")
Back to Journals » Drug Design, Development and Therapy » Volume 17
Therapeutic Efficacy of Boric Acid Treatment on Brain Tissue and Cognitive Functions in Rats with Experimental Alzheimer’s Disease
Authors Özdemir Ç , Arslan M , Küçük A , Yığman Z, Dursun AD
Received 3 March 2023
Accepted for publication 6 May 2023
Published 17 May 2023 Volume 2023:17 Pages 1453—1462
DOI https://doi.org/10.2147/DDDT.S405963
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Çağrı Özdemir,1 Mustafa Arslan,2– 4 Ayşegül Küçük,5 Zeynep Yığman,6,7 Ali Doğan Dursun8
1Mamak State Hospital Department of Anesthesiology and Reanimation, Ankara, Turkey; 2Gazi University Faculty of Medicine Department of Anesthesiology and Reanimation, Ankara, Turkey; 3Gazi University, Life Sciences Application and Research Center, Ankara, Turkey; 4Gazi University, Laboratory Animal Breeding and Experimental Research Center (GÜDAM), Ankara, Turkey; 5Kutahya Health Sciences University Faculty of Medicine, Department of Physiology, Kutahya, Turkey; 6Gazi University Faculty of Medicine, Department of Histology and Embryology, Ankara, Turkey; 7Gazi University Neuroscience and Neurotechnology Center of Excellence (NÖROM), Ankara, Turkey; 8Atılım University Faculty of Medicine Department of Physiology, Ankara, Turkey
Correspondence: Mustafa Arslan, Gazi University Faculty of Medicine, Department of Anesthesiology and Reanimation, Ankara, 06510, Türkiye, Email [email protected]
Introduction: Oxidative stress has an important role in the pathophysiology of Alzheimer’s disease (AD), the most common type of dementia. Boric acid (BA) contributes significantly to the protection of the brain by reducing lipid peroxidation and supporting antioxidant defense. We aimed to evaluate the therapeutic potential of BA treatment in AD rats.
Materials and Methods: Four groups were formed as Control (C), Alzheimer’s (A), Alzheimer’s + Boric acid (ABA), Boric acid (BA). Intracerebroventricular injection of Streptozotocin (STZ) was preferred to create an AD. After 4 weeks, BA was applied 3 times every other day. The Radial Arm Maze Test (RAMT) was used to evaluate memory and learning abilities. Biochemical and histopathological evaluations were made in the hippocampus.
Results: Initial RAMT inlet/outlet (I/O) numbers were similar. Two weeks after STZ injection, I/O numbers decreased in group A and ABA compared to group C and BA (p< 0.05). After the second BA application, I/O numbers increased in the ABA group compared to the A group (p< 0.05). In group A, PON-1, TOS and OSI levels were higher and TAS levels were lower than in groups BA and C. After BA treatment, PON-1 and OSI levels were lower in the ABA group than in the A group (p< 0.05). Although there was an increase in TAS value and a decrease in TOS, this did not make a statistical difference. The thickness of the pyramidal cell in CA1 and the granular cell layers in the dentate gyrus, and the number of intact and degenerated neurons in the pyramidal cell layer were similar between the groups.
Discussion: Significant improvement in learning and memory abilities after BA application is promising for AD.
Conclusion: These results show that BA application positively affects learning and memory abilities, and reduces oxidative stress. More extensive studies are required to evaluate histopathological efficacy.
Keywords: Alzheimer’s disease, boric acid, cognitive function, oxidative stress
Introduction
Alzheimer’s disease (AD) is the most common type of dementia that progresses with impairments in memory, personality, and cognitive functions. It is estimated that there are 50 million dementia patients worldwide, and this number is expected to rise to 152 million by 2050.1 Many experimental and clinical studies are carried out for the solution of this disease, which brings many financial and moral difficulties.
Due to the complex and multifactorial nature of the disease, its pathophysiology has not been clearly defined.1 Its important pathological markers are senile plaques containing amyloid β peptide (Aβ), neurofibrillary tangle and neuron death.2 The contribution of reactive oxygen species (ROS) in the pathogenesis of AD has also been emphasized. In fact, this is true for all neurodegenerative diseases. Inadequate antioxidant defense is as important as ROS production in disease progression. Neuronal collapse and oxidative stress are closely related to disease-specific Aβ deposition.3 Mutations associated with AD trigger oxidative stress by increasing both cerebral Aβ load and Aβ1-42 production.4 Imbalance in the clearance of Ap1-42 is also closely related to the oxidation of macromolecules such as protein, lipid and nucleic acid.5 Accumulation of this peptide at synapse junctions initiates the formation of ROS and reactive nitrogen species and causes an increase in malondialdehyde levels, a lipid peroxidation product.6 It has been reported that nuclear and mitochondrial DNA damage increases with increasing oxidation in AD patients.7
Boron-containing compounds are very common in nature and are most commonly found in nature as borate and boric acid (BA). BA, a weak Lewis acid, is the most common boron derivative found in biological systems due to its water solubility.8 It takes part in cell membrane integrity, signal transmission, mineral and hormone metabolism and enzymatic reactions.9–11 Its deficiency is important in the development of a number of diseases such as heart disease, stroke, glucose-related disorders and the aging process.11 BA can act as a protective agent in apoptotic processes by regulating the mitochondrial membrane potential as well as oxidative and inflammatory processes.12 Currently, there is no FDA or EMA approved therapeutic indication. However, as far as we know, there are very few studies evaluating the neuroprotective effects of BA on neurodegenerative diseases.
