")
Back to Journals » Journal of Inflammation Research » Volume 15
Therapeutic Effects of Retinoic Acid in Lipopolysaccharide-Induced Cardiac Dysfunction: Network Pharmacology and Experimental Validation
Authors Wang X , Kong C, Liu P, Zhou B, Geng W , Tang H
Received 26 January 2022
Accepted for publication 1 August 2022
Published 30 August 2022 Volume 2022:15 Pages 4963—4979
DOI https://doi.org/10.2147/JIR.S358374
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Xi Wang,1,2,* Chang Kong,1,3,* Pan Liu,1,2 Baofeng Zhou,1,2 Wujun Geng,1,2 Hongli Tang1,2
1Department of Anesthesia, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, People’s Republic of China; 2Wenzhou Key Laboratory of Perioperative Medicine, Wenzhou, People’s Republic of China; 3Department of Anesthesiology and Critical Care Medicine, Tianjin Nankai Hospital, Tianjin Medical University, Tianjin, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hongli Tang; Wujun Geng, Doctor’s Degree, Department of Anesthesia, The First Affiliated Hospital of Wenzhou Medical University, Nanbaixiang, Ouhai District, Wenzhou, Zhejiang, 325000, People’s Republic of China, Tel +86 13587436057; +86 15325502139, Fax +86 0577-88069555, Email [email protected]; [email protected]
Purpose: Sepsis, which is deemed as a systemic inflammation reaction syndrome in the face of infectious stimuli, is the primary cause of death in ICUs. Sepsis-induced cardiomyopathy (SIC) may derive from systemic inflammation reaction and oxidative stress. Retinoic acid (RA) is recognized by its beneficial roles in terms of the immunoresponse to infections and antioxygen actions. However, the treatment efficacy and potential causal links of RA in SIC are still elusive.
Methods: By virtue of the STITCH database, we identified the targets of RA. Differentially expressed genes in SIC were acquired from the GEO database. The PPI network of intersected targets was established. GO and KEGG pathway enrichment analysis was completed. Hub genes were analyzed by cytoHubba plug-in. In the process of experimental validation, a mouse sepsis model was established by lipopolysaccharide (LPS), and the treated mice were intraperitoneally injected with RA or Dexamethasone (DEX) 60 min prior to LPS injections. Survival conditions, cardiac functions and antioxidant levels of the mice were assessed. Cardiac inflammation and injury were detected by HE and TUNEL. The levels of key genes and signal pathway expression were analyzed by RT-PCR and Western blot.
Results: PPARA, ITGAM, VCAM-1, IGF-1 and IL-6 were identified as key therapeutic targets of RA by network pharmacology. PI3K-Akt signaling pathway is the main regulatory pathway of RA. In vivo researches unraveled that RA can improve the survival rate and cardiac function of LPS-treated mice, inhibit inflammatory factors and myocardial injury, and regulate the expression of key therapeutic targets and key pathways, which is PI3K-Akt signaling pathway.
Conclusion: Network pharmacological method offers a predicative strategy to explore the treatment efficacy and causal links of RA in endotoxemic myocarditis. Through experimental verification, we discover that RA can reduce lipopolysaccharide-induced cardiac dysfunction by regulating the PI3K-Akt signaling pathway and key genes.
Keywords: retinoic acid, lipopolysaccharide, sepsis-induced cardiomyopathy, network pharmacology, inflammatory response
Introduction
Sepsis is a lethal critical disease, identified as a main health concern across the world. In the past 10 years, hospital mortalities from sepsis and septic shock every year registered 17% and 26%, separately, causing about 8 million deaths each year.1,2 Cardiac function disorder induced by sepsis, often referred to as SIC, is commonly seen and has long been an intriguing topic.3 As a pathophysiological syndrome caused by infection rather than a specific disease, the specific identification of targeted SIC is essential for minimizing the mortality and morbidity in this regard. Despite the fact that the indications for supervising and healing SIC are clinical and directed toward restoring tissue perfusion, a deeper comprehension of the important gene signatures and underlying pathogenesis of SIC can assist in optimizing treatments and ameliorating clinical results.4–7
Gram-negative bacterial endotoxin (lipopolysaccharide, LPS) serves as a key sepsis mediator for septicemia-associated multiple organ dysfunction or mortality.8 After undergoing LPS insult, both macrophages and cardiomyocytes can release substantial inflammatory mediators, such as MCP-1, GM-CSF, IL-1α, IL-1β, IL-6, IL-7, IL-8, and IL-12.9,10 These inflammatory cytokines could give rise to the imbalance of calcium homeostasis,11 disturbance of energy metabolism,12 impairment of adrenergic signaling, and excess production of nitric oxide,13 all of which facilitate decreased contractility, diastolic dysfunction, impaired ejection fraction and reduced cardiac index. Cytokines, especially IL‐1β, IL‐6, and TNF‐α, are major contributors in the initiation of SIC.14
RA is a powerful derivant of vitamin A, and is pivotal for body developmental process and organ genesis via regulating cellular proliferative and differentiative activities. Substantial researches on animal models and clinic tests have verified its capability of preventing infections and enhance immunosystems.15,16 Austenaa et al demonstrated that RA inhibited LPS-triggered stimulation in mice and mankind monoblasts.17 In addition, Martire-Greco D et al displayed that all-trans retinoic acid (ATRA), which could improve functional immunoresponses in LPS-exposed mice, could serve as a novel underlying method for the healing of the immune suppressive status of sepsis.18 Considering the increasing evidence that RA can improve immune function and reduce inflammation, we speculated that RA might exert a beneficial effect on LPS-induced heart function disorder. The present research aimed to determine the roles of RA in LPS-induced cardiac dysfunction and explore its underlying mechanisms.
