")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
Time to onset and time to resolution of extrapyramidal symptoms in patients with exacerbated schizophrenia treated with 3-monthly vs once-monthly paliperidone palmitate
Authors Mathews M, Nuamah I, Savitz AJ, Hough DW, Najarian D , Kim E , Gopal S
Received 25 May 2018
Accepted for publication 15 August 2018
Published 25 October 2018 Volume 2018:14 Pages 2807—2816
DOI https://doi.org/10.2147/NDT.S175364
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Maju Mathews,1 Isaac Nuamah,1 Adam J Savitz,1 David W Hough,1 Dean Najarian,2 Edward Kim,2 Srihari Gopal1
1Janssen Research and Development, LLC, Titusville, NJ, USA; 2Janssen Scientific Affairs, Titusville, LLC, NJ, USA
Objective: The aim of this study was to evaluate the safety of 3-monthly paliperidone palmitate (PP3M) vs once-monthly paliperidone palmitate (PP1M) treatment with regard to extrapyramidal symptom (EPS)-related treatment-emergent adverse events (TEAEs) in patients with schizophrenia, previously stabilized on PP1M treatment.
Patients and methods: Data on overall incidence, time to onset (TTO), and time to resolution (TTR) of EPS-related TEAEs (overall, subclasses such as dyskinesia, dystonia, hyperkinesia, parkinsonism, and tremor) from a randomized double-blind (DB) non-inferiority study were compared between PP3M and PP1M. Subgroup analysis was performed by age (18–25, 26–50, and 50+ years) and final open-label (OL) dose (50/75, 100, and 150 mg eq.).
Results: Overall incidence of spontaneously reported EPS-related TEAEs decreased from 12.6% (PP1M) in OL phase to 8.3% (PP3M) and 7.4% (PP1M) in the DB phase; overall median TTO and TTR values were comparable between both groups. Among patients with reported EPS-related TEAEs, the median TTO for all EPS-related TEAEs was 17 days (PP1M) in OL phase and 115 days (PP3M) and 98.5 days (PP1M) in DB phase; median TTR was 36.5 days (PP1M) in OL phase and 91 days (PP3M) and 85.5 days (PP1M) in DB phase. No clear dose- or age-related differences in TTO and TTR of EPS-related TEAEs were noted.
Conclusion: Despite differences in apparent half-life and pharmacokinetic profiles (peak plasma exposure of PP3M formulation is 70% higher than that of PP1M formulation), both PP3M and PP1M formulations exhibited comparable incidence of EPS-related TEAEs, TTO, and TTR in patients with schizophrenia.
Keywords: extrapyramidal symptoms, once-monthly paliperidone palmitate, 3-monthly paliperidone palmitate, time to onset, time to resolution
Introduction
Management strategies for schizophrenia typically involve pharmacological intervention, managing side effects of these medications, and psychosocial treatment. Antipsychotic drugs are the mainstay of treatment in schizophrenia and other psychotic disorders; conventional antipsychotics have been associated with extrapyramidal symptoms (EPSs) in patients with schizophrenia.1,2 These neuroleptic-induced movement disorders include akathisia (a sense of restlessness manifested as fidgeting and restlessness), dystonia, and parkinsonism. These EPS-related adverse events (AEs) negatively affect the quality of life of both patients and their families, and thus minimizing these events is critical from the perspectives of clinician, patient, and family, and they are linked to long-term treatment adherence.2
Incidences of EPS-related treatment-emergent AEs (TEAEs) due to atypical antipsychotics are low, and these antipsychotics have been widely used as a drug therapy to improve adherence and quality of life of patients with schizophrenia.3 The once-monthly paliperidone palmitate (PP1M) formulation is approved for the treatment of schizophrenia and schizoaffective disorders.4 PP1M has demonstrated a lower incidence of EPS-related TEAEs in patients with schizophrenia compared with oral paliperidone.1 The 3-monthly paliperidone palmitate (PP3M) formulation, approved in the US,5 the European Union,6 and other countries for the maintenance treatment of schizophrenia, has a distinct advantage of reduced frequency of administration (only four times a year) over the PP1M formulation. Following intramuscular administration, PP3M is released slowly into the bloodstream, where it is rapidly hydrolyzed to paliperidone, and is absorbed into the systemic circulation; the absorbed drug release starts as early as day 1 and lasts for up to 18 months.7 Peak paliperidone concentration is achieved between 23 and 34 days, with an apparent half-life of 2–4 months, which is longer (approximately 70% more peak plasma exposure) than that usually observed after PP1M treatment; this substantiates the higher dosing (3.5 times PP1M dose) and prolonged dosing interval of PP3M; however, the total exposure (mean area under the curve) and maximum serum concentration remain similar between PP1M and PP3M (dose equivalency with longer dosing interval).7,8 The efficacy of PP3M, determined by the percentage of patients who remained relapse free, was confirmed in two Phase III studies.9,10 PP3M was non-inferior to PP1M in terms of efficacy, and the safety and tolerability profiles of PP3M and PP1M were comparable over the 48-week double-blind (DB) phase.10 Although the incidence of EPS-related TEAEs following PP3M treatment was similar to that following PP1M treatment in patients with schizophrenia,10 little is known regarding the time to onset (TTO) and time to resolution (TTR) of EPS-related TEAEs in patients with schizophrenia treated with either PP1M or PP3M. The current post hoc analysis was therefore undertaken to compare the overall incidence of EPS-related TEAEs as well as the TTO and TTR of these EPS-related TEAEs following treatment with PP3M vs PP1M in patients with schizophrenia.