Different models have been described in the literature to create experimental AD (Intracerebroventricular (icv) Streptozotocin (STZ) model, Aluminum Chloride in the Rat Model, Cholinergic Dysfunction in Mouse Models, Transgenic mouse models, Cell culture models). Studies show that icv STZ injections are a valid model for impairing learning, memory and cognitive functions.13 Neuroinflammation, tangles, amyloid plaque deposition, and oxidative stress formation were demonstrated in this model.14,15 Therefore, STZ application has been widely used for the AD model.16
In this study, we aimed to evaluate the effect of BA treatment on learning function, memory level, oxidative stress and neuronal damage in rats in AD model.
Materials and Methods
Animals and Ethics Committee Permission
This research was conducted with the ethical approval of Gazi University Animal Experiments Ethics Committee (Ref. No. GUET-19-071). Accepted standards of the Guide for the Care and Use of Laboratory Animals were followed at all stages of the study. All phases of the study were conducted in agreement with the UK Animals (Scientific Procedures) Act 1986, ARRIVE guidelines and EU Directive for animal testing. Twenty-four male Wistar Albino rats weighing 400–500 gr, supplied by Gazi University Experimental Research Center, were used. Rats were kept under a reverse light dark cycle. Room temperature was maintained at 21±1°C and humidity at 45–55%. Rats were fed with standard pellets throughout the study. Drinking water was available ad libitum. Four groups were formed with 6 rats in each; Control (C), Boric acid group (BA), Boric acid-Alzheimer group (ABA) and Alzheimer group (A).
Alzheimer Model
The rats in Groups A and BA were treated under ketamine (50 mg/kg i.p., Ketalar, Parke-Davis, Eczacıbası, İstanbul, Turkey) and xylazine hydrochloride (10 mg/kg, i.p 2%; Alfazyne, Ege Vet, İzmir, Turkey) anesthesia. If there was a reaction to the painful stimulus, 20 mg.kg−1 ketamine was repeated. During the surgical procedure, rats were wrapped with cotton to maintain body temperature. A stereotactic hood was inserted under anesthesia. After the cranium was wiped with iodine solution, the skin was dissected to reach the bregma. The dura was entered 0.8 mm posterior to the bregma, ±1.4 mm lateral to the sagittal line, and 3.4 mm below the skull surface, guided by Paxinos and Watson’s brain atlas. The AD model was induced by icv injection of 3 mg/kg STZ, 10 µL per side.
Radial Arm Maze Test (RAMT) and Boric Acid Treatment
The device consisted of an eight-armed radial labyrinth made of plexiglass. A dimly lit and quiet room was preferred for the test. Each arm is 60 cm long, 10 cm wide and 15 cm high and extends radially from the central starting platform (35 cm in diameter). Four arms had food on the distal end and no food on the other four arms. Animals were placed one by one in the middle of the maze. During the RAMT trial, an animal must receive food from all four arms of the maze.17,18 Each test took 5 minutes. RAMT was applied to all groups at the beginning of the study and baseline data were recorded. This was repeated once a week for four weeks. Four weeks after the STZ injection, 200 mg/kg BA was injected intraperitoneally (i.p) into Group A and Group ABA every other day for 3 days. RAMT was repeated for all groups 1 day after each BA application to Group A and ABA. Following the completion of all cognitive tests, the rats were sacrificed by taking intracardiac blood under ketamine-xylazine anesthesia. The hippocampus was evaluated biochemically and histopathologically in the brain tissue.
Histopathological Analysis
Left hemispheres of rat brains were fixed in 10% neutral buffered formalin, and processed for paraffin embedding. The hemispheres were cut into sections five microns thick by means of a microtome. All sections were stained with hematoxylin and eosin (H&E) for histological evaluation. H&E-stained brain tissues were evaluated under a light microscope (Leica DM 4000B, Germany). For histopathological evaluation, images of the CA1 (Cornu Ammonis 1) area of the hippocampus and the dentate gyrus were captured at 400x magnification. The thickness of the pyramidal cell layer in the CA1 area and the granular cell layer of the dentate gyrus were measured. Also, intact and degenerated pyramidal cells in the CA1 area were counted.19,20
Biochemical Evaluations
Tissue Homogenization
Brain tissue collected in an Eppendorf tube was frozen in liquid nitrogen and stored at −80°C. Brain tissues were cut into pieces of 80–100 mg. The tissues were crushed in a bowl in the presence of liquid nitrogen, and the pulverized tissue was transferred to the homogenization tube (099C S3, Glas-Col). KCl solution was added to achieve 1/10 (w/v) dilution. A Glas-Col homogenizer was used for 2 minutes at 50 rpm to complete the process. The homogenates were transferred to eppendorf tubes and centrifuged at 3000 rpm for 10 minutes.
Assessment of TOS/TAS/OSI/PON
Brain TAS level was measured by fully automatic spectrophotometric method. Rel Assay Diagnostics kit (RelAssay Diagnostic®, Turkey) was used in the measurement. TAS level was calculated using the formula provided in the kit and expressed as mmol Trolox Eq/L.
Brain TOS level was measured by fully automatic spectrophotometric method. Rel Assay Diagnostics kit (RelAssay Diagnostic®, Turkey) was used in the measurement. TOS level was calculated using the formula provided in the kit and expressed as μmol H2O2 Equiv./L.