Currently, the network pharmacological approach can be employed to forecast the correlation between targets and diseases.19,20 The network pharmacological approach was deemed as a new method of medicine design.21,22 Network pharmacology methods are developing rapidly and have been leveraged to find new treatment methods, ameliorating the approved drugs and expanding the application scenarios of clinical medicines. They can also be utilized to effectively search for undeveloped targets for compounds or natural products.23 The purpose of network construction was to reveal the interaction between bioactive compounds and target proteins as well as the interaction between various target proteins. We identified and verified key nodes through network analysis and verification.21 Systematic or network pharmacology combined with multiomics analysis showed unique advantages in predicting and explaining the pharmacological principles of drugs and mechanisms of action in treating various diseases.24,25 For that reason, herein, network pharmacological approach was employed to discover treatment targets and associated signaling pathways of RA against SIC.
The primary aims of our research were 1) to select the underlying targets of Retinoic acid and DEGs in SIC heart tissues; 2) to study the potential causal links of Retinoic acid against SIC via bioinformatic analysis; 3) to confirm the anti-inflammation, antioxidant levels and potential signaling pathways of Retinoic acid in LPS-induced cardiac dysfunction. Our research might offer a novel treatment method for improving sepsis-induced cardiomyopathy.
Materials and Methods
Animals
Male C57BL/6 mice (8–10 weeks) were kept at the Experiment Animal Center of Wenzhou Medical University. Those animals were kept at 23±1℃ with a 12-hour light/dark period in a specially disinfected environment with free access to bacteria-free water and food. Every animal assay, with a minimum sacrificed mice according to our design, was accepted by the Animal Assay Ethical Board of our university (ID: WYYY-AEC-2021-301). All experiment procedures were completed blindly, such as the animal models and following assays. The animals were stochastically separated into these groups: (1) Saline (i.p., n = 6), (2) LPS (10 mg/kg i.p., n = 6), (3) LPS (10 mg/kg) plus RA (1 mg/kg, i.p., n = 6), (4) LPS (10 mg/kg) plus RA (3 mg/kg, i.p., n = 6), (5) LPS (10 mg/kg) plus Dexamethasone (DEX) (2.5 mg/kg, i.p., n = 6). The administration of RA was arranged 60 min prior to LPS injections. Subsequently, after 24 hours of supervision, the animals were euthanised via exsanguination with overdosage of sodium pentobarbital and all our efforts aimed to minimise pains of the animals. Meanwhile, all heart samples were collected for histology analyses or immediately frozen in liquid nitrogen, preserved under −80 ℃ for future biochemistry examination. Lipopolysaccharide (LPS), Retinoic Acid (RA) and Dexamethasone (Dex) were bought from Sigma (St Louis, MO, USA).
Identification of DEGs
The gene expression dataset GSE44363 related to endotoxemic myocarditis was acquired from the GEO database.26 The GSE44363 data collection, which involved 4 normal and 4 endotoxemic myocardium that were treated for 24 hours with either saline or LPS, was based on wild-type mice. All RNA information of the chosen specimens was acquired for future analysis. The limma package27 was employed to calculate the difference between two groups of patients, and gene screening conditions with p.adj<0.05 and | Log2FC | > 1 were used for filtering DEGs in SIC.
Retinoic Acid Target Prediction
The chemistry structure and SMILES of Retinoic acid was acquired from PubChem web site.28 The target forecast of Retinoic acid was completed via the STITCH data base (http://stitch.embl.de/),29 while the species was limited to “Mus musculus”.
GO and KEGG Pathway Enrichment Assays
GO function analysis (CC, BP, and MF) was a potent biological information method to categorize genetic expressing and its performances,30,31 while KEGG pathway analysis was adopted to determine which cellular pathway may participate in the variations of DEGs.32,33 The visualization of GO enriching assay (p.adj<0.05) and KEGG pathway assay (p.adj<0.05) was realized via the R ggplot2 package.
