")
Back to Journals » International Journal of General Medicine » Volume 14
Three Novel Homozygous Mutations of the SLC12A3 Gene in a Gitelman Syndrome Patient
Authors Zhong M, Zhai Z, Zhou X , Sun J, Chen H, Lu W
Received 24 February 2021
Accepted for publication 30 April 2021
Published 24 May 2021 Volume 2021:14 Pages 1999—2002
DOI https://doi.org/10.2147/IJGM.S308246
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Mei Zhong,1,* Zhenwei Zhai,2,* Xing Zhou,1 Jingxia Sun,1 Hui Chen,1 Wensheng Lu1
1Department of Endocrinology, The Peoples Hospital of Guangxi Zhuang Autonomous Region, Guangxi, People’s Republic of China; 2Department of Endocrinology, Tongde Hospital of Yuncheng, Changzhi Medical College, Shanxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wensheng Lu Tel +86 17634045555
Email [email protected]
Aim: Gitelman syndrome (GS) is an autosomal recessive disorder characterized by hypokalemic metabolic alkalosis. In this study, we investigated the clinical presentation and sequenced 26 exons of SLC12A3 gene in a patient with a clinical suspicion of GS.
Methods: Clinical work-up including clinical examination, electrocardiography (ECG), chest X-ray, bone mineral density (BMD), and ultrasound examination was conducted and all exons of SLC12A3 gene were analyzed by whole-exome sequencing.
Results: The patient showed hypokalemia, hypomagnesemia, and metabolic alkalosis and was found to have four novel homozygous missense mutations including one known mutation (c.1456 G>A in exon 12) and three novel mutations (c.366A > G in exon 2, c.791C > G in exon 6 and c.1027C > T in exon 8).
Conclusion: Four mutation sites of SLC12A3 gene were found in the patient, three of which have not been reported before. These results may be useful for better understanding the function of this gene and can assist clinicians with treatment decision-making.
Keywords: Gitelman syndrome, clinical characteristics, SLC12A3 gene, exome sequencing, gene mutation
Introduction
Gitelman syndrome (GS) is an autosomal recessive inherited disease initially reported in 1996 and is a rare renal tubular disorder with a prevalence of 1:40,000.1,2 GS is closely associated with the mutations of SLC12A3 gene coding for the thiazide-sensitive sodium-chloride cotransporter (NCCT) of the distal convoluted tubule (DCT).3 To date, more than 400 mutations of SLC12A3 have been identified in GS.4–8 Most of the mutations are missense, splice-site, nonsense, frameshift, deletions, and insertion mutations.9
The main clinical manifestations of GS are hypokalemic metabolic alkalosis, hypomagnesemia, hypocalciuria and renin-angiotensin-aldosterone system (RAAS) activation. Besides, patients with GS may present with spasticity, muscle weakness, paresthesia, numbness, polyuria, and palpitation.10 However, GS lacks specific clinical manifestations and is easily confused with other diseases such as Bartter syndrome, renal tubular acidosis, and primary aldosteronism.11,12 In addition to history taking and clinical work-up, genetic testing is an important and effective tool for diagnosing GS in clinical practice.13 Here, our present study aimed to analyze the clinical features and genetic characteristics of a Chinese female patient with GS.
Methods
Subject and Clinical Characteristics
A 60-year-old female patient was admitted to the Department of Endocrinology in Tongde Hospital of Shanxi province because of recurrent hypokalemia. She had no obvious clinical symptoms such as palpitations, shortness of breath, muscle weakness of the lower limbs, vomiting, nausea, anorexia, diarrhea, polyphagia and emaciation and denied a history of chronic kidney disease. She was treated with potassium citrate or potassium chloride, but she had no history of other drugs such as diuretics, proton pump inhibitors, and anti-tumor agents.
Clinical Examinations
The patient underwent detailed systemic physical examinations. Fasting blood samples and urine samples were obtained. The electrolytes of the blood and urine, plasma angiotensin, plasma renin activity, and plasma aldosterone were analyzed at the central chemistry laboratory of Tongde Hospital of Shanxi province. Besides, ECG, chest X-ray, bone mineral density (BMD) examination and ultrasound examination were conducted.