OSI is the ratio of TOS level to TAS level. Specifically, OSI (Arbitrary Unit) = TOS (μmol H2O2 Eq/L)/TAS (mmol Trolox Eq/L)*100. When calculating the OSI of the samples, the TAS levels to equalize TOS levels and units were multiplied by 100.21 The results were expressed as arbitrary unit (AU).
PON enzyme activity was determined spectrophotometrically. The enzyme assay was based on the prediction of p-nitrophenol at 412 nm. An enzyme unit was defined as the amount of enzyme that catalyzes the hydrolysis of 1 µmol of substrate at 25 °C. PON level was calculated and expressed as U/L.
Statistical Analysis
SPSS 20 (SPSS Inc, Chicago, IL, USA) software was used for the statistical analysis. The Shapiro–Wilk test and Q–Q plot test were used to check whether the data were normally distributed. When multiple comparisons were required, one-way ANOVA followed by the Bonferroni-adjusted post hoc test was used. The results were expressed as the mean ± standard deviation (SD). Statistical significance was defined as P-value <0.05.
Results
RAM Test
The number of RAM inputs and outputs, which was similar in all groups at baseline, decreased significantly in Group A and Group ABA 3 weeks after the onset of Alzheimer’s (P<0.05). In the 3rd and 4th weeks, the difference between the groups gradually widened and at the end of 30 days, it was observed that the Alzheimer’s model was successful in the rats in Group A and ABA. While the results did not change in the test performed one day after the first BA application, there was an increase in Group ABA input and output data compared to Group A after the second application (P<0.05) (Table 1). This showed that boric acid positively affected the learning and memory functions of rats with AD, but still lagged behind Group C and BA. After the third application, the data of this group were not different from Groups C and BA. These results suggested that the application of BA in Alzheimer’s model may have therapeutic potential.
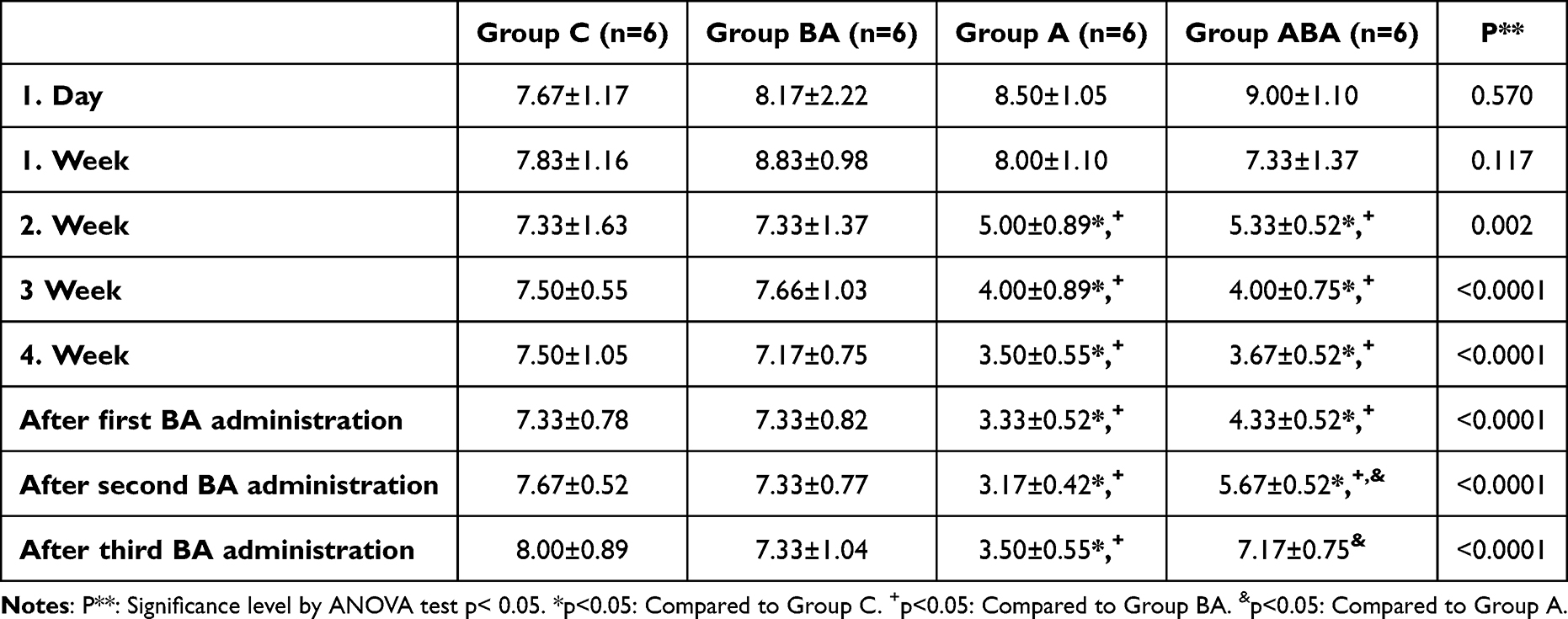 |
Table 1 RAM Input-Output Data of Rats [Mean ± SD] |
Biochemical Markers
The levels of biochemical markers in the brain were analyzed after the RAM test. In this way, the role of oxidative stress was tried to be revealed. Table 2 shows the effect of BA on TAS, TOS, OSI and PON-1 in rat brain after STZ-induced AD. Group A’s TOS level increased significantly compared to the other groups (p<0.05). In Group ABA, however, boric acid application did not make a significant difference compared to Group A, the values were similar with the other 3 groups (p>0.05). In terms of TAS, the results were consistent with TOS and the antioxidant level was significantly lower in Group A (p<0.05) and the values in Group ABA were similar to the other groups (p>0.05). OSI and PON-1 were significantly higher in rats in Group A (p<0.05). In Group ABA, these values were lower with the antioxidant effect of boric acid compared to Group A (p<0.05), and were similar to the control and BA groups (p > 0.05).