Patients and methods
This post hoc analysis used data from the 48-week, DB, parallel-group, multicenter, Phase III non-inferiority study of PP3M vs PP1M in patients aged 18–70 years with schizophrenia (ClinicalTrials.gov identifier: NCT01515423; www.ClinicalTrials.gov).10 The details of the primary study are published elsewhere.10 In brief, the study enrolled patients with a confirmed diagnosis of schizophrenia (Diagnostic and Statistical Manual of Mental Disorders, fourth Edition [DSM-IV-TR], total Positive and Negative Syndrome Scale [PANSS] score between 70 and 120 at screening and at baseline) and with worsening of symptoms of schizophrenia. Patients who discontinued the current antipsychotic therapy due to inadequate efficacy, safety, or tolerability, or patient’s preferences for injectable medications, were also eligible to participate in the study.10
An independent ethics committee or institutional review board (listed in Supplementary material) at each study site approved the study protocol. The study was conducted in accordance with the ethical principles originating in the Declaration of Helsinki and International Conference on Harmonization Good Clinical Practice guidelines, applicable regulatory requirements, and in compliance with the protocol. All patients (or their legally acceptable representatives, if required by local regulations) provided written informed consent.
Following a ≤3-week screening period, and a 17-week, flexible-dose, open-label (OL) phase (PP1M: day 1 [150 mg eq. deltoid], day 8 [100 mg eq. deltoid], weeks 5, 9, and 13 [50, 75, 100, or 150 mg eq. deltoid/gluteal]), clinically stable patients (defined as PANSS total score <70, PANSS item [P1, P2, P3, P6, P7, G8, G14] scores ≤4, and reduction in the Clinical Global Impression – Severity (CGI-S) score by ≥1 from OL baseline) entered the 48-week DB phase and were randomly assigned to fixed doses of PP3M or PP1M (Figure 1).10
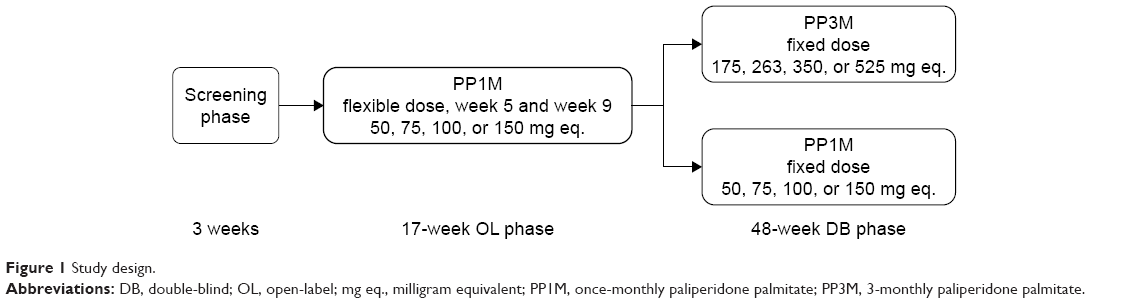 | Figure 1 Study design. |
EPS-related TEAE
Parameters of interest included spontaneously reported EPS-related TEAEs recorded at every visit. A TEAE was defined as an event that first occurred or worsened after study entry. Incidences of spontaneously reported EPS-related TEAEs assessed using Medical Dictionary for Regulatory Activities (MedDRA) preferred terms (version 17.1) were grouped under five prespecified categories (referred as EPS group) of parkinsonism, tremor, dystonia, hyperkinesia (which includes akathisia), and dyskinesia. Each EPS group represents predefined EPS-related TEAEs using MedDRA preferred terms: 1) hyperkinesia: akathisia, hyperkinesia, periodic limb movement disorder, restless legs syndrome, restlessness; 2) parkinsonism: stiffness, akinesia, hypokinesia, nuchal rigidity, parkinsonian gait, parkinsonian rest tremor, parkinsonism, muscle rigidity, muscle tightness, glabellar reflex abnormal, on and off phenomenon, Parkinson’s disease, parkinsonian crisis, extrapyramidal disorder, masked facies; 3) tremor: tremor, essential tremor, intention tremor; 4) dyskinesia: dyskinesia, muscle contractions involuntary, movement disorder, muscle twitching, athetosis, chorea, choreoathetosis, tardive dyskinesia, myoclonus, protrusion tongue, rabbit syndrome, buccoglossal syndrome; 5) dystonia: oculogyration, oculogyric crisis, trismus, tongue spasm, tongue paralysis, cervical spasm, emprosthotonus, myotonia, pleurothotonus, risus sardonicus, muscle spasms, blepharospasm, dystonia, opisthotonus, torticollis, facial spasm, muscle contracture.
The TTO was defined as the minimum value of the time to onset for any EPS-related TEAEs in the particular EPS group, while the TTR of an EPS-related TEAE was defined as the number of days from the onset date of the first EPS-related TEAE to its resolution date. The TTO and TTR values of EPS-related TEAEs were also assessed in subgroups of patients stratified by age (18–25, 26–50, and 50+ years) and the final OL dose (50/75, 100, and 150 mg eq.).