Genetic Analysis
The diagnosis of Gitelman syndrome was based on the clinical symptoms, biochemical measurements and analysis of genetic mutations of SLC12A3 gene. Genomic DNA was extracted from peripheral blood samples of the female patient using a nucleic acid extraction kit (NO. BST01051, BaiO Technology Co., Ltd, Shanghai, China) according to the manufacturer’s protocol. The SLC12A3 gene was screened for mutations using Sanger sequencing. The nucleotide sequences of the PCR products were aligned to the UCSC database using SnapGee software (v3.2.1)
Results
Baseline Clinical Characteristics of the Subject
Blood pressure, heart rate, and body mass index (BMI) of the patient were 128/70 mmHg, 76 beats per minute, and 26.7 kg/m2, respectively. Systemic physical examinations showed no abnormalities. The results of laboratory tests of the patient are listed in Table 1. Laboratory assays revealed hypokalemia, hypomagnesemia, hypercalcemia, and metabolic alkalosis. Furthermore, the patient had elevated parathyroid hormone (PTH) and plasma renin activity and angiotensin II. 24-h urine analysis showed that the levels of urinary potassium and urinary calcium were normal. ECG revealed normal sinus rhythm, ST segment and T-waves abnormality but had no prolongation of the QT interval. The BMD data showed there was a significant decrease at the left forearm. Chest X-ray and ultrasound (thyroid, carotid, heart, abdomen, and pelvic cavity) did not show any obvious abnormalities (data not shown).
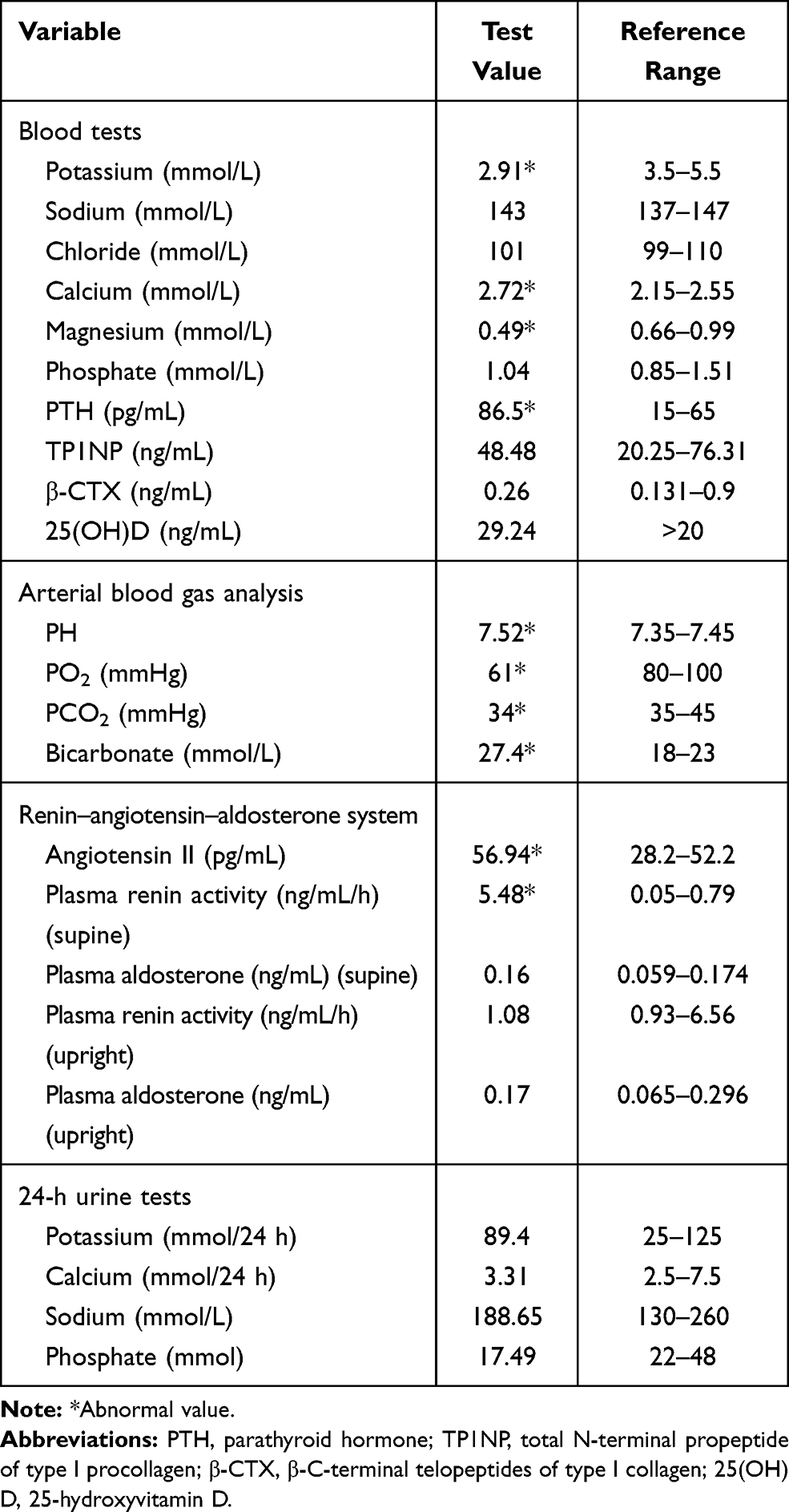 |