PPI Network Analysis
The PPI net of targeted genes was acquired via from STRING 11.0 database,34 with minimal needed interactive score ≥0.7. The visualization of the PPI net was realized via Cytoscape 3.7.2.35 In the net, nodal points denoted targeted protein, and edges denoted the forecasted or verified mutual effect between protein. Topology analyses of targeted genes were completed via the Cytohubba plug-in of Cytoscape. Targeted protein was subjected to filtration, respectively, as per the BottleNeck, Betweenness, Stress and Radiality subnetworks, which were computed via Cytohubba plug-in. Top 10 genes of every sub-net were searched, and overlapping genes were chosen as critical targets herein.
Survival Condition
Another 50 mice were stochastically separated into the following groups: (1) Saline (i.p., n = 10), (2) LPS (10 mg/kg i.p., n = 10), (3) LPS (10 mg/kg) plus RA (1 mg/kg, i.p., n = 10), (4) LPS (10 mg/kg) plus RA (3 mg/kg, i.p., n = 10), (5) LPS (10 mg/kg) plus Dexamethasone (DEX) (2.5 mg/kg, i.p., n = 10). Administration of saline, RA, or DEX was scheduled 60 minutes before LPS injection and then administered for 7 consecutive days. The intervention and modeling methods of mice in each group were the same as before. After modeling, the 7-day survival rates of the five groups of mice were observed.
CK-MB and LDH in Serum
CK-MB (MEIMIAN, China) and LDH (LEAGENE, China) levels in serum were quantified using kits according to the manufacturer's instructions.
Assessment of Oxidative Stress
Our team prepared samples according to the test kit specifications. The levels of catalase (CAT),36 superoxide dismutase (SOD)37 and GSH/GSSG38 in the heart samples were determined by colorimetry according to the kits in previous studies mentioned above. The results of CAT and SOD were expressed in units of protein per mg (U/mg prot).
Echocardiography
Echocardiography was implemented by a Vevo 3100 ultrasonic equipment with a 10-MHz linear array ultrasonic transducer (Fujifilm, VisualSonics, USA) after mice were anesthetized by 1.5% isoflurane. As medial echocardiographic readings were collected from 3–5 heart cycles, heart function indexes, such as fractional shortening (FS), ejection fraction (EF), etc., were documented.
Total RNA Extraction and qauantitative real-time PCR
Overall RNA from all frozen cardiac tissues was abstracted via TRIzol reagent (Invitrogen). Overall RNA (2 μg) was converted to cDNA through reverse transcription by cDNA synthesis kit (TaKaRa). qPCR was completed by toroivd SYBR Green qPCR Master Mix (20 μL). The cycling status were stated below: denaturalization under 95℃ for 60s, 40 cycles under 95℃ for 10s, 60℃ for 30s, and 72℃ for 45s. The RNA quantity was computed via the comparative threshold cycle approach, with every primer customized by Sangon Biotechnology Co., Ltd. (Shanghai, China). Eventually, each primer sequence is presented in Table 1.
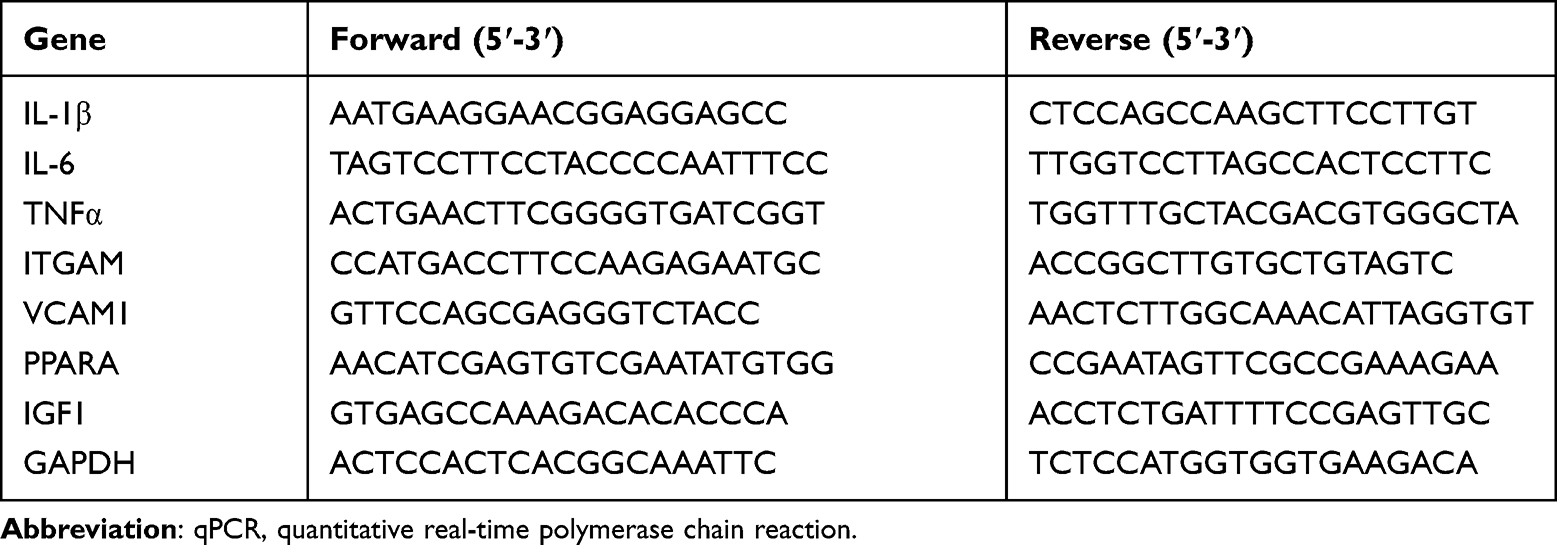 |
Table 1 Primers Used for qPCR of Genes from Mouse |
Western Blot