 |
Table 2 Brain Tissue TAS, TOS, OSI and PON-1 Levels of Rats [Mean ± SD] |
Hippocampal Histopathology
The thicknesses of the CA1 pyramidal cell layer and the dentate gyrus granular cell layer, and the numbers of intact and degenerated neurons in the pyramidal cell layer of the CA1 field (cell/1000 µm2) were examined [Mean ± SD]. In the histopathological evaluation, there was no difference between groups in cell layer thicknesses, and in numbers of intact and degenerated cells of CA1 pyramidal cells layer (ANOVA, p>0.05) (Table 3 and Figure 1). Although it was not statistically significant, cell layer thicknesses and intact cell number decreased in Group A. These findings suggest that duration of experiment may not be sufficient for the occurrence of histopathological changes. Atrophy, apoptosis, and glutaminergic neuron damage, which are characteristically seen in patients with AD, are more likely the outcomes to occur in the chronic process. When the efficiency of BA is examined, numerical improvements were observed in these values in Group ABA.
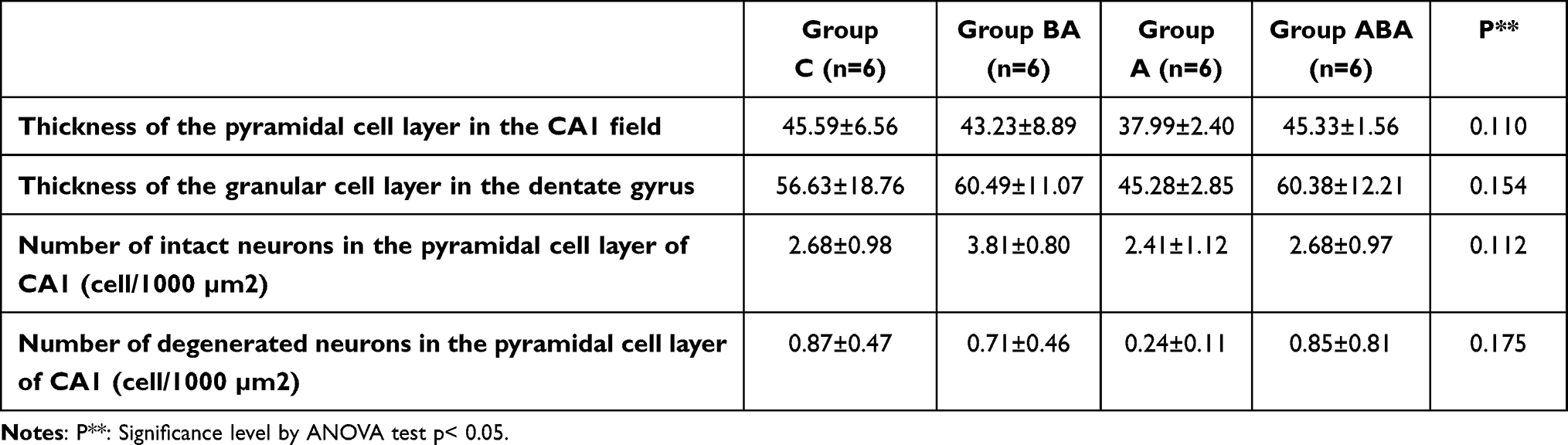 |
Table 3 Data of Pyramidal Cell Layer in Hippocampus CA1 (Cornu Ammonis 1) and Granular Cell Layer in Dentate Gyrus of Rats [Mean ± SD] |
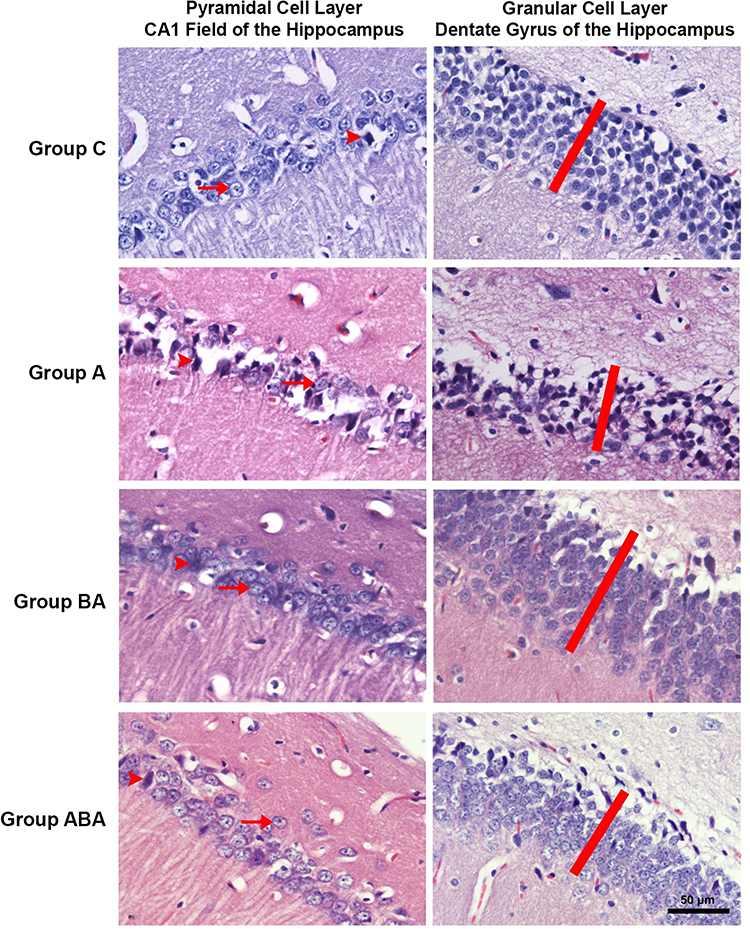 |
Figure 1 CA1 pyramidal cell layer and dentate gyrus granular cell layer are seen in H&E stained preparations of hippocampus sections. The heads of the arrows indicate degenerated neurons, and the arrows indicate the vesicular nuclei of intact neurons. The red bar shows the granular cell layer thickness of the dentate gyrus. |
Discussion
Alzheimer’s Disease is a progressive disease characterized by a decline in memory, language, and other cognitive functions. The pathophysiology of AD is complex, multifactorial and not fully known.1 The characteristic findings of AD include senile plaques (containing Aβ peptide), neurofibrillary tangles (containing tau), and progressive neuronal death.2 Although the Aβ peptide is known to be neurotoxic, Aβ accumulation facilitated in transgenic mice did not result in the expected neuron death. This indicates that the role of other damage pathways, such as oxidative stress, in disease progression is more important than expected. The brain is responsible for 20% of total body oxygen consumption, which explains why neurons are more susceptible to oxidative damage than other cells.21 When ROS exceeds its antioxidant defense capacity, it damages macromolecules, especially the phospholipid layer of the cell membrane. Lipid peroxidation and protein oxidation have been associated with various neurodegenerative disorders, including AD, Parkinson’s disease, amyotrophic lateral sclerosis, tardive dyskinesia, Huntington’s disease, and multiple sclerosis.22 BA can act as a protective agent in apoptotic processes by regulating oxidative and inflammatory processes as well as mitochondrial membrane potential.12 Few studies have been published investigating the neuroprotective potential of BA on neurodegenerative diseases. Thanks to the hydroxyl and