Table 1 Laboratory Tests of the Subject |
Genetic Analysis
Sanger sequencing of SLC12A3 gene was performed in the subject. Genetic analysis showed 4 mutations in the exons of SLC12A3 gene: c.366A > G in exon 2, c.791C > G in exon 6, c.1027C > T in exon 8, and c1456G>A in exon 12 (Figure 1).
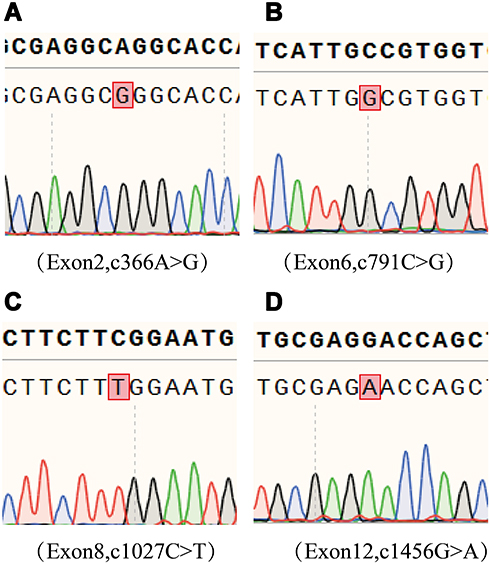 |
Figure 1 Genetic analysis of the SLC12A3 gene. (A) c.366A > G in Exon 2. (B) c.791C > G in Exon 6. (C) c.1027C > T in Exon 8 and (D) c.1456 G > A in Exon 12. The mutant nucleotides are marked in the red frames. |
Clinical Treatment of the Subject
After the diagnosis, spironolactone and potassium citrate were used to treat hypokalemia and potassium magnesium aspartate was used to treat hypomagnesaemia. The patient was discharged with normokalemia and no other discomfort following one week of treatment.
Discussion
In the present study, we reported the case of a 60-year-old Chinese female patient with GS. The biochemical examination showed hypokalemia, metabolic alkalosis, hypomagnesemia, hypercalcemia, hyperreninemia, elevated angiotensin II, and PTH levels. ECG showed normal sinus rhythm, ST segment and T-waves abnormality but had no prolongation of the QT interval. Besides, BMD was decreased in the left forearm. Genetic analysis identified four mutations of SLC12A3 gene, c.366A > G in exon 2, c.791C > G in exon 6, c.1027C > T in exon 7, and c1456G>A in exon 12. The treatment with supplements of potassium and magnesium improved hypokalemia and hypomagnesemia.