Rating-based assessment of EPS-related events
In addition to spontaneously reported EPS-related TEAEs, other safety parameters related to EPS severity were assessed using the Abnormal Involuntary Movement Scale (AIMS), Simpson-Angus Scale (SAS), and Barnes Akathisia Rating Scale (BARS) scores at baseline, every 12 weeks, and end of the study (EOS). The incidence rates of these rating-based EPS (RB-EPS) were assessed at DB end point and compared in patients in the PP3M and PP1M treatment groups for 1) parkinsonism as the percentage of patients with an SAS total score of ≥0.3 at any time; 2) akathisia as the percentage of patients with a BARS global clinical rating score of ≥2 at any time; and 3) dyskinesia as the percentage of patients with a score of ≥3 on any of the first seven items or a score of ≥2 on two or more of any of the first seven items of AIMS. The identification of RB-EPS was based on an EPS rating scale score that occurred after the first injection during each phase meeting the EPS criteria as defined earlier.
Use of anti-EPS medication
Use of anti-EPS medication to treat EPS is an indirect measure of clinically relevant EPS-related TEAEs. Data on concomitant medications, reasons for treatment discontinuation, and reported AEs were used to identify onset and to identify the occurrence of any acute dystonic reactions. In the current post hoc analysis, it was calculated as the proportion of patients in each treatment group who had at least one dose of an anti-EPS medication (eg, trihexyphenidyl, benztropine, and biperiden) or an antihistamine with ancillary anticholinergic activity (eg, diphenhydramine and hydroxyzine) during the study.
Statistical analyses
Overall incidences of EPS-related TEAEs, TTO, and TTR were analyzed using descriptive statistics and Kaplan–Meier estimates. For patients who had >1 event for a specific EPS-related TEAE, only the event for that specific EPS-related TEAE with the earliest onset was included in the TTO analysis. For patients who had >1 event for a specified group of EPS-related TEAEs, only the event for that specified EPS-related TEAE with longest TTR was included in the TTR analysis. For any TEAE that did not resolve during a phase, the TTR was censored using the end date of that phase. The TTO and TTR of EPS-related TEAEs of patients in the OL phase were calculated using the OL intent-to-treat (OL-ITT) analysis set (all patients who received at least one dose of PP1M during the OL phase), and for patients in the DB phase they were calculated using the safety analysis set (all patients who received at least one dose of PP3M or PP1M during the DB phase).
Results
Demographic and baseline characteristics
The demographic and baseline characteristics of patients participating in the study have been described in detail previously.10 Overall, 1,429 patients with schizophrenia were enrolled and dosed in the OL phase, and of these, 1,016 (71%) were randomized (PP3M: n=504; PP1M: n=512) in the DB phase (safety analysis set). Similar percentages of patients in both groups completed the DB phase, with withdrawal of consent being the most common reason for discontinuation in the DB phase.10 The demographic and baseline characteristics were similar between PP3M- and PP1M-treated patients in the DB phase.10
Spontaneously reported EPS-related TEAEs
There were 180 patients who had at least one spontaneously reported EPS-related TEAE during the OL phase. During the DB phase, there were 42 patients in the PP3M treatment group and 38 patients in the PP1M treatment group with spontaneously reported EPS-related TEAEs. The most common EPS-related TEAEs in the OL phase were hyperkinesia (n=92 [6.4%]) and parkinsonism (n=68 [4.8%]; Table 1). In the DB phase, the incidence of EPS-related TEAEs was similar between the PP3M and PP1M groups (PP3M: n=42 [8.3%]; PP1M: n=38 [7.4%]; Table 1), except tremor (PP3M: n=9 [1.8%]; PP1M: n=3 [0.6%]) and dystonia (PP3M: n=0; PP1M: n=4 [0.8%]; Table 1).
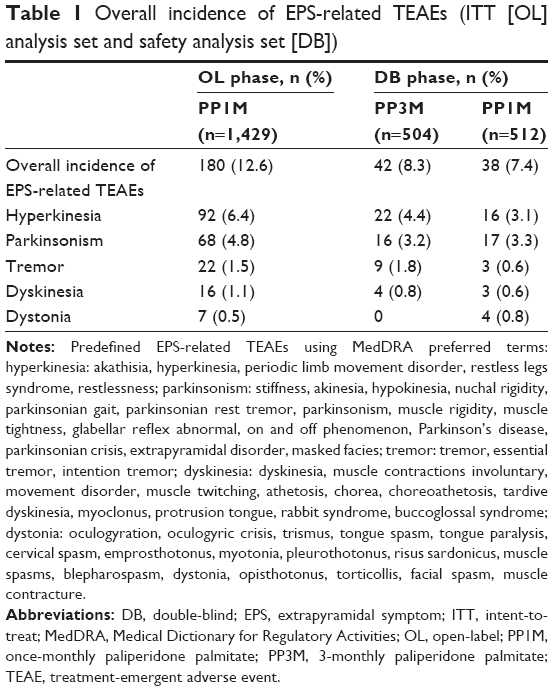 | Table 1 Overall incidence of EPS-related TEAEs (ITT [OL] analysis set and safety analysis set [DB]) |
Rating-based EPS-related TEAEs
During the OL phase, 53 (4%) patients experienced parkinsonism (as determined by SAS global score), 49 (3%) patients experienced akathisia (as determined by BARS), and nine (1%) patients experienced dyskinesia (as determined by AIMS). During the DB phase, both the PP3M and PP1M treatment groups had similar rates of RB-EPS symptoms assessed using the rating scales: parkinsonism (patients with SAS score >0.3): PP3M, 15 (3%); PP1M, 15 (3%); akathisia (patients with BARS score ≥2): PP3M, 22 (4%); PP1M, 18 (4%); and dyskinesia (patients with AIMS ≥2): 4 (1%), each group.