benzyl groups it contains, BA interacts with lipids and proteins. In addition to supporting antioxidant mechanisms, it prevents lipid peroxidation and protects the integrity of tissues.23 BA has been reported that BA also reduces oxidative stress by increasing glutathione reserves,24 and strongly inhibits Aβ aggregation via hydroxyl groups.25 In rats with cyclophosphamide-induced cardiotoxicity, 200 mg/kg BA has been reported to reduce damage with antioxidant and membrane stabilizing properties.26 In a model of necrotizing enterocolitis, it increased the antioxidant level by preventing the reduction of GSH reserves.27 The biochemical activity of hepatocyte damage and oxidative stress in hepatocellular carcinoma was also able to be reversed.28 BA added to the diet prevented lipid peroxidation by supporting and strengthening the antioxidant defense system.29 Boron is thought to prevent apoptosis and strengthen antioxidant defense by reducing intracellular oxygen radicals and calcium levels.12,23 The 200mg/kg BA dose used in our study was preferred because it was used safely in experimental studies before. In biochemical analyzes, the acute LD50 value of boric acid was determined as 2660 mg/kg for rats and 3450 mg/kg for mice. Since the dose we used was much lower than the toxic doses, toxicology analysis was not performed. Our hypothesis is that boric acid can improve cognitive function and histopathological outcomes by reducing oxidative stress in rats with STZ-induced Alzheimer’s Disease.
Creating an Alzheimer’s model with STZ is one of the models frequently used in experimental studies.30 Injection of STZ into cerebral ventricles causes learning-memory disorders and neuronal losses. Similar to previous reports, regression in learning and memory was detected in our study, thus exhibiting the neurodegenerative effect of STZ exposure. Elevated markers of oxidative stress observed in rats exposed to STZ suggest that cognitive dysfunction is a combined effect of biochemical changes with histopathological damage.31,32
Assessment of learning and memory is necessary to reveal any possible CNS-related depression or stimulant effects of interventions on experimental animals. In our study, after the STZ-induced Alzheimer’s model was carried out, we performed a RAMT to evaluate cognitive functions. RAMT was designed by Olton & Samuelson.33 The Radial Arm Maze Test is widely used by researchers to measure spatial learning and memory in rodents; allows the evaluation of spatial working and reference memory.17 In the RAM test, we observed the number of inputs and outputs in the arms. In the 3rd week after STZ exposure, the number of entries and exits in Alzheimer’s groups decreased significantly. After the second BA application, the input-outputs numbers were lower in Group ABA compared to the control group and group BA, but increased compared to Group A. After the third administration of BA, these values were similar in Group C, BA and ABA. This shows the positive effect of BA on cognitive functions in 3 doses of 200 mg/kg in Alzheimer’s model.