GS is a rare inherited salt-wasting disorder characterized by hypokalemic metabolic alkalosis with hypomagnesemia, hypocalciuria and RAAS activation. GS is caused by mutations in SLC12A3 gene coding for the thiazide-sensitive NCCT of the DCT, which leads to a decrease in sodium reabsorption, increases potassium and hydrogen excretion and therefore develop hypokalemic metabolic alkalosis.14 The enhanced passive Ca2+ transport in the proximal tubule leads to hypocalciuria15 and the downregulation of the epithelial Mg2+ channel transient receptor potential channel subfamily M, member 6 (Trpm6) is a possible mechanism involved in hypomagnesemia.16 The mechanism of hypocalciuria is uncertain, but some studies have found that one reason for it may be hypovolemia. Meanwhile, hypovolemia activates RAAS.17,18 The patient had hypokalemia, metabolic alkalosis, hypomagnesemia, hyperreninemia, and elevated angiotensin II level, which was consistent with the clinical manifestations of GS. In the study, the elderly female had decreased BMD. Postmenopausal women are prone to develop hypocalcemia and postmenopausal osteoporosis, which is caused by estrogen deficiency after menopause. Long-term hypocalcemia may overstimulate the parathyroid gland and lead to secondary hyperparathyroidism.
Genetic identification is the golden standard for the diagnosis of GS. We identified compound mutations of SLC12A3, c.366A > G in exon 2, c.791C > G in exon 6, c.1027C > T in exon 7, and c1456G>A in exon 12. The c1456G>A in exon 12 is reported as a hotspot mutation of SLC12A3. A heterozygous mutation, c.366A > G in the gene has been reported. But c.366A > G in exon 2 was a homozygous mutation in this study. Moreover, c.791C > G and c.1027C > T are two novel mutations. Hence, we performed a complementary study on the mutations of SLC12A3.
GS and Bartter syndrome (BS) show extremely similar clinical and laboratory manifestations including hypokalemia, metabolic alkalosis, hyperreninemia, and hyperaldosteronemia. But BS presents with an early age of onset and exhibits apparent clinical symptoms.12 Simultaneously, BS is closely related to the mutations of CLCNKB gene. They can be differentiated by clinical manifestation and genetic tests.19,20
Our present study has several limitations. First, the mutation was detected in only one patient but not in pedigree. Further research should be performed in the pedigree. Second, a further exploration is needed to find the correlation between genotype and phenotype and then provide better understanding of GS. Moreover, more experiments are needed to reveal the underlying molecular mechanism of GS.
Conclusions
Overall, our study identified four mutations of SLC12A3 gene in a Chinese female patient and three of the mutations were novel. These findings might be useful for better understanding the function of this gene and aid with diagnosis and treatment decisions.
Ethics Approval and Consent to Participate
The study was conducted in compliance with the Declaration of Helsinki. The protocol was approved by the Ethics Committee of Tongde Hospital of Shanxi province. The patient provided informed consent for the case details to be published.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from the Chinese National Natural Science Foundation [No. 81560044, No. 30860113], the Appropriate Technology for Medical Health Research and Development Projects of Guangxi [S201422-01] and Health Research Project of Shanxi [2019165].
Disclosure
The authors declare that they have no competing interests.
References
1. Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Phys. 1966;79:221–235.
2. Knoers NV, Levtchenko EN. Gitelman Syndrome. Orphanet J Rare Dis. 2008;3:22. doi:10.1186/1750-1172-3-22
3. Mastroianni N, Bettinelli A, Bianchetti M, et al. Novel molecular variants of the Na-cl cotransporter gene are responsible for Gitelman syndrome. Am J Hum Genet. 1996;59(5):1019–1026.
4. Glaudemans B, Yntema HG, San-Cristobal P, et al. Novel NCC mutants and functional analysis in a new cohort of patients with Gitelman syndrome. Eur J Hum Genet. 2012;20:263–270. doi:10.1038/ejhg.2011.189
5. Ma J, Ren H, Lin L, et al. Genetic features of Chinese patients with Gitelman syndrome: sixteen novel SLC12A3 mutations identified in a new cohort. Am J Nephrol. 2016;44:113–121. doi:10.1159/000447366
6. Gug C, Mihaescu A, Mozos I. Two mutations in the thiazide sensitive NaCl co-transporter gene in a Romanian Gitelman syndrome patient: case report. Ther Clin Risk Manag. 2018;14:149–155. doi:10.2147/TCRM.S150483
7. Vargas-Poussou R, Dahan K, Kahila D, et al. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol. 2011;22:693–703. doi:10.1681/ASN.2010090907
8. Reissinger A, Ludwig M, Utsch B, et al. Novel NCCT gene mutations as a cause of Gitelman’s syndrome and a systematic review of mutant and polymorphic NCCT alleles. Kidney Blood Press Res. 2002;25(6):354–362. doi:10.1159/000068695
9. Lu Q, Zhang Y, Song C, et al. A novel SLC12A3 gene homozygous mutation of Gitelman syndrome in an Asian pedigree and literature review. J Endocrinol Investig. 2016;39(3):333–340. doi:10.1007/s40618-015-0371-y
10. Munoz EV, Chang Q, Bindels RJ, et al. Gitelman syndrome: towards genotypephenotype correlations. Pediatr Nephrol. 2007;22(3):326–332. doi:10.1007/s00467-006-0321-1
11. Fedeli GGC, Cosmai ML, Badalamenti S, et al. Gitelman syndrome: pathophysiological and clinical aspects. Q J Med. 2010;103(10):741–748. doi:10.1093/qjmed/hcq123
12. Matsunoshita N, Nozu K, Shono A, et al. Differential diagnosis of Bartter syndrome, Gitelman syndrome, and pseudo-Bartter/Gitelman syndrome based on clinical characteristics. Genet Med. 2016;18(2):180–188. doi:10.1038/gim.2015.56
13. Blanchard A, Bockenhauer D, Bolignano D, et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO)controversies conference. Kidney Int. 2017;91:24–33. doi:10.1016/j.kint.2016.09.046
14. Güvercin B, Kaynar K, Güler Ö, et al. In the presence of hypokalemia and hypomagnesemia; remember Gitelman syndrome. Hippokratia. 2019;23.
15. Reilly RF, Huang CL. The mechanism of hypocalciuria with NaCl cotransporter inhibition. Nat Rev Nephrol. 2011;7:669–674. doi:10.1038/nrneph.2011.138
16. Shahzad MA, Mukhtar M, Ahmed A, et al. Gitelman Syndrome: a rare cause of seizure disorder and a systematic review. Case Rep Med. 2019;2019:4204907. doi:10.1155/2019/4204907
17. Tseng MH, Yang SS, Hsu YJ, et al. Genotype, phenotype, and follow-up in Taiwanese patients with salt-losing tubulopathy associated with SLC12A3 mutation. J Clin Endocrinol Metab. 2012;97(8):E1478–1482. doi:10.1210/jc.2012-1707
18. Hsu YJ, Yang SS, Cheng CJ, et al. Thiazide-sensitive Na+−cl− Cotransporter (NCC) gene inactivation results in increased duodenal Ca2+ absorption,enhanced osteoblast differentiation and elevated bone mineral density. J Bone Miner Res. 2015;30(1):116–127. doi:10.1002/jbmr.2306
19. Simon DB, Karet FE, Hamdan JM, et al. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet. 1996;13(2):183–188. doi:10.1038/ng0696-183
20. Shaer AJ. Inherited primary renal tubular hypokalemic alkalosis: a review of Gitelman and Bartter syndromes. Am J Med Sci. 2001;322(6):316–332. doi:10.1097/00000441-200112000-00004
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.