The median AIMS total scores and the median SAS global scores were 0 (none) at DB baseline and at DB end point in both the PP3M and PP1M treatment groups. The percentages of patients with akathisia rated as absent at DB baseline and DB end point in both the treatment groups were similar (DB baseline: 92.9% in both groups; DB end point: 92.9% and 92.1% in the PP3M and PP1M groups, respectively). A similar observation was noted for patients having questionable or mild akathisia at both DB baseline and DB end point. No patient in either treatment group had marked akathisia or severe akathisia at DB baseline or end point.
One patient treated with PP1M (75 mg eq. during both the OL [after the initiation doses] and DB phases) experienced a TEAE of dyskinesia of mild severity on day 83 that resolved on day 121 and a second TEAE of dyskinesia of mild severity on day 274 that resolved on day 363. The AIMS total score was 0 at screening and baseline, 2 on day 92, 1 on day 99 and 120, 0 on day 205, 5 on day 274, 0 on day 372, and 2 on day 457 (the day the patient completed the study without a relapse). The AIMS item 8 (severity of abnormal movements, 0–4 score) score was 0 at screening, baseline, day 66, and day 205 and was 1 on day 92, day 99, day 120, day 288, and day 457. The dyskinesia was resolved prior to the end of the study.
EPSs based on the use of anticholinergic medication
In the OL phase, 254 out of 1,429 patients (18%) used anticholinergic medication. During the DB phase, both the PP3M and PP1M treatment groups had similar use of anticholinergic medication (90/504 [18%] vs 83/512 [16%]).
TTO and TTR of EPS-related TEAEs
Among patients who had at least one spontaneously reported EPS-related TEAE, the overall median TTO and TTR values were comparable between the PP3M and PP1M formulations during the DB phase. The median TTO of all EPS-related TEAEs was 17 days (range: 1–120) in the OL phase and was 115 days (range: 1–323) and 98.5 days (range: 1–322) in the PP3M- and PP1M-treated groups, respectively, during the DB phase (Table 2). The median TTR of all EPS-related TEAEs was 36.5 days (range: 1–127 days) in the OL phase; median TTR was 91 days (range: 1–336) in PP3M-treated patients and was 85.5 days (range: 1–337 days) in the PP1M-treated patients in the DB phase (Table 2). There was no clear age-related trend or dose-related trend in the TTO and TTR of EPS-related TEAEs in subgroups of patients receiving either PP1M or PP3M (Tables 3 and 4).
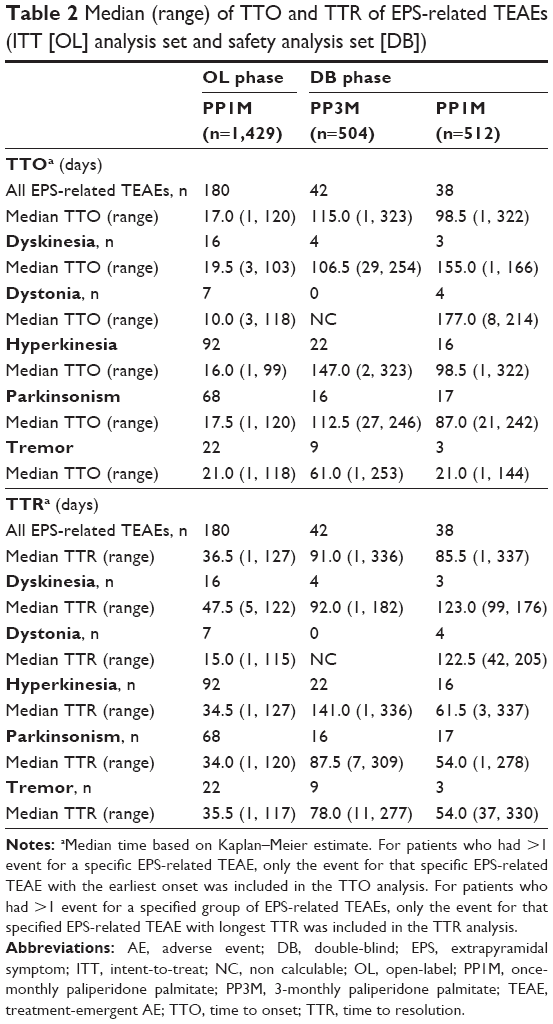 | Table 2 Median (range) of TTO and TTR of EPS-related TEAEs (ITT [OL] analysis set and safety analysis set [DB]) |
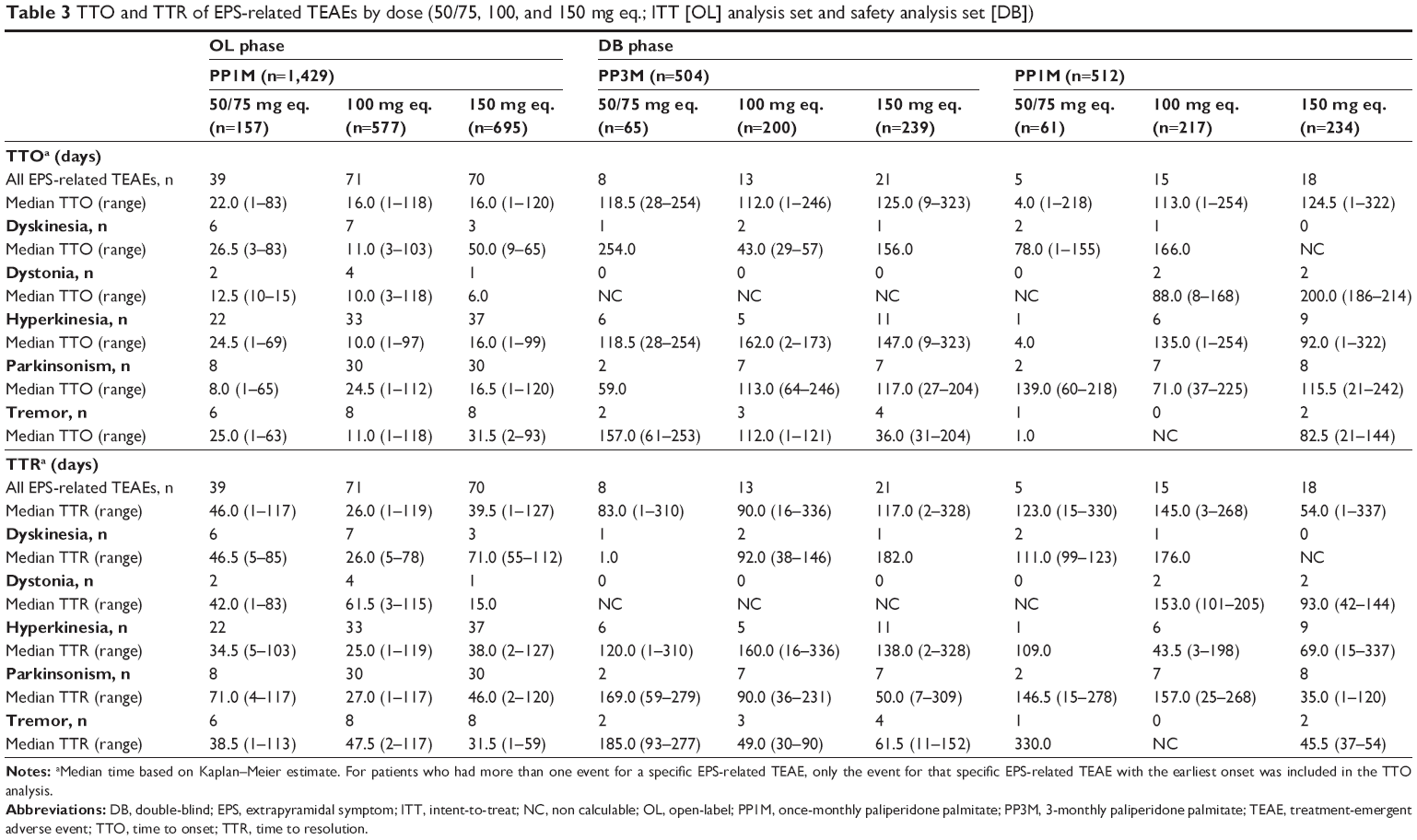 | Table 3 TTO and TTR of EPS-related TEAEs by dose (50/75, 100, and 150 mg eq.; ITT [OL] analysis set and safety analysis set [DB]) |
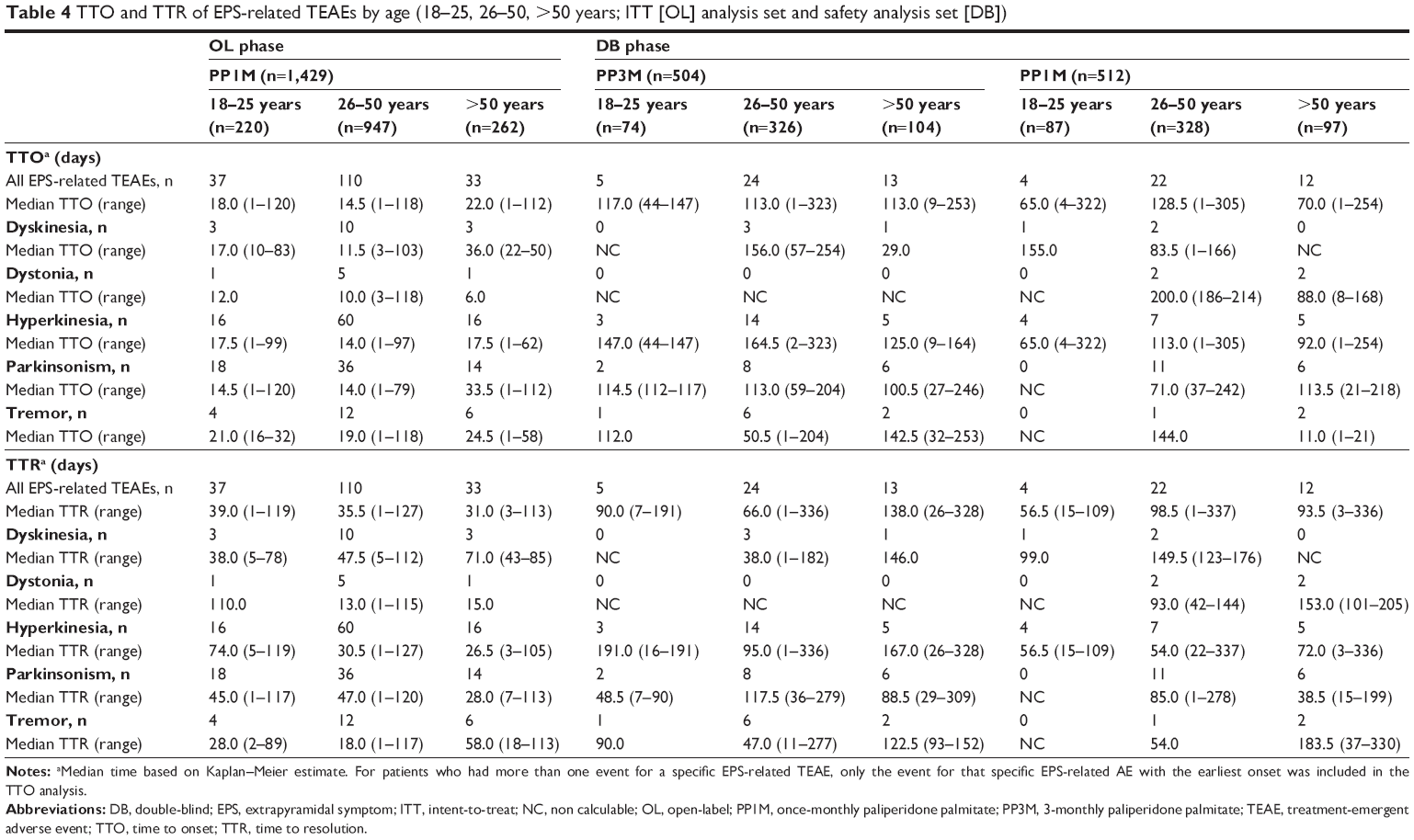 | Table 4 TTO and TTR of EPS-related TEAEs by age (18–25, 26–50, >50 years; ITT [OL] analysis set and safety analysis set [DB]) |
EPS-related TEAEs leading to study drug discontinuation
Seven patients (0.5%) discontinued the study during the OL PP1M phase due to akathisia, two patients discontinued the study due to dyskinesia, while one patient discontinued the study due to restlessness and muscle rigidity (both events in same patient). One patient randomized to PP1M treatment had EPS-related TEAE of restlessness during the DB phase that led to study discontinuation. One patient in the PP3M group and two patients in the PP1M group discontinued the study due to akathisia, while in the PP3M group one patient discontinued the study due to extrapyramidal disorder and tardive dyskinesia during the DB phase.
The patient who was discontinued due to tardive dyskinesia was treated with PP3M (263 mg eq. during the DB phase) had TEAE of tardive dyskinesia of moderate severity on day 373. The patient had an AIMS total score of “0” reported for all visits until day 373: AIMS total score on day 373 was 6. The AIMS total score was reported as “0” at the follow-up visit on day 465 when the TEAE of tardive dyskinesia was considered resolved. This patient was withdrawn from the study due to the AE on day 373. The tardive dyskinesia was resolved prior to the end of the study, suggesting that the event may not be tardive dyskinesia in nature due to the rapid resolution while the patient was still exposed to significant levels of medication.
Another patient had tardive dyskinesia of moderate severity reported on day 9 of the OL phase, which continued until the end of the study (the patient was treated with 150 mg eq. of PP1M during the DB phase and completed the study without relapse on day 450). The patient had an AIMS total score of 4 at screening and baseline and a score of 1 for all subsequent visits through the end of the study.