Proteins were abstracted from the entire frozen cardiac tissues via RIPA lysis buffering solution with 1% protease suppressor mixture. The BCA approach was employed to compute the protein level. Equal amounts of proteins were separated by 10–15% SDS-PAGE. Samples were moved onto PVDF films, subjected to blockade via 5% dry skimmed milk, and incubated with primary antibodies including anti-ITGAM (Abcam, Ab133357, 1:1000), anti-VCAM1 (Abcam, Ab134047, 1:1000), anti-PPARA (Abcam, Ab61182, 1:1000), anti-IGF1 (Abcam, Ab9572, 1:1000), anti-IL-6 (Abcam, Ab259341, 1:1000), anti-GAPDH (Abcam, Ab181602, 1:2000), anti-phospho-Akt S473 (Cell Signaling Technology, 4060, 1:1000) and anti-Akt (Cell Signaling Technology, 4691, 1:1000) separately, at 4℃ nightlong. Posterior to the cleaning in TBST, the blots were cultivated with antirabbit or antimouse second antisubstances for 60 min under ambient temperature. Afterwards, the outcomes were identified via the ECL identification reagents. Proteins in Western blot were quantified via Image Lab software. All assays were completed in triplicate.
Histological Analysis
Cardiac specimens were subjected to 4% neutral PFA fixation, paraffin embedment, and sectioning. For H&E dyeing (Solarbio, Beijing, China), 5 μm slices were dyed in hematoxylin for 600 s, and afterwards cleaned and dyed in 0.5% eosin for 300 s. Posterior to the cleaning in water, the samples were subjected to dehydration in 70%, 85%, 95%, and 100% ethyl alcohol and afterwards in xylene. Heart injuries were analyzed by microscopic fields of every tissular specimen, which was stochastically chosen. The morphological status of myofilament and inflammation cell infiltration were evaluated as standard.
For IHC, paraffin slices were subjected to deparaffinization via xylene and subjected to rehydration via the concentration gradient of ethyl alcohol. Subsequently, antigen repair was completed and the specimens were cultivated with anti-CD68 (CST, D4B9C, 1:200) nightlong at 4 ℃. Eventually, the slices were cultivated by an antirabbit EnVisionTM +/HRP reagent for sixty minutes at 37 ℃ for the observation via a light microscopy (Nikon, H550L, Tokyo, Japan).
Terminal Deoxynucleotidyl Transferase (TdT)-Mediated dUTP Nick
End Labeling (TUNEL) Assay
By virtue of the TUNEL approach, heart slices were dyed for identifying DNA fragmentation, which could reflect cell apoptosis. Slices were cultivated in TdT-reaction liquor and the visualization of nuclei was realized via TUNEL reagents (Promega, America) and DAPI nuclear dye. Fluorescent pictures were captured via a Nikon microscopic device. Quantitation of TUNEL-positive cells was completed via identifying the corresponding proportion (%) (green) in several high-power fields (n = 3 slices every mouse strain and treatment group).
Statistical Analysis
The experiment data were studied via GraphPad Prism 8.0. The entire experiment data were described as average ± SD. Student's t-test was used for comparison between the two groups, and the diversities between the groups were compared by one-way ANOVA. P < 0.05 had significance on statistics. Survival rate was evaluated by Kaplan–Meier analysis.
Results
Target Screening of Retinoic Acid and Sepsis-Induced Cardiomyopathy
The 2D structure of RA was acquired from PubChem (Figure 1A). An overall 500 genes were identified as targeted genes of RA from the STITCH database. In addition, 1035 DEGs were selected from GSE44363 dataset, and 547 genes were regulated upward, with 488 regulated downward (Figure 1B). By pairing DEGs with RA targets (Figure 1C), 54 genes were chosen as underlying targeted genes in septic cardiac dysfunction. The thermograph of those 54 genes was presented by Figure 1D.
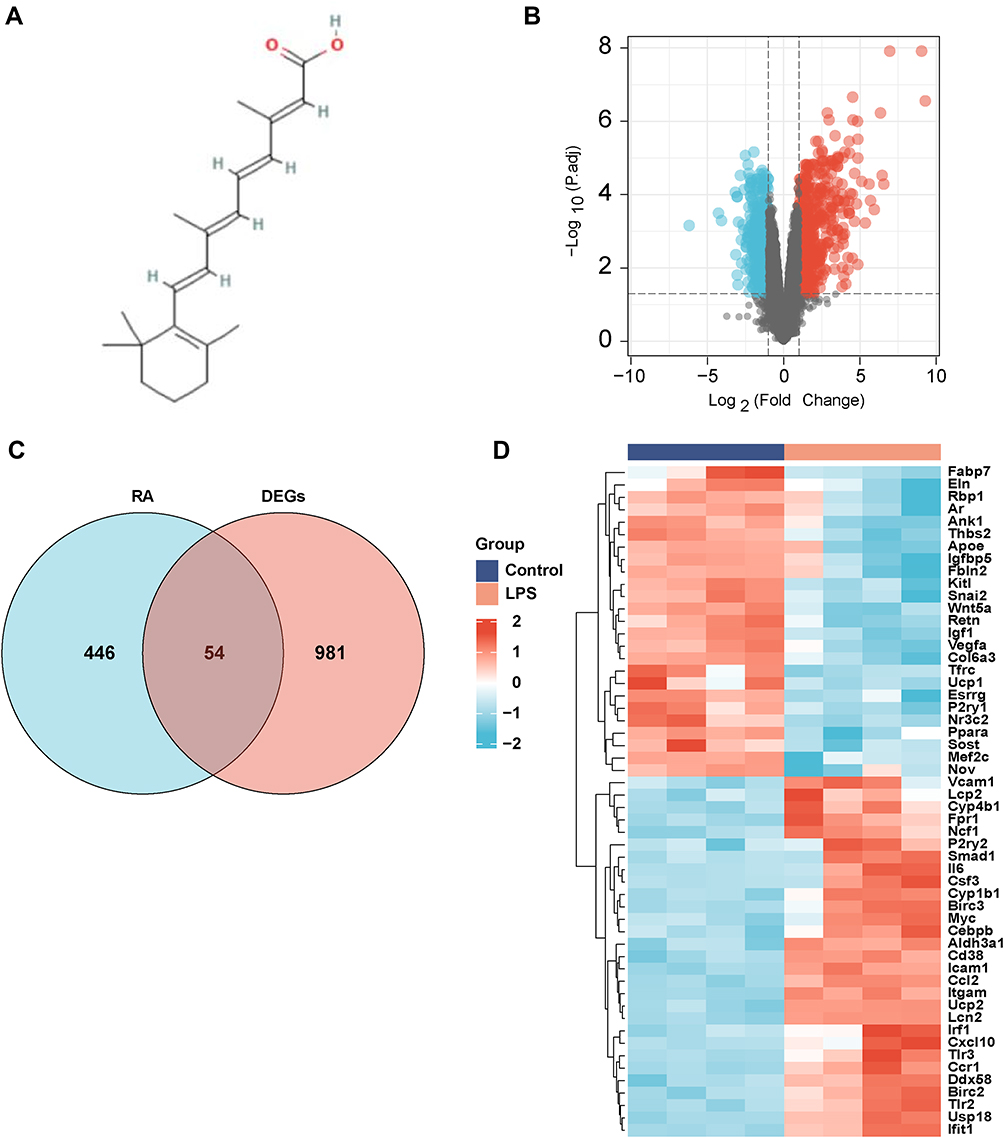 |
Figure 1 Target genes of RA and DEGs in GSE44363. (A) Chemical structure of Retinoic acid; (B) DEGs in GSE44363 (Upregulated genes were marked in red and downregulated genes were marked in blue). (C) Venn diagram of Retinoic acid target genes and DEGs. (D) Clustered heat map of overlapped genes . |
Enrichment Analysis of Overlapped Target
GO analyses of the 54 underlying treatment target genes were completed via the DAVID database. Targeted genes were primarily enriched in the regulation of ossification, myeloid leukocyte differentiation and epithelial cell proliferation in BP enrichment analysis, and they were also enriched in extracellular matrix, collagen-containing extracellular matrix and membrane raft in CC analysis. In MF analysis, they were enriched in glycosaminoglycan binding, heparin binding, and cytokine activity (Figure 2A). The outcome of KEGG pathway enriching analyses revealed that targeted genes were remarkably enriched in the PI3K-Akt signaling pathway, transcriptional misregulation in cancer, and TNF signaling pathway, etc. (Figure 2B and C).
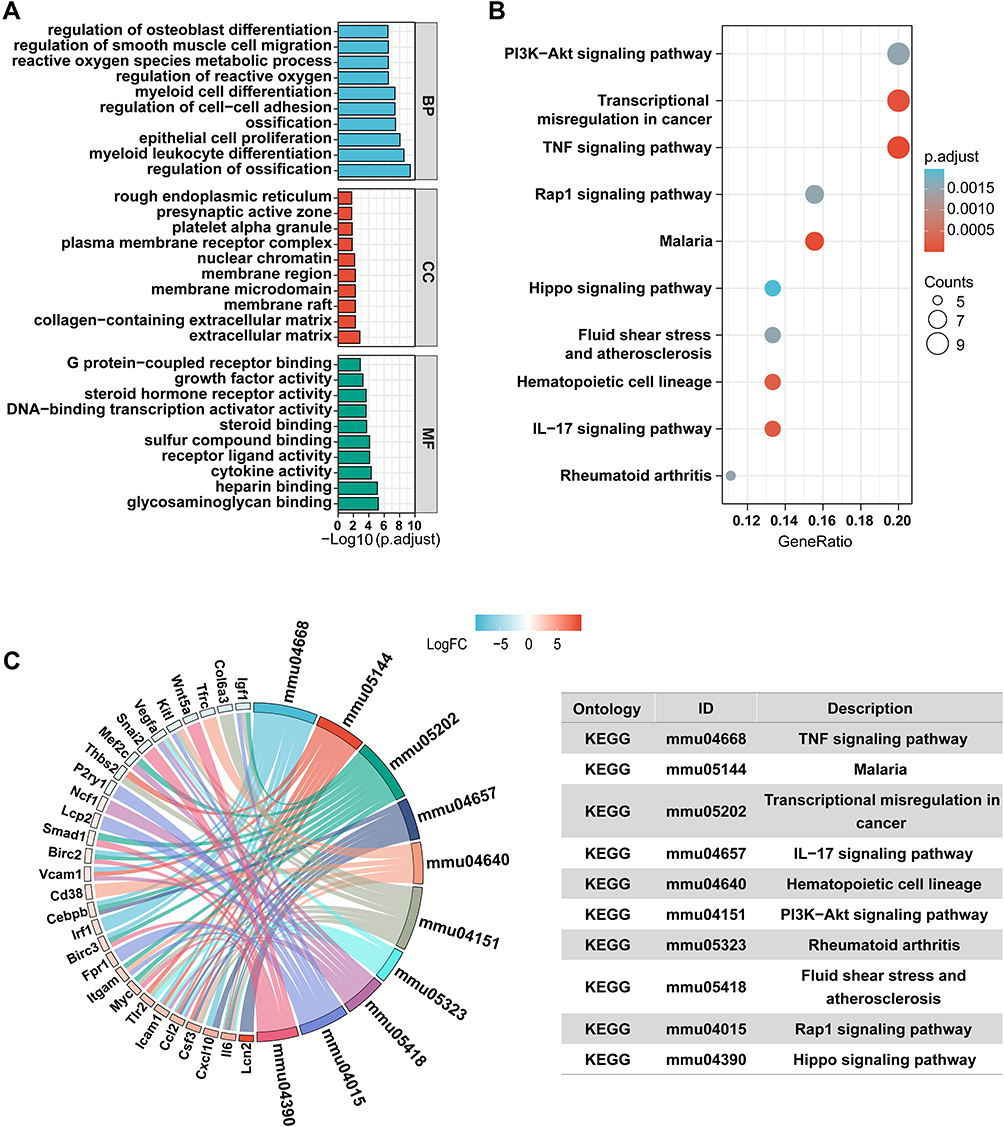 |
Figure 2 Enrichment Analysis of Overlapped Target. (A) Gene ontology (GO) enrichment analysis for key targets (Top 10 were listed). (B) KEGG pathway enrichment analysis of key targets (Top 10 were listed); the abscissa label represents GeneRatio. (C) KEGG pathway analysis and related genes (Top ten were listed). |
PPI Network Establishment
The PPI net of aforesaid targeted proteins was established via STRING and the visualization of the network was realized via Cytoscape. The PPI net comprised 54 nodal points and 266 edges (Figure 3A). BottleNeck, Betweenness, Stress and Radiality of targeted protein were computed via topology analyses (Figure 3B–E). Top 10 hub nodes of BottleNeck, Betweenness, Stress and Radiality sub-nets were searched, and we discovered 5 overlapping genes: Peroxisome proliferator activated receptor alpha (PPARA), Integrin Subunit Alpha M (ITGAM), Vascular cell adhesion molecule-1 (VCAM1), Insulin-like growth factor 1 (IGF-1), and Interleukin-6 (IL-6) (Figure 3F).
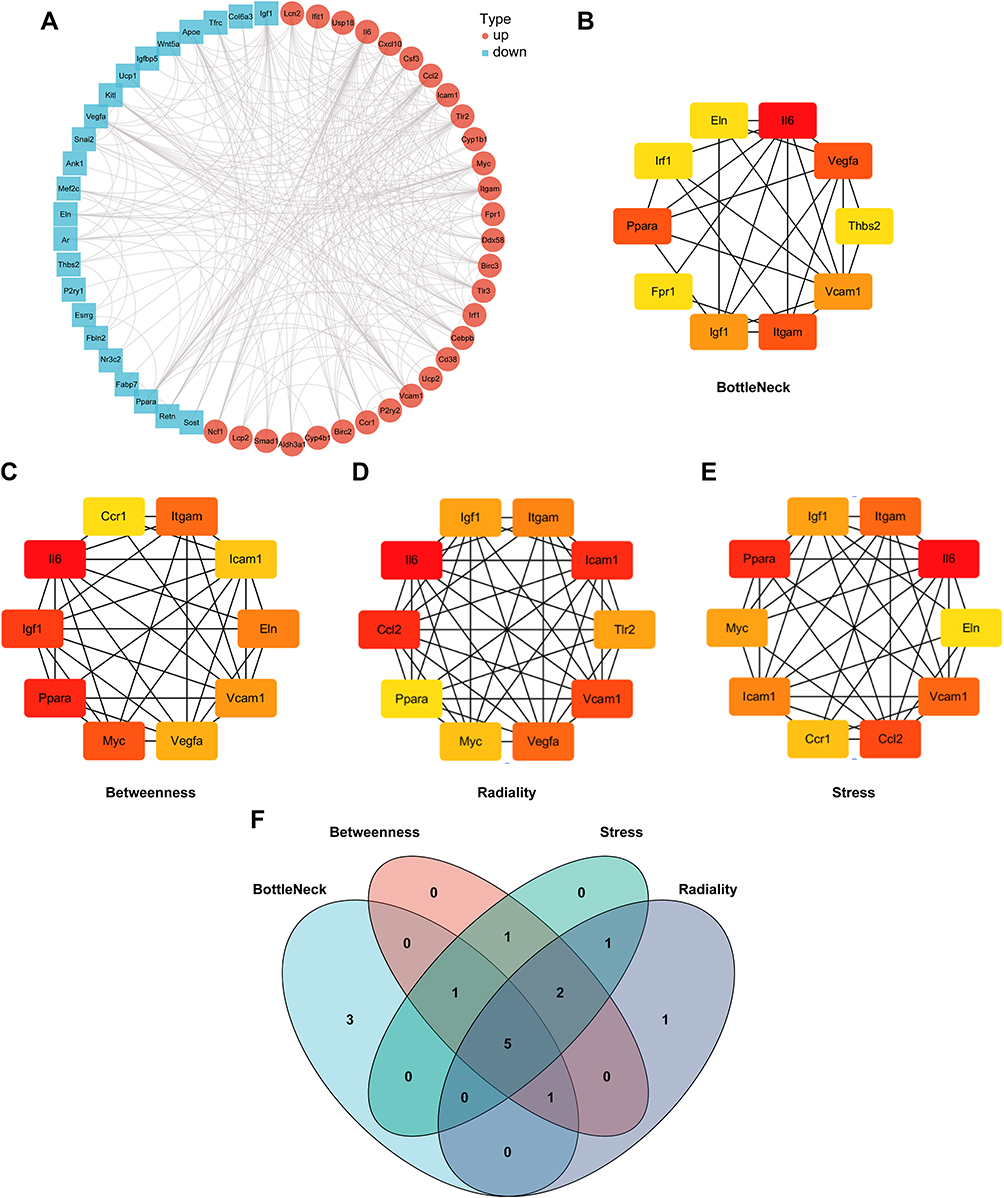 |
Figure 3 PPI network construction. (A) PPI network construction of overlapped genes (Retinoic acid target genes and DEGs). (B–E) Top 10 genes with the highest BottleNeck, Betweenness, Stress and Radiality. (F) Venn diagram summarizing overlapped genes in four sections. |
The Effective RA Concentration Was Determined According to the Survival Rate and Serum Levels of CK-MB and LDH
Our team studied the role of RA in survival condition by intraperitoneally injecting male C57BL/6 mice with LPS (10mg/kg), LPS (10 mg/kg) plus RA (1 mg/kg), LPS (10 mg/kg) plus RA (3 mg/kg), LPS (10 mg/kg) plus DEX (2.5 mg/kg) or an equal volume of saline for 7 days. As presented in Figure 4A, our team computed the 7-day survival rate in the five groups below: the Sham group, LPS group, LPS + RA (1 mg/kg) group, LPS + RA (3 mg/kg) group, LPS + DEX (2.5 mg/kg) group. The 7-day survival rate in the Sham group was nearly a hundred percent, whereas 7 days posterior to LPS treatment, the survival rate of LPS group dropped notably to 40%. RA (1 mg/kg) pretreatment enhanced the survival rate to 50% in LPS-exposed animals. RA (3 mg/kg) pretreatment elevated the survival rate to 70% in LPS-exposed animals. DEX (2.5 mg/kg) pretreatment elevated the survival rate to 60% in LPS-exposed animals.
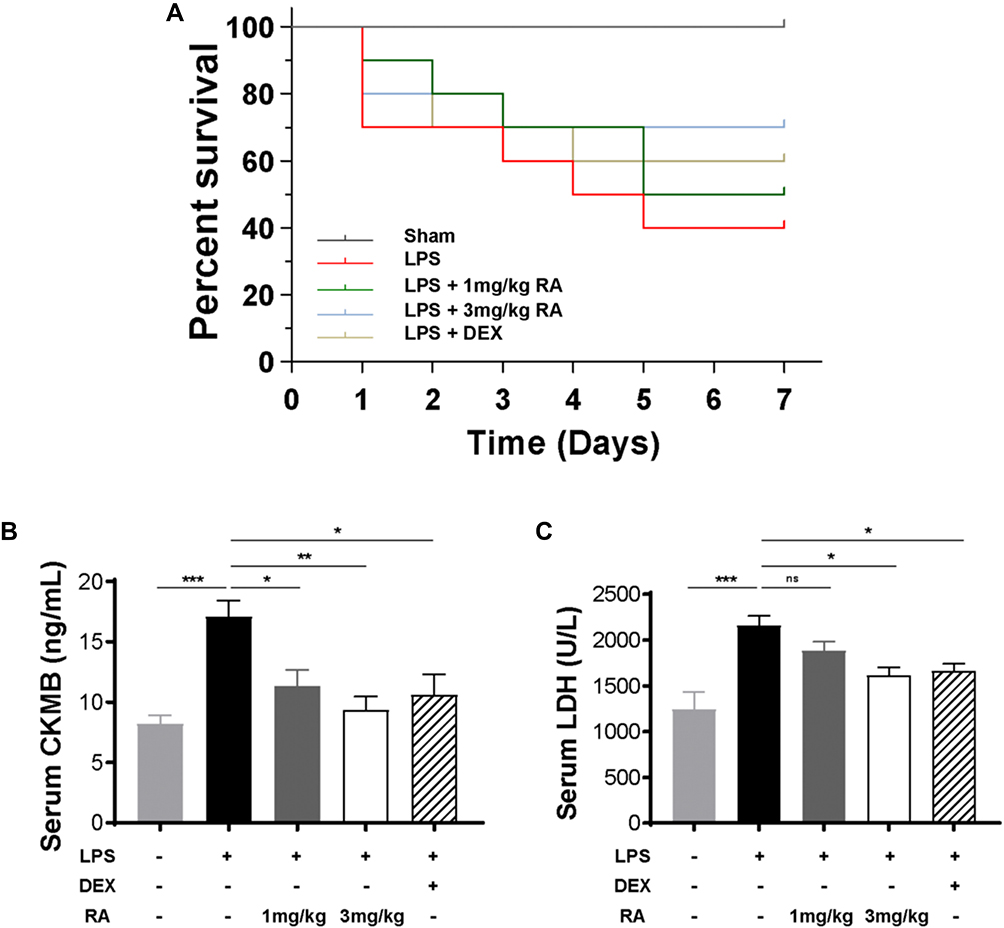 |
Figure 4 Determine the effective concentration of RA. (A) Survival curve of mice treated with saline, LPS (10 mg/kg), LPS (10 mg/kg) plus RA (1 mg/kg), LPS (10 mg/kg) plus RA (3 mg/kg), LPS (10 mg/kg) plus DEX (2.5 mg/kg). Observe and record the mortality of mice within 1 week (n = 10) .(B and C) The serum levels of CK-MB and LDH in mice treated with saline, LPS (10 mg/kg), LPS (10 mg/kg) plus RA (1 mg/kg), LPS (10 mg/kg) plus RA (3 mg/kg), LPS (10 mg/kg) plus DEX (2.5 mg/kg) for 24h were determined. *P < 0.05, **P < 0.01, ***P < 0.001 vs LPS. ns: no significant difference. |
Since LDH and CK-MB are sensitive biomarkers of cardiac injury, our team measured LDH and CK-MB levels in serum. We found that using RA (1 mg/kg), RA (3 mg/kg) and DEX (2.5 mg/kg) significantly reduced the level of CK-MB in LPS-injected mice, and the administration of RA (3 mg/kg) displayed the most significant effect (Figure 4B). Meanwhile, the administration of RA (3 mg/kg) and DEX (2.5 mg/kg) significantly reduced LDH levels in LPS-injected mice. However, the administration of RA (1 mg/kg) also reduced LDH in LPS-injected mice, whereas it was not statistically significant (Figure 4C). As high-dose group showed a more significant efficacy when it came to the improvement of mortality and myocardial injury in mice, the dose of RA was 3 mg/kg in the subsequent experiments.
RA Suppressed Oxidative Stress, Cardiac Inflