: FAK, focal adhesion kinase; GEF-H1, guanine exchange factor H1; IL, interleukin; pSrc, phosphorylated Src. |
Inhibition of Src Kinase Signaling
SFKs represent a family of nine non-receptor tyrosine kinases,64 involved in vascular epithelial growth factor and angiogenesis.65,66 SFKs expression is frequently elevated in several epithelial tumors compared with the adjacent normal tissues.64 Elevated Src expression has been observed in AK and SCC, suggesting that increased signaling is necessary for keratinocyte migration, and squamous carcinoma invasion.67–69 In particular, to examine the role of SFKs in cell invasion, Mariotti et al have challenged epidermoid squamous carcinoma cells with Src chemical inhibitors, showing that tumor invasion in-vitro was heavily affected.69 Metastatic SFKs activity has been associated to the activation of integrins-mediated signaling of focal adhesion kinase (FAK), paxillin, p130, and p120.70 Moreover, a study on a mouse model of cutaneous carcinogenesis linked increased SFKs activity in keratinocytes to Fyn-dependent NOTCH1 downregulation, suggesting that this pathway activation may be necessary for promoting neoplasia.71 Thus, since Src play a significant role in the progression of many cancers, it is a likely target for drug discovery efforts.72,73 It is known that topical tyrosine kinase inhibitors can induce regression of cutaneous SCCs in in-vivo models. Inhibition of SFKs and downstream oncogenic signaling pathways determined lesion regression in an SCC mouse model treated with topical dasatinib.74 Significant down-regulated phospho-Src (p-Src) and Src signaling molecules and activity were observed in tumor xenograft mouse models treated with tirbanibulin.59,63 In particular, tirbanibulin was able to disrupt SFKs signaling in various cancer cell lines.59 In addition, tirbanibulin affected Src signaling, as demonstrated by the reduced levels of its downstream target FAK,59,63 known to be involved in MT dynamics, cell proliferation, differentiation, and survival.75,76 Cell culture experiments demonstrated that MT can regulate active Src mediating Src intracellular trafficking,77,78 via FKA, integrins signaling, Rho, and GEF-H1.70,79 Other MT-targeting agents, such as colchicine80 and paclitaxel81 have been shown to inhibit the phosphorylation of the FAK/Src complex and paxillin, suppressing cell invasion dynamics in human cancer cell lines. Thus, FAK inhibition could result from MTs suppression. However, the lack of FAK expression, in turn, can delay MT polymerization.82 Whether tirbanibulin exerts its antiproliferative activity primarily through FAK/Src or via MT inhibition remains to be shown. Moreover, even though tirbanibulin was found to directly bind Src,53,83 its inhibition could also result from an indirect effect, likely due to the disruption of the MT network and perturbation of intracellular trafficking signaling pathways.
Tirbanibulin: Summary of Clinical Development
Clinical trials have successfully confirmed the safety and efficacy of tirbanibulin 1% ointment. Phase I (NCT02337205) and Phase II (NCT02838628) trials evaluated the safety, tolerability, and pharmacokinetics of tirbanibulin ointment 1%. These studies confirmed a significant AK lesion clearance rate, safety profile, and tolerability as LSRs were mild and resolved quickly. No deaths, serious adverse events (SAEs), or discontinuations due to treatment were reported. Importantly, the Phase I trial has shown that all subjects had quantifiable but low plasma concentrations of tirbanibulin. On day 5, overall mean maximum concentration was 0.26 ng/mL (or 0.60 nM), and the mean area under the plasma concentration–time curve from 0 to 24 hours was 4.09 ng · h/mL, demonstrating that, under maximal use conditions, tirbanibulin ointment 1% for five days over an area of 25 cm2 in the treatment of AK on the face or scalp was well tolerated and resulted in low systemic exposure with sub-nanomolar plasma concentrations.84 Tirbanibulin also underwent two identical double-blinded Phase III studies involving 702 adults with 4–8 AK lesions on the face or scalp.85 More than 99% of enrolled patients completed the treatment. The studies evaluated self-applied tirbanibulin ointment 1% or vehicle once daily for five consecutive days on 702 subjects (351 per study). Successfully, subjects who received tirbanibulin ointment compared with vehicle showed significantly greater complete (100%) clearance rates at Day 57: 44% vs 5% (P<0.0001) and 54% vs 13% (P<0.0001). Similarly, partial (≥75%) clearance rates at Day 57 were significantly higher for tirbanibulin ointment than vehicle (Figure 5): 68% vs 16%, P<0.0001 (KX01-AK-003), and 76% vs 20%, P<0.0001 (KX01-AK-004). Pooled median percent reduction in AK lesion count from baseline was significantly greater in tirbanibulin at Day 57 when compared to vehicle (87.5% vs 20%).86 Efficacy was consistent across subgroups including age, gender, baseline AK lesion count, and Fitzpatrick Skin Type. In patients with complete (100%) clearance, 47% had lesion recurrence at 1-year post Day 57. LSRs were mostly mild-to-moderate. Signs of LSRs were assessed using a 4-point scale ranging from absent to severe reaction, and the sum of each score for all LSR identified a composite score. In the tirbanibulin group, LSRs peaked on Day 8 with a maximum mean composite LSR score of 4.1 (possible range, 0 to 18), but significantly diminished by Day 15 (Figure 6). LSRs resolved by Day 29–57 and mean composite LSR scores were similar between tirbanibulin (0.6 and 0.4, respectively) and vehicle groups (0.6 and 0.5).87 Severe LSRs were infrequent among tirbanibulin ointment-treated subjects in both studies and resolved quickly. For each of the 6 assessed LSRs, severe cases were observed in less than 10% of patients who received tirbanibulin. The incidence of treatment emergent adverse events (TEAEs) was similar between the tirbanibulin ointment 1% (35%) and vehicle (36%) groups in the pooled analysis. The incidence of treatment-related TEAEs in the tirbanibulin ointment 1% group (16%) was higher than the vehicle ointment group (10%). Treatment-related adverse events included tenderness, stinging, or burning sensation and were reported in 11‒20% in the tirbanibulin-treated group, compared with 9‒11% in the vehicle-treated group, in both studies. Most adverse events (AEs) were mild or moderate in severity, and only 9 subjects experienced severe AEs (3 subjects in the tirbanibulin ointment 1% group and 6 subjects in the vehicle group). The incidence of SAEs was no more than 2% in either treatment group, with a total of 7 subjects having SAEs (1 subject in the tirbanibulin ointment 1% group and 6 subjects in the vehicle group) with none deemed treatment-related.85 In summary, the results from the two double-blinded Phase III studies confirm that tirbanibulin ointment 1% represent a valuable treatment option for AK in terms of safety, efficacy, tolerability profile, and short treatment regimen.
 |
Figure 5 Number of lesions by visit and treatment group up to Day 57: assessment of number of lesions at baseline and by visit on Day 8, 15, 29 and 57 in tirbanibulin vs vehicle-controlled subjects. *P<0.0001. Abbreviations: AK, actinic keratosis; BL, baseline; D, day; SE, standard error. Notes: Adapted from Blauvelt A, Kempers S, Schlesinger T, Lain E, Wang H, Cutler D et al. Tirbanibulin Ointment 1% for Actinic Keratosis (AK): Pooled Data from Two Phase 3 Studies. J of Skin. 2020;4(6):s121. Open Access.86 |
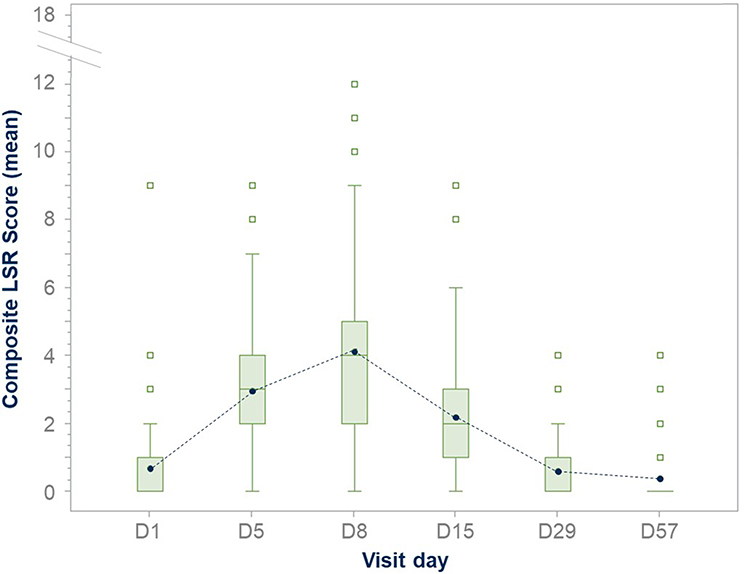 |
Figure 6 Tirbanibulin LSRs composite score from baseline to Day 57: LSR composite score assessment in the ITT population. On D8, tirbanibulin showed a maximum mean composite LSR score of 4.1 (possible range, 0 to 18), decreased significantly by D15, and resolved by D29-D57. The length of the box represents the interquartile range (the distance between the 25th and 75th percentiles). The symbol in the box interior represents the group mean. The horizontal line in the box interior represents the group median. Abbreviations: D, day; LSR, local skin reaction. Notes: Adapted from Schlesinger T, Bhatia N, Berman B, Grada A, Torra A, Cutler D et al. Favorable Safety Profile of Tirbanibulin Ointment 1% for Actinic Keratosis: Pooled Results from Two Phase III Studies. J of Skin. 2020;4(6):s120. Open Access.87 |
Further post-approval evidence of the effect of tirbanibulin in the treatment of AK is currently being tested in four registered ongoing studies in EudraCT/ClinicalTrials.gov. Three of them are interventional studies, of which one is a Phase III study (NCT05279131) and two are Phase IV studies (NCT05387525 and EudraCT number: 2022–001251-16). The fourth one ongoing trial is a non-interventional study of tirbanibulin in real world, which is being conducted in the United States (NCT05260073).
Discussion and Future Directions
Tirbanibulin mechanism of action has been extensively investigated in preclinical studies showing tubulin polymerization inhibition and cell cycle arrest in rapidly dividing cells. Preclinical data over 40 days in nude mice treated with tirbanibulin showed decreased tumor mass and proto-oncogene SFKs activity in tumor xenografts. Moreover, tirbanibulin could induce cell death in human cancer cells, by targeting the MTs and p53 trafficking. Tirbanibulin determined apoptosis by activating both intrinsic and extrinsic pathways and showed potent anti-proliferative activity against various cancer cell lines, including melanoma, SCC, and multi-drug resistant cancer cell lines. Because apoptosis is associated with less inflammation when compared to necrosis, tirbanibulin mechanism of action may explain the milder LSRs observed in clinical trials. Finally, tirbanibulin achieved high efficacy in clearing AK lesions, yet with a shorter regimen and less inflammation compared to topical AK treatments, such as 5-FU and imiquimod. Tirbanibulin has been considered a SFKs inhibitor. Unlike other Src inhibitors which bind to the ATP-binding site, tirbanibulin is thought to bind to the peptide-substrate binding site of Src. However, more experiments are needed to clarify whether tirbanibulin acts through direct Src inhibition, which may disrupt MT cytoskeleton functions. To our knowledge, no data suggest that tirbanibulin inhibits tubulin polymerization solely because of its kinase inhibitory properties. What is known is that tirbanibulin binds to β-tubulin and thereby inhibits tubulin polymerization causing apoptosis. By blocking the downstream signaling pathways of Src, cancerous cell migration, proliferation, and survival are reduced. Moreover, tirbanibulin binds the colchicine-binding site of tubulin but unlike colchicine and other tubulin-binding agents, the binding is totally reversible.54 This likely explains low cytotoxicity for cultured cells incubated with tirbanibulin and reported mild application site reactions in clinical trials for AKs. In contrast, other chemotherapeutic tubulin-binding drugs, such as paclitaxel and vinblastine bind irreversibly causing severe toxicity and limited clinical use. Tirbanibulin also reduces FAK expression although it is unclear if this inhibition results from a direct effect or follows MT suppression. Finally, future investigation is needed to elucidate the role of tirbanibulin in targeting the tumor suppressor p53.
Conclusions
Tirbanibulin is a novel synthetic chemical drug with potent antitumor and antiproliferative activity, which may be explained through the tirbanibulin’s ability to bind to tubulin, inhibiting its polymerization and promoting microtubule disruption, as well as indirectly altering Src tyrosine kinase signaling. Considering that AK is associated with cell hyperproliferation, tirbanibulin represents a good option for AK treatment, with a simple dosage regimen that favors adherence to therapy. Moreover, tirbanibulin does not induce a pronounced release of pro-inflammatory cytokines in keratinocytes in vitro, unlike other treatments for AK such as 5-FU. This is associated with a favorable safety profile in clinical practice.
Acknowledgments
Medical writing support was provided by Stefania Ippati, PhD, TFS HealthScience and Eva Mateu, PhD, TFS HealthScience, and was funded by Almirall S.A., in accordance with Good Publication Practice (GPP3) guidelines. We thank Yahao Bu, PhD, and Murray Cutler, PhD (Athenex) for revising the manuscript. We also thank Emilio Fumero, MD, PhD, Ayman Grada, MD, MS, and Ana Blanco, DVM, MS, DACVP (Almirall) for the valuable inputs and for extensively revising the manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This publication was funded by Almirall S.A., Barcelona, Spain.
Disclosure
Brian Berman has received grants or research funding from Biofrontera, LEO Pharma, and Sirnaomics. He is a consultant to Aiviva, Almirall, Biofrontera, Castle Biosciences, Inc., LEO Pharma, Mediwound, PHD Biosciences, Sirnaomics, and Sun Pharma. He has attended speakers’ bureaus for Almirall, and Sun Pharma, and served on advisory boards for Almirall, Biofrontera, Castle Biosciences, Inc., LEO Pharma, and Sun Pharma. Todd Schlesinger has received grants or research funding from AbbVie, Aclaris Therapeutics, Inc., Akros Pharma, Inc., Allergan, AOBiome Therapeutics, Arcutis Biotherapeutics, Astellas Pharma US, Inc., Bioderma Laboratories, Biofrontera, BioPharmX, Boehringer Ingelheim, Bristol-Myers Squibb, Cara Therapeutics, Castle Biosciences, Inc., Celgene, ChemoCentryx, Coherus BioSciences, Corrona, Dermavant, Dermira, Inc., DT Collagen & DT Pharmacy, Eli Lilly & Co., Almirall, Galderma, Genentech, Inc., Janssen/Centocor Ortho Biotech, Inc., Kinex, Kiniksa Pharmaceuticals, LEO Pharma, Merz Pharma, Novartis, Pfizer, Inc., Regeneron Pharmaceuticals, Sanofi Genzyme, SiSaf, Ltd., Tetraderm Group, LLC, and Trevi Therapeutics. He is a consultant to AbbVie, Aclaris Therapeutics, Inc., Allergan, Almirall, Biofrontera, Bristol-Myers Squibb, Castle Biosciences, Inc., Eli Lilly & Co., EPI Health, Evolus, Foundation for Research and Education in Dermatology, Galderma, LEO Pharma, MED Learning Group CME Program, Merz Pharma, MJH Associates ONCLive SCC Filming, Novartis, Ortho Dermatologics, Pfizer, Inc., Prolacta Bioscience, Regeneron Pharmaceuticals, SiSaf, Ltd., Sun Pharma, Suneva Medical, Inc., and UCB Pharma. He has attended speakers’ bureaus for Aclaris Therapeutics, Inc., Almirall, Dermira, Inc., DUSA Pharmaceuticals, EPI Health, Regeneron Pharmaceuticals, Sanofi-Aventis, Sanofi Genzyme, Sun Pharma, and Suneva Medical, Inc.; and served on advisory boards for Allergan, Almirall, Bioderma Laboratories, Biofrontera, Celgene, Greenway Therapeutix, Remedly, and Suneva Medical, Inc. He is also co-chair committee and/or performance measurement, corporate relations committee member for American Academy of Dermatology Actinic Keratosis Guideline and Actinic Keratosis Performance Measurement Work Group. Eggert Stockfleth has received grants or research funding from Almirall, Pierre Fabre, Aresus and Dr Pfleger. Ayman Grada is the Former Head of R&D and Medical Affairs at Almirall (US). The authors report no other conflicts of interest in this work.
References
1. Fania L, Didona D, Di Pietro FR, et al. Cutaneous squamous cell carcinoma: from pathophysiology to novel therapeutic approaches. Biomedicines. 2021;9(2):171. doi:10.3390/biomedicines9020171
2. Cockerell CJ. Pathology and pathobiology of the actinic (solar) keratosis. Br J Dermatol. 2003;149(Suppl 66):34–36. doi:10.1046/j.0366-077X.2003.05625.x
3. Shen Y, Ha W, Zeng W, Queen D, Liu L. Exome sequencing identifies novel mutation signatures of UV radiation and trichostatin A in primary human keratinocytes. Sci Rep. 2020;10(1):4943. doi:10.1038/s41598-020-61807-4
4. Lee JH, Pyon JK, Kim DW, et al. Elevated c-Src and c-Yes expression in malignant skin cancers. J Exp Clin Cancer Res. 2010;29(1):116. doi:10.1186/1756-9966-29-116
5. Ayli EE, Li W, Brown TT, Witkiewicz A, Elenitsas R, Seykora JT. Activation of Src-family tyrosine kinases in hyperproliferative epidermal disorders. J Cutan Pathol. 2008;35(3):273–277. doi:10.1111/j.1600-0560.2007.00807.x
6. Thomson J, Bewicke-Copley F, Anene CA, et al. The genomic landscape of actinic keratosis. J Invest Dermatol. 2021;141:1664–1674.e7. doi:10.1016/j.jid.2020.12.024
7. Marks R, Rennie G, Selwood TS. Malignant transformation of solar keratoses to squamous cell carcinoma. Lancet. 1988;1(8589):795–797. doi:10.1016/S0140-6736(88)91658-3
8. Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the veterans affairs topical tretinoin chemoprevention trial. Cancer. 2009;115(11):2523–2530. doi:10.1002/cncr.24284
9. Kim YS, Shin S, Jung SH, et al. Genomic progression of precancerous actinic keratosis to squamous cell carcinoma. J Invest Dermatol. 2021;S0022–202X(21):2162–X.
10. South AP, Purdie KJ, Watt SA, et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J Invest Dermatol. 2014;134(10):2630–2638. doi:10.1038/jid.2014.154
11. Das Mahapatra K, Pasquali L, Søndergaard JN, et al. A comprehensive analysis of coding and non-coding transcriptomic changes in cutaneous squamous cell carcinoma. Sci Rep. 2020;10(1):3637. doi:10.1038/s41598-020-59660-6
12. Nindl I, Dang C, Forschner T, et al. Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol Cancer. 2006;5(1):30. doi:10.1186/1476-4598-5-30
13. Fernandez Figueras MT. From actinic keratosis to squamous cell carcinoma: pathophysiology revisited. J Eur Acad Dermatol Venereol. 2017;31(Suppl 2):5–7. doi:10.1111/jdv.14151
14. Rosen T, Lebwohl MG. Prevalence and awareness of actinic keratosis: barriers and opportunities. J Am Acad Dermatol. 2013;68(1 Suppl 1):S2–9. doi:10.1016/j.jaad.2012.09.052
15. Memon AA, Tomenson JA, Bothwell J, Friedmann PS. Prevalence of solar damage and actinic keratosis in a Merseyside population. Br J Dermatol. 2000;142(6):1154–1159. doi:10.1046/j.1365-2133.2000.03541.x
16. Landis ET, Davis SA, Taheri A, Feldman SR. Top dermatologic diagnoses by age. Dermatol Online J. 2014;20(4):22368. doi:10.5070/D3204022368
17. Bickers DR, Lim HW, Margolis D, et al. The burden of skin diseases: 2004 a joint project of the American Academy of dermatology association and the society for investigative dermatology. J Am Acad Dermatol. 2006;55(3):490–500. doi:10.1016/j.jaad.2006.05.048
18. Gupta AK, Cooper EA, Feldman SR, Fleischer AB. A survey of office visits for actinic keratosis as reported by NAMCS, 1990–1999. National Ambulatory Med Care Survey Cutis. 2002;70(2 Suppl):8–13.
19. Frost C, Williams G, Green A. High incidence and regression rates of solar keratoses in a Queensland community. J Invest Dermatol. 2000;115(2):273–277. doi:10.1046/j.1523-1747.2000.00048.x
20. Flohil SC, van der Leest RJT, Dowlatshahi EA, Hofman A, de Vries E, Nijsten T. Prevalence of actinic keratosis and its risk factors in the general population: the Rotterdam Study. J Invest Dermatol. 2013;133(8):1971–1978. doi:10.1038/jid.2013.134
21. Hensen P, Müller ML, Haschemi R, et al. Predisposing factors of actinic keratosis in a North-West German population. Eur J Dermatol. 2009;19(4):345–354. doi:10.1684/ejd.2009.0706
22. Dirschka T, Gupta G, Micali G, et al. Real-world approach to actinic keratosis management: practical treatment algorithm for office-based dermatology. J Dermatolog Treat. 2017;28(5):431–442. doi:10.1080/09546634.2016.1254328
23. Werner RN, Stockfleth E, Connolly SM, et al. Evidence- and consensus-based (S3) guidelines for the treatment of actinic keratosis - international league of dermatological societies in cooperation with the European dermatology forum - short version. J Eur Acad Dermatol Venereol. 2015;29(11):2069–2079. doi:10.1111/jdv.13180
24. Khanna R, Bakshi A, Amir Y, Goldenberg G. Patient satisfaction and reported outcomes on the management of actinic keratosis. Clin Cosmet Investig Dermatol. 2017;10:179–184. doi:10.2147/CCID.S121323
25. de Berker D, McGregor JM, Mohd Mustapa MF, Exton LS, Hughes BR. British association of dermatologists’ guidelines for the care of patients with actinic keratosis 2017. Br J Dermatol. 2017;176(1):20–43. doi:10.1111/bjd.15107
26. Di Nuzzo S, Cortelazzi C, Boccaletti V, et al. Comparative study of trichloroacetic acid vs. photodynamic therapy with topical 5-aminolevulinic acid for actinic keratosis of the scalp. Photodermatol Photoimmunol Photomed. 2015;31(5):233–238. doi:10.1111/phpp.12164
27. Sidiropoulou P, Gregoriou S, Rigopoulos D, Kontochristopoulos G. Chemical peels in skin cancer: a review. J Clin Aesthet Dermatol. 2020;13(2):53–57.
28. Ericson MB, Wennberg AM, Larkö O. Review of photodynamic therapy in actinic keratosis and basal cell carcinoma. Ther Clin Risk Manag. 2008;4(1):1–9. doi:10.2147/TCRM.S1769
29. Dodds A, Chia A, Shumack S. Actinic keratosis: rationale and management. Dermatol Ther. 2014;4(1):11–31. doi:10.1007/s13555-014-0049-y
30. Krawtchenko N, Roewert-Huber J, Ulrich M, Mann I, Sterry W, Stockfleth E. A randomised study of topical 5% imiquimod vs. topical 5-fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1-year follow-up. Br J Dermatol. 2007;157(Suppl 2):34–40. doi:10.1111/j.1365-2133.2007.08271.x
31. Pomerantz H, Hogan D, Eilers D, et al. Long-term efficacy of topical fluorouracil cream, 5%, for treating actinic keratosis: a randomized clinical trial. JAMA Dermatol. 2015;151(9):952–960. doi:10.1001/jamadermatol.2015.0502
32. Nelson CG. Diclofenac gel in the treatment of actinic keratoses. Ther Clin Risk Manag. 2011;7:207–211. doi:10.2147/TCRM.S12498
33. Cramer P, Stockfleth E. Actinic keratosis: where do we stand and where is the future going to take us? Expert Opin Emerg Drugs. 2020;25(1):49–58. doi:10.1080/14728214.2020.1730810
34. Sauder DN. Immunomodulatory and pharmacologic properties of imiquimod. J Am Acad Dermatol. 2000;43(1):S6–11. doi:10.1067/mjd.2000.107808
35. Full prescribing information of Imiquimod, FDA. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/020723s022lbl.pdf.
36. Uhlenhake EE. Optimal treatment of actinic keratoses. Clin Interv Aging. 2013;8:29–35. doi:10.2147/CIA.S31930
37. Goldenberg G. Treatment considerations in actinic keratosis. J Eur Acad Dermatol Venereol. 2017;31(Suppl 2):12–16. doi:10.1111/jdv.14152
38. Grada A, Feldman SR, Bragazzi NL, Damiani G. Patient-reported outcomes of topical therapies in actinic keratosis: a systematic review. Dermatol Ther. 2021;34(2):e14833. doi:10.1111/dth.14833
39. Wilson L, Jordan MA. Microtubule dynamics: taking AIM at a moving target. Chem Biol. 1995;2(9):569–573. doi:10.1016/1074-5521(95)90119-1
40. McLoughlin EC, O’Boyle NM. Colchicine-binding site inhibitors from chemistry to clinic: a review. Pharmaceuticals. 2020;13(1):8. doi:10.3390/ph13010008
41. Arnst KE, Banerjee S, Chen H, et al. Current advances of tubulin inhibitors as dual acting small molecules for cancer therapy. Med Res Rev. 2019;39(4):1398–1426. doi:10.1002/med.21568
42. Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23(16):2825–2837. doi:10.1038/sj.onc.1207528
43. Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7(5):637–651. doi:10.1016/j.devcel.2004.09.002
44. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;Apr(4):253–265. doi:10.1038/nrc1317
45. Rohena CC, Mooberry SL. Recent progress with microtubule stabilizers: new compounds, binding modes and cellular activities. Nat Prod Rep. 2014;31(3):335–355. doi:10.1039/C3NP70092E
46. Bhattacharyya B, Panda D, Gupta S, Banerjee M. Anti-mitotic activity of colchicine and the structural basis for its interaction with tubulin. Med Res Rev. 2008;28(1):155–183. doi:10.1002/med.20097
47. Owellen RJ, Hartke CA, Dickerson RM, Hains FO. Inhibition of tubulin-microtubule polymerization by drugs of the Vinca alkaloid class. Cancer Res. 1976;36(4):1499–1502.
48. Gutiérrez-Gutiérrez G, Sereno M, Miralles A, Casado-Sáenz E, Gutiérrez-Rivas E. Chemotherapy-induced peripheral neuropathy: clinical features, diagnosis, prevention and treatment strategies. Clin Transl Oncol. 2010;12(2):81–91. doi:10.1007/S12094-010-0474-z
49. Yan W, Yang T, Yang J, et al. SKLB060 reversibly binds to colchicine site of tubulin and possesses efficacy in multidrug-resistant cell lines. Cell Physiol Biochem. 2018;47(2):489–504. doi:10.1159/000489983
50. Fojo T, Menefee M. Mechanisms of multidrug resistance: the potential role of microtubule-stabilizing agents. Ann Oncol. 2007;18(Suppl 5):v3–8. doi:10.1093/annonc/mdm172
51. Čermák V, Dostál V, Jelínek M, et al. Microtubule-targeting agents and their impact on cancer treatment. Eur J Cell Biol. 2020;99(4):151075. doi:10.1016/j.ejcb.2020.151075
52. Dumontet C, Jordan MA. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov. 2010;9(10):790–803. doi:10.1038/nrd3253
53. Smolinski MP, Bu Y, Clements J, et al. Discovery of novel dual mechanism of action src signaling and tubulin polymerization inhibitors (KX2-391 and KX2-361). J Med Chem. 2018;61(11):4704–4719. doi:10.1021/acs.jmedchem.8b00164
54. Niu L, Yang J, Yan W, et al. Reversible binding of the anticancer drug KXO1 (tirbanibulin) to the colchicine-binding site of β-tubulin explains KXO1’s low clinical toxicity. J Biol Chem. 2019;294(48):18099–18108. doi:10.1074/jbc.RA119.010732
55. Gilaberte Y, Fernández-Figueras MT. Tirbanibulin: review of its novel mechanism of action and how it fits into the treatment of actinic keratosis. [Tirbanibulina: revisión de su mecanismo de acción novedoso y de cómo encaja en el tratamiento de la queratosis actínica. Actas Dermosifiliogr]. Actas Dermo-Sifiliográficas. 2022;113(1):T58–66. doi:10.1016/j.ad.2021.07.016
56. Chughtai K, Gupta R, Upadhaya S, Al Hadidi S. Topical 5-Fluorouracil associated skin reaction. Oxf Med Case Reports. 2017;2017(8):omx043. doi:10.1093/omcr/omx043
57. Gross O, Yazdi AS, Thomas CJ, et al. Inflammasome activators induce interleukin-1α secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36(3):388–400. doi:10.1016/j.immuni.2012.01.018
58. Wooff Y, Man SM, Aggio-Bruce R, Natoli R, Fernando N. IL-1 family members mediate cell death, inflammation and angiogenesis in retinal degenerative diseases. Front Immunol. 2019;10:1618. doi:10.3389/fimmu.2019.01618
59. Kim S, Min A, Lee KH, et al. Antitumor effect of KX-01 through inhibiting src family kinases and mitosis. Cancer Res Treat. 2017;49(3):643–655. doi:10.4143/crt.2016.168
60. Fernández-Figueras MT, Carrato C, Sáenz X, et al. Actinic keratosis with atypical basal cells (AK I) is the most common lesion associated with invasive squamous cell carcinoma of the skin. J Eur Acad Dermatol Venereol. 2015;29(5):991–997. doi:10.1111/jdv.12848
61. Giannakakou P, Sackett DL, Ward Y, Webster KR, Blagosklonny MV, Fojo T. p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat Cell Biol. 2000;2(10):709–717. doi:10.1038/35036335
62. Giannakakou P, Nakano M, Nicolaou KC, et al. Enhanced microtubule-dependent trafficking and p53 nuclear accumulation by suppression of microtubule dynamics. PNAS. 2002;99(16):10855–10860. doi:10.1073/pnas.132275599
63. Anbalagan M, Ali A, Jones RK, et al. Peptidomimetic Src/pretubulin inhibitor KX-01 alone and in combination with paclitaxel suppresses growth, metastasis in human ER/PR/HER2-negative tumor xenografts. Mol Cancer Ther. 2012;11(9):1936–1947. doi:10.1158/1535-7163.MCT-12-0146
64. Frame MC. Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta. 2002;1602(2):114–130. doi:10.1016/s0304-419x(02)00040-9
65. Munshi N, Groopman JE, Gill PS, Ganju RK. c-Src mediates mitogenic signals and associates with cytoskeletal proteins upon vascular endothelial growth factor stimulation in Kaposi’s sarcoma cells. J Immunol. 2000;164(3):1169–1174. doi:10.4049/jimmunol.164.3.1169
66. Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19(49):5636–5642. doi:10.1038/sj.onc.1203912
67. Serrels B, Serrels A, Mason SM, et al. A novel Src kinase inhibitor reduces tumour formation in a skin carcinogenesis model. Carcinogenesis. 2009;30(2):249–257. doi:10.1093/carcin/bgn278
68. Ainger SA, Sturm RA. Src and SCC: getting to the FAKs. Exp Dermatol. 2015;24(7):487–488. doi:10.1111/exd.12725
69. Mariotti A, Kedeshian PA, Dans M, Curatola AM, Gagnoux-Palacios L, Giancotti FG. EGF-R signaling through Fyn kinase disrupts the function of integrin α6β4 at hemidesmosomes. J Cell Biol. 2001;155(3):447–458. doi:10.1083/jcb.200105017
70. Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23(48):7928–7946. doi:10.1038/sj.onc.1208080
71. Zhao L, Li W, Marshall C, et al. Srcasm inhibits Fyn-induced cutaneous carcinogenesis with modulation of Notch 1 and p53. Cancer Res. 2009;69(24):9439–9447. doi:10.1158/0008-5472.CAN-09-2976
72. Lee D, Gautschi O. Clinical development of SRC tyrosine kinase inhibitors in lung cancer. Clin Lung Cancer. 2006;7(6):381–384. doi:10.3816/CLC.2006.n.020
73. Summy JM, Gallick GE. Treatment for advanced tumors: SRC reclaims center stage. Clin Cancer Res. 2006;12(5):1398–1401. doi:10.1158/1078-0432.CCR-05-2692
74. Yang X, Daifallah AEM, Shankar S, et al. Topical kinase inhibitors induce regression of cutaneous squamous cell carcinoma. Exp Dermatol. 2019;28(5):609–613. doi:10.1111/exd.13902
75. Bershadsky A, Chausovsky A, Becker E, Lyubimova A, Geiger B. Involvement of microtubules in the control of adhesion-dependent signal transduction. Current Biology. 1996;6(10):1279–1289. doi:10.1016/S0960-9822(02)70714-8
76. Palazzo AF, Eng CH, Schlaepfer DD, Marcantonio EE, Gundersen GG. Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science. 2004;303(5659):836–839. doi:10.1126/science.1091325
77. Suter DM, Schaefer AW, Forscher P. Microtubule dynamics are necessary for SRC family kinase-dependent growth cone steering. Curr Biol. 2004;14(13):1194–1199. doi:10.1016/j.cub.2004.06.049
78. Wu B, Decourt B, Zabidi MA, et al. Microtubule-mediated Src tyrosine kinase trafficking in neuronal growth cones. MBoC. 2008;19(11):4611–4627. doi:10.1091/mbc.e08-06-0603
79. Arnette C, Frye K, Kaverina I. Microtubule and actin interplay drive intracellular c-Src Trafficking. PLoS One. 2016;11(2):e0148996. doi:10.1371/journal.pone.0148996
80. Cho JH, Joo YH, Shin EY, Park EJ, Kim MS. Anticancer effects of colchicine on hypopharyngeal cancer. Anticancer Res. 2017;37(11):6269–6280. doi:10.21873/anticanres.12078
81. McGrail DJ, Khambhati NN, Qi MX, et al. Alterations in ovarian cancer cell adhesion drive taxol resistance by increasing microtubule dynamics in a FAK-dependent manner. Sci Rep. 2015;17(5):9529. doi:10.1038/srep09529
82. Rea K, Sensi M, Anichini A, Canevari S, Tomassetti A. EGFR/MEK/ERK/CDK5-dependent integrin-independent FAK phosphorylated on serine 732 contributes to microtubule depolymerization and mitosis in tumor cells. Cell Death Dis. 2013;4(10):e815–e815. doi:10.1038/cddis.2013.353
83. Moy FJ, Lee A, Gavrin LK, et al. Novel synthesis and structural characterization of a high-affinity paramagnetic kinase probe for the identification of non-ATP site binders by nuclear magnetic resonance. J Med Chem. 2010;53(3):1238–1249. doi:10.1021/jm901525b
84. Yavel R, Overcash JS, Cutler D, Fang J, Zhi J. Phase 1 maximal use pharmacokinetic study of tirbanibulin ointment 1% in subjects with actinic keratosis. Clin Pharmacol Drug Dev. 2021;11:16. doi:10.1002/cpdd.1027
85. Blauvelt A, Kempers S, Lain E, et al. Phase 3 trials of tirbanibulin ointment for actinic keratosis. N Engl J Med. 2021;384(6):512–520. doi:10.1056/NEJMoa2024040
86. Blauvelt A, Kempers S, Schlesinger T, et al. Tirbanibulin ointment 1% for Actinic Keratosis (AK): pooled data from two phase 3 studies. J of Skin. 2020;4(6):s121. doi:10.25251/skin.4.supp.121
87. Schlesinger T, Bhatia N, Berman B, et al. Favorable safety profile of tirbanibulin ointment 1% for actinic keratosis: pooled results from two phase III studies. J of Skin. 2020;4(6):s120. doi:10.25251/skin.4.supp.120
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.