Discussion
The PP1M formulation has robust therapeutic evidence for efficacy in alleviating symptoms and delaying time to relapses in schizophrenia.11–15 In two large Phase III studies, the PP3M formulation has demonstrated low relapse rates (PP3M: n=37, 8%; PP1M: n=45, 9%) and non-inferiority to PP1M formulation and significantly delayed time to relapse vs placebo.9,10 Although atypical antipsychotics have a lower propensity to cause EPS-related TEAEs, available agents have heterogeneous safety and tolerability profiles, and the incidences of EPS events have been reported with several atypical antipsychotic agents, including paliperidone formulations.16
The objective of this post hoc descriptive analysis was to compare EPS-related safety data between PP3M and PP1M formulations. The results suggest that PP3M, which is dosed at 3.5 times the corresponding PP1M dosage strength, is not associated with higher EPS rates compared with PP1M in patients with schizophrenia. This is consistent with PK modeling that demonstrated similar plasma exposure between equivalent doses of the two formulations.17,18 During the DB phase, EPS-related TEAE rates were generally numerically lower for both PP3M and PP1M compared with the first 17 weeks (OL phase), consistent with previous observations of EPS-related TEAE rates for PP1M.9,12,19 There were no consistent, substantial, or significant differences in the overall incidence of EPS-related TEAEs, TTO, and TTR between the PP3M- and PP1M-treated groups. The higher rates of dystonia in the PP1M group could likely be attributed to the difference in peak plasma level which is attained faster in PP1M vs PP3M. On the other hand, the higher incidence of tremor in the PP3M group can be correlated with the higher and longer-lasting plasma levels of PP3M vs PP1M. Study withdrawal rates due to EPS-related TEAEs were low and comparable for both treatments. This is noteworthy as PP3M has a longer apparent half-life (approximately 2–4 months) with plasma exposure almost 70% more than that of PP1M.8 Peak drug plasma concentrations were achieved within 30–33 days after a single injection of PP3M over the dose range of 175–525 mg eq.17 Yet, the incidence of EPS-related TEAEs was similar between the PP3M and PP1M treatment groups; furthermore, TTO and TTR of EPS were also not meaningfully longer for PP1M.
No clear dose-related, age, or racial differences in TTO and TTR of EPS-related TEAEs were identified between the two treatments. Typically, dystonia and parkinsonism are expected at higher rates in younger patients with less total antipsychotic exposure, while TD is associated with older age and longer total antipsychotic exposure. However, in the current study, EPS-related TEAEs were observed at low rates in a wide range of ages in both the PP3M and PP1M treatment groups; it was thus difficult to obtain the characteristics of the expression age in this population.
One limitation is that prior antipsychotic medication use was not controlled and could have been different between patients treated with PP3M and PP1M. In addition, putative risk factors for EPS-related TEAEs, such as family history of primary movement disorders, duration of exposure to antipsychotics, and substance abuse, were not explored in this analysis.20–22 Hence, the EPS tolerability data in the current study cannot specify an individual patient’s risk for developing EPS in clinical settings. Another likely limitation is that since EPS-related TEAEs were categorized based on MedDRA terms, it was not possible to distinguish benign essential tremors from parkinsonian tremors, which may have very distinct clinical and physiologic implications. For assessing the use of anti-EPS medication, only anticholinergic and antihistamine medications were examined in this analysis. Other medications, such as beta-blockers and benzodiazepines, while commonly used to treat akathisia, are also used to treat other conditions, making it difficult to interpret the rate of use of these medications for treating EPS-related TEAEs. Furthermore, this post hoc analysis did not address the severity of the EPS-related TEAEs examined and was not powered to address the issue of dose response. Patients with earlier and more severe EPS-related TEAEs may have discontinued the study during the first 17-week OL phase (0.5%) and not been included in the analysis. In addition, these patients who entered the DB treatment phase were clinically stabilized and tolerated PP1M for 4 months and were potentially less likely to experience EPS-related TEAE. Formal assessment of EPS with rating scales only occurred every 12 weeks during the DB period. This occurred during the simultaneous troughs of PP1M and PP3M. Although spontaneous reports of TEAEs occurred at any point in the study, it is possible that more EPS-related TEAEs would have been detected if rating scales were used around peak levels (1 week after PP1M dosing, 4 weeks after PP3M dosing). Finally, unlike the primary study, the current analysis was not powered and did not define a margin for determining the non-inferiority of PP3M to PP1M in terms of EPS-related safety.
Conclusion
The overall incidence of EPS-related TEAEs and their TTO and TTR were similar in patients treated with PP3M and PP1M. Subgroup analyses did not reveal any effect of dose or age on the TTO or TTR of the EPS-related TEAEs for any of the two treatments. Having similar or lower EPS-related TEAEs and less frequent dosing may encourage the use of PP3M to increase the overall adherence rates in patients with schizophrenia and will allow more time for physicians, caregivers, and patients to treat other issues frequently constraining a good quality of life for these patients, such as psychosocial rehabilitation, substance abuse treatment, health maintenance, and vocational rehabilitation.
Acknowledgments
Shruti Shah, PhD, and Priya Ganpathy, MPharm, ISMPP CMPP™ (SIRO Clinpharm Pvt. Ltd.), provided writing assistance and Ellen Baum, PhD (Janssen Research & Development, LLC), provided additional editorial support for this manuscript. The study was supported by funding from Janssen Research & Development, LLC. The sponsor also provided funding for the development of this manuscript. The authors also thank the study participants, without whom this study would never have been accomplished, and all the investigators for their participation in this study. Posters were presented at the fifth Biennial Schizophrenia International Research Society (SIRS) Conference, Florence, Italy, April 2–6, 2016; Society of Biological Psychiatry’s (SOBP) 71st annual meeting, May 12–14, 2016, Atlanta, GA, USA; 30th CINP World Congress, International College of Neuropsychopharmacology, Seoul, Republic of Korea, July 3–5, 2016.
Author contributions
MM, IN, AJS, DWH, DN, EK, and SG contributed to study design, data collection, analysis, and interpretation. IN was responsible for the statistical analyses. All the authors had full access to all the data in the study and took responsibility for the integrity of the data and the accuracy of the data analysis. All the authors meet the ICMJE criteria, and all those who fulfilled those criteria are listed as authors. All the authors provided direction and comments on the manuscript, critically revised the proof, made the final decision about where to publish these data, and approved submission to this journal.
Disclosure
Drs MM, IN, AJS, DWH, and SG are employees of Janssen Research & Development, LLC (a Johnson & Johnson company). Drs DN and EK are employees of Janssen Scientific Affairs, LLC. All the authors hold stock in Johnson & Johnson, parent company of Janssen. The authors report no other conflicts of interest in this work.
References
Gopal S, Liu Y, Alphs L, Savitz A, Nuamah I, Hough D. Incidence and time course of extrapyramidal symptoms with oral and long-acting injectable paliperidone: a posthoc pooled analysis of seven randomized controlled studies. Neuropsychiatr Dis Treat. 2013;9:1381–1392. | ||
Leo RJ, Regno PD. Atypical Antipsychotic Use in the Treatment of Psychosis in Primary Care. Prim Care Companion J Clin Psychiatry. 2000;2(6):194–204. | ||
Barnes TR, Mcphillips MA. Critical analysis and comparison of the side-effect and safety profiles of the new antipsychotics. Br J Psychiatry Suppl. 1999;38(38):34–43. | ||
Invega Sustenna™ Prescribing Information; 2015. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207946s000lbl.pdf. Accessed August 17, 2017. | ||
Invega Trinza™ Prescribing Information; 2015. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207946s000lbl.pdf. Accessed August 17, 2017. | ||
Trevicta [homepage on the Internet]. Summary of Product Characteristics. Available from: https://www.medicines.org.uk/emc/product/7230/smpc. Accessed August 17, 2017. | ||
Carpiniello B, Pinna F. Critical appraisal of 3-monthly paliperidone depot injections in the treatment of schizophrenia. Drug Des Devel Ther. 2016;10:1731–1742. | ||
Ravenstijn P, Remmerie B, Savitz A, et al. Pharmacokinetics, safety, and tolerability of paliperidone palmitate 3-month formulation in patients with schizophrenia: A phase-1, single-dose, randomized, open-label study. J Clin Pharmacol. 2016;56(3):330–339. | ||
Berwaerts J, Liu Y, Gopal S, et al. Efficacy and Safety of the 3-Month Formulation of Paliperidone Palmitate vs Placebo for Relapse Prevention of Schizophrenia: A Randomized Clinical Trial. JAMA Psychiatry. 2015;72(8):830–839. | ||
Savitz AJ, Xu H, Gopal S, et al. Efficacy and Safety of Paliperidone Palmitate 3-Month Formulation for Patients with Schizophrenia: A Randomized, Multicenter, Double-Blind, Noninferiority Study. Int J Neuropsychopharmacol. 2016;19(7):pyw018. | ||
Gopal S, Vijapurkar U, Lim P, Morozova M, Eerdekens M, Hough D. A 52-week open-label study of the safety and tolerability of paliperidone palmitate in patients with schizophrenia. J Psychopharmacol. 2011;25(5):685–697. | ||
Hough D, Gopal S, Vijapurkar U, Lim P, Morozova M, Eerdekens M. Paliperidone palmitate maintenance treatment in delaying the time-to-relapse in patients with schizophrenia: a randomized, double-blind, placebo-controlled study. Schizophr Res. 2010;116(2–3):107–117. | ||
Marcus SC, Zummo J, Pettit AR, Stoddard J, Doshi JA. Antipsychotic Adherence and Rehospitalization in Schizophrenia Patients Receiving Oral Versus Long-Acting Injectable Antipsychotics Following Hospital Discharge. J Manag Care Spec Pharm. 2015;21(9):754–769. | ||
Pandina GJ, Lindenmayer JP, Lull J, et al. A randomized, placebo-controlled study to assess the efficacy and safety of 3 doses of paliperidone palmitate in adults with acutely exacerbated schizophrenia. J Clin Psychopharmacol. 2010;30(3):235–244. | ||
Zhang F, Si T, Chiou CF, et al. Efficacy, safety, and impact on hospitalizations of paliperidone palmitate in recent-onset schizophrenia. Neuropsychiatr Dis Treat. 2015;11:657–668. | ||
Cha DS, Mcintyre RS. Treatment-emergent adverse events associated with atypical antipsychotics. Expert Opin Pharmacother. 2012;13(11):1587–1598. | ||
Magnusson MO, Samtani MN, Plan EL, et al. Population Pharmacokinetics of a Novel Once-Every 3 Months Intramuscular Formulation of Paliperidone Palmitate in Patients with Schizophrenia. Clin Pharmacokinet. 2017;56(4):421–433. | ||
Samtani MN, Vermeulen A, Stuyckens K. Population pharmacokinetics of intramuscular paliperidone palmitate in patients with schizophrenia: a novel once-monthly, long-acting formulation of an atypical antipsychotic. Clin Pharmacokinet. 2009;48(9):585–600. | ||
Fu DJ, Turkoz I, Simonson RB, et al. Paliperidone palmitate once-monthly reduces risk of relapse of psychotic, depressive, and manic symptoms and maintains functioning in a double-blind, randomized study of schizoaffective disorder. J Clin Psychiatry. 2015;76(3):253–262. | ||
Kasten M, Brüggemann N, König IR, et al. Risk for antipsychotic-induced extrapyramidal symptoms: influence of family history and genetic susceptibility. Psychopharmacology. 2011;214(3):729–736. | ||
Lencer R, Eismann G, Kasten M, et al. Family history of primary movement disorders as a predictor for neuroleptic-induced extrapyramidal symptoms. Br J Psychiatry. 2004;185:465–471. | ||
Potvin S, Blanchet P, Stip E. Substance abuse is associated with increased extrapyramidal symptoms in schizophrenia: a meta-analysis. Schizophr Res. 2009;113(2–3):181–188. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.