In the case of oxidative stress, antioxidants interact with each other and a collective antioxidant effect occurs. For this reason, instead of measuring each parameter in the evaluation of the oxidant-antioxidant balance in the human body, it is more useful to examine the markers showing the global status such as TOS and TAS.34 OSI, which is presented as the ratio of both markers, can be considered as the net relationship of oxidant-antioxidant status. In this study, we found that TOS increased and TAS decreased in rats with AD. OSI increased in Alzheimer’s groups due to the shift of oxidant-antioxidant balance in favor of oxidative stress. After BA administration, it increased TAS by increasing the antioxidant effect, and as a result, TOS and OSI decreased. These results support the view that emphasizes the role of oxidative stress in the development of AD. According to our results, there was a negative relationship between learning-memory function and TOS, and a positive relationship between learning-memory function and TAS. These results support the relationship between decreased cognitive performance and increased oxidant/antioxidant balance. In a study investigating the effects of BA in ethanol-induced renal damage, it reduced the damage by lowering high TOS and OSI and increasing TAS.35 In the study investigating the protective effect of BA on liver ischemia reperfusion (IR) damage in cholestatic rats, TAS was highest in the IR group administered with BA, while TOS was highest in the IR group and lower in the BA+IR group.36 An association between PON-1 and AD has been demonstrated.37 PON-1 is associated with high-density lipoprotein (HDL).38 PON-1 is the main antiatherosclerotic component of HDL.39,40 PON-1, an antioxidant enzyme, is known to delay low-density lipoprotein (LDL) oxidation by preventing the formation of lipid peroxides.41 PON-1 polymorphism has been associated with neurodegenerative diseases.42–44 Several studies have shown that PON-1 level may play important roles in oxidative stress and many neurodegenerative diseases.42,45–47 PON-1 level increased in Group A similar to previous publications. In the study, in which Alzheimer’s model was created with the same methodology, PON-1 level was higher in the treated AD groups.48 This confirmed the PON1-AD relationship. Group ABA PON-1 decreased significantly after boric acid administration.
The hippocampus is one of the most affected brain areas in patients with AD.49 This region is subdivided into the dentate gyrus (DG) and CA1, CA2, CA3/4. The classical hippocampal trisynaptic circuit (EC-DG-CA3-CA1) is crucial for learning and memory.50 Interestingly, patients with AD51,52 and animal models of this disease53 show morphological changes in the dendritic trees and spines of CA1 and CA3 pyramidal neurons. CA1 is the first hippocampal subarea where neurofibrillary pathology arises,54 and neuropathological studies have shown CA1 atrophy in Alzheimer’s dementia,55,56 but not in preclinical AD57 In fact, CA1 hypertrophy was reported in preclinical AD in one study.58 AD patients with severe dementia show a decrease in dentate granule cell (DGC), density of dendritic spines and total dendritic length.59–61 As a result of adult hippocampal neurogenesis (AHN), new DGCs are introduced into the hippocampal circuit throughout life. The number of immature DGCs gradually decreases throughout AD progression.62 In addition, fewer immature neurons are seen in patients with cognitive impairment.63 In contrast, the number of mature DGCs remains constant in these patients.62 Therefore, it has been suggested that AHN disorders underlie cognitive dysfunction in AD.64 In the light of this information, the pyramidal cell layer in CA1 and the thickness of the granular cell layer in the dentate gyrus and the number of intact and degenerated neurons (cell/1000 µm2) in the pyramidal cell layer in CA1 were evaluated in order to examine the histopathological damage in the AD model created in the light of this information. As a result of the evaluation, although there was a decrease in the cell layer thickness and the number of intact cells in Group A, it did not make a statistically significant difference. The results of the BA applied groups were similar to the group C. Although this indicates that BA did not cause any toxic damage, the absence of significant damage in the Alzheimer group precluded us from commenting on the therapeutic potential of BA in this model. In our opinion there was not sufficient time for the emergence of histopathological changes in the Alzheimer’s model. Atrophy, apoptosis, and glutaminergic neuron damage, which are characteristically seen in these patients, are more likely outcomes to occur in the chronic process. When the BA efficiency is examined, numerical improvements were observed in Group ABA in these values.
Limitation
There are no significant limitations in our study.
Conclusion
Our results showed that BA reduced damage to learning and memory functions and significantly lowered oxidative stress markers in the AD model. Since no significant damage was observed in the histopathology of Alzheimer’s rat brains after modeling, we cannot tell about the negative or positive effect of BA.
Disclosure
No conflict of interest was declared by the authors.
References
1. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer’s disease. Lancet. 2021;397(10284):1577–1590.
2. Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2):312–339.
3. Bhatia V, Sharma S. Role of mitochondrial dysfunction, oxidative stress and autophagy in progression of Alzheimer’s disease. J Neurol Sci. 2021;421:117253.
4. Xiong Z, Hongmei Z, Lu S, Yu L. Curcumin mediates presenilin-1 activity to reduce β-amyloid production in a model of Alzheimer’s Disease. Pharmacol Rep. 2011;63(5):1101–1108.
5. Sultana R, Mecocci P, Mangialasche F, Cecchetti R, Baglioni M, Butterfield DA. Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer’s disease: insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this dementing disorder. J Alzheimers Dis. 2011;24(1):77–84.
6. Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging. 2002;23(5):655–664.
7. Butterfield DA, Swomley AM, Sultana R. Amyloid β-peptide (1-42)-induced oxidative stress in Alzheimer disease: importance in disease pathogenesis and progression. Antioxid Redox Signal. 2013;19(8):823–835.
8. Barranco WT, Eckhert CD. Cellular changes in boric acid-treated DU-145 prostate cancer cells. Br J Cancer. 2006;94(6):884–890.
9. Samman S, Naghii MR, Lyons Wall PM, Verus AP. The nutritional and metabolic effects of boron in humans and animals. Biol Trace Elem Res. 1998;66(1–3):227–235.
10. Hunt CD. Regulation of enzymatic activity. Biol Trace Elem Res. 1998;66(1):205–225.
11. Naghii MR, Samman S. The role of boron in nutrition and metabolism. Prog Food Nutr Sci. 1993;17(4):331–349.
12. Sogut I, Oglakci A, Kartkaya K, et al. Effect of boric acid on oxidative stress in rats with fetal alcohol syndrome. Exp Ther Med. 2015;9(3):1023–1027.
13. Ravelli KG, Rosário B, Camarini R, Hernandes MS, Britto LRG. Intracerebroventricular Streptozotocin as a Model of Alzheimer’s Disease: neurochemical and Behavioral Characterization in Mice. Neurotox Res. 2016;31:327–333.
14. Wu C, Yang L, Tucker D, et al. Beneficial Effects of Exercise Pretreatment in a Sporadic Alzheimer’s Rat Model. Med Sci Sports Exerc. 2018;50(5):945–956.
15. Sharma M, Gupta YK. Intracerebroventricular injection of streptozotocin in rats produces both oxidative stress in the brain and cognitive impairment. Life Sci. 2001;68(9):1021–1029.
16. Salkovic-Petrisic M, Knezovic A, Hoyer S, Riederer P. What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer’s disease, about the therapeutic strategies in Alzheimer’s research. J Neural Transm. 2013;120(1):233–252.
17. Kim H, Park JY, Kim KK. Spatial learning and memory using a radial arm maze with a head-mounted display. Psychiatry Investig. 2018;15(10):935–944.
18. Mizuno M, Yamada K, Olariu A, Nawa H, Nabeshima T. Involvement of brain-derived neurotrophic factor in spatial memory formation and maintenance in a radial arm maze test in rats. J Neurosci. 2000;20(18):7116–7121.
19. Saffari PM, Alijanpour S, Takzaree N, et al. Metformin loaded phosphatidylserine nanoliposomes improve memory deficit and reduce neuroinflammation in streptozotocin-induced Alzheimer’s disease model. Life Sci. 2020;255:117861.
20. Harakeh S, Ramadan W, Al Muhayawi M, Al Jaouni S, Mousa S, Hakeem K. Pomegranate peel extract lessens histopathologic changes and restores antioxidant homeostasis in the hippocampus of rats with aluminium chloride-induced Alzheimer’s disease. Asian Pac J Trop Med. 2020;13(10):456–463.
21. Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proce National Acad Sci. 1993;90(17):7915–7922.
22. Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32(11):1050–1060.
23. Sogut I, Paltun SO, Tuncdemir M, Ersoz M, Hurdag C. The antioxidant and antiapoptotic effect of boric acid on hepatoxicity in chronic alcohol-fed rats. Can J Physiol Pharmacol. 2018;96(4):404–411.
24. Cao J, Jiang L, Zhang X, et al. Boric acid inhibits LPS-induced TNF-alpha formation through a thiol-dependent mechanism in THP-1 cells. J Trace Elem Med Biol. 2008;22(3):189–195.
25. Lu CJ, Hu J, Wang Z, et al. Discovery of boron-containing compounds as Aβ aggregation inhibitors and antioxidants for the treatment of Alzheimer’s disease. Medchemcomm. 2018;9(11):1862–1870.
26. Cengiz M. The Prevention Effects Boric Acid on Cyclophosphamide induced Cardiotoxicity in Rats. Bitlis Eren Üniversitesi Fen Bilimleri Dergisi. 2018;7:113–118.
27. Yazıcı S, Akşit H, Korkut O, Sunay B, Çelik T. Effects of boric acid and 2-aminoethoxydiphenyl borate on necrotizing enterocolitis. J Pediatr Gastroenterol Nutr. 2014;58(1):61–67.
28. Zafar H, Ali S. Boron inhibits the proliferating cell nuclear antigen index, molybdenum containing proteins and ameliorates oxidative stress in hepatocellular carcinoma. Arch Biochem Biophys. 2013;529(2):66–74.
29. Ince S, Kucukkurt I, Cigerci IH, Fatih Fidan A, Eryavuz A. The effects of dietary boric acid and borax supplementation on lipid peroxidation, antioxidant activity, and DNA damage in rats. J Trace Elem Med Biol. 2010;24(3):161–164.
30. Gao C, Liu Y, Jiang Y, Ding J, Li L. Geniposide Ameliorates Learning Memory Deficits, Reduces Tau Phosphorylation and Decreases Apoptosis via GSK 3β Pathway in Streptozotocin‐Induced A lzheimer Rat Model. Brain Pathology. 2014;24(3):261–269.
31. Bhosale UA, Yegnanarayan R, Pophale PD, Zambare MR, Somani RS. Study of central nervous system depressant and behavioral activity of an ethanol extract of Achyranthes aspera (Agadha) in different animal models. Int J Appl Basic Med Res. 2011;1(2):104–108.
32. Gupta G, Kazmi I, Afzal M, et al. Sedative, antiepileptic and antipsychotic effects of Viscum album L. (Loranthaceae) in mice and rats. J Ethnopharmacol. 2012;141(3):810–816.
33. Olton DS, Samuelson RJ. Remembrance of places passed: spatial memory in rats. J Exp Psychol Anim Behav Process. 1976;2(2):97.
34. Erel O. A novel automated method to measure total antioxidant response against potent free radical reactions. Clin Biochem. 2004;37(2):112–119.
35. Cikler-Dulger E, Sogut I. Investigation of the protective effects of boric acid on ethanol induced kidney injury. Biotech Histochem. 2020;95(3):186–193.
36. Güler S, Aslaner A, Ellidağ HY, Yildirim Ş, Çakir T. The Protective Effect of Boric Acid on Cholestatic Rats Liver Ischemia Reperfusion Injury. Turk J Med Sci. 2021;51(5):2716–2726.
37. Paragh G, Balla P, Katona E, Seres I, Égerházi A, Degrell I. Serum paraoxonase activity changes in patients with Alzheimer’s disease and vascular dementia. Eur Arch Psychiatry Clin Neurosci. 2002;252(2):63–67.
38. Tomás M, Latorre G, Sentí M, Marrugat J. The antioxidant function of high density lipoproteins: a new paradigm in atherosclerosis. Revista Española de Cardiología. 2004;57(6):557–569.
39. Getz GS, Reardon CA. Paraoxonase, a cardioprotective enzyme: continuing issues. Curr Opin Lipidol. 2004;15(3):261–267.
40. Mackness M, Mackness B. Paraoxonase 1 and atherosclerosis: is the gene or the protein more important? Free Radic Biol Med. 2004;37(9):1317–1323.
41. Paragh G, Seres I, Balogh Z, et al. The Serum Paraoxonase Activity in Patients with Chronic Renal Failure and Hyperlipidemia. Nephron. 1998;80(2):166–170.
42. Sand PG. Paraoxonase genes and the susceptibility to ischemic stroke. Int J Stroke. 2013;8(6):E39–E.
43. Rothstein L, Jickling GC. Ischemic stroke biomarkers in blood. Biomark Med. 2013;7(1):37–47.
44. Xing C, Arai K, Lo EH, Hommel M. Pathophysiologic cascades in ischemic stroke. Int J Stroke. 2012;7(5):378–385.
45. Zhang G, Li W, Li Z, et al. Association between paraoxonase gene and stroke in the Han Chinese population. BMC Med Genet. 2013;14(1):1–10.
46. Androutsopoulos VP, Kanavouras K, Tsatsakis AM. Role of paraoxonase 1 (PON1) in organophosphate metabolism: implications in neurodegenerative diseases. Toxicol Appl Pharmacol. 2011;256(3):418–424.
47. Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8):a006239.
48. Arslan M, Küçük A, Kıran MM, Köksal Z, Kurtipek Ö, Kavutçu M. Evaluation of the Effects of Recurrent Dexmedetomidine on Cognitive Functions and Brain Tissue in Streptozotocin-Induced Rats with Alzheimer’s Disease. Gazi Med J. 2022;1:5–9.
49. Braak H, Braak E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16(3):271–278.
50. Hainmueller T, Bartos M. Dentate gyrus circuits for encoding, retrieval and discrimination of episodic memories. Nat Rev Neurosci. 2020;21(3):153–168.
51. Kerchner GA, Deutsch GK, Zeineh M, Dougherty RF, Saranathan M, Rutt BK. Hippocampal CA1 apical neuropil atrophy and memory performance in Alzheimer’s disease. Neuroimage. 2012;63(1):194–202.
52. Merino-Serrais P, Benavides-Piccione R, Blazquez-Llorca L, et al. The influence of phospho-tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer’s disease. Brain. 2013;136(6):1913–1928.
53. Mehder RH, Bennett BM, Andrew RD. Morphometric analysis of hippocampal and neocortical pyramidal neurons in a mouse model of late onset Alzheimer’s disease. J Alzheimer’s Dis. 2020;74(4):1069–1083.
54. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259.
55. Lim HK, Hong SC, Jung WS, et al. Automated hippocampal subfield segmentation in amnestic mild cognitive impairments. Dement Geriatr Cogn Disord. 2012;33(5):327–333.
56. La Joie R, Fouquet M, Mézenge F, et al. Differential effect of age on hippocampal subfields assessed using a new high-resolution 3T MR sequence. Neuroimage. 2010;53(2):506–514.
57. West MJ, Kawas CH, Stewart WF, Rudow GL, Troncoso JC. Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol Aging. 2004;25(9):1205–1212.
58. Iacono D, Markesbery WR, Gross M, et al. The Nun study: clinically silent AD, neuronal hypertrophy, and linguistic skills in early life. Neurology. 2009;73(9):665–673.
59. De Ruiter J, Uylings H. Morphometric and dendritic analysis of fascia dentata granule cells in human aging and senile dementia. Brain Res. 1987;402(2):217–229.
60. Flood DG, Buell SJ, Horwitz GJ, Coleman PD. Dendritic extent in human dentate gyrus granule cells in normal aging and senile dementia. Brain Res. 1987;402(2):205–216.
61. Llorens-Martin M, Fuster-Matanzo A, Teixeira CM, et al. GSK-3β overexpression causes reversible alterations on postsynaptic densities and dendritic morphology of hippocampal granule neurons in vivo. Mol Psychiatry. 2013;18(4):451–460.
62. Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, et al. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med. 2019;25(4):554–560.
63. Tobin MK, Musaraca K, Disouky A, et al. Human hippocampal neurogenesis persists in aged adults and Alzheimer’s disease patients. Cell Stem Cell. 2019;24(6):974–82. e3.
64. Young JK. Neurogenesis makes a crucial contribution to the neuropathology of Alzheimer’s disease. J Alzheimer’s Dis Rep. 2020;4(1):365–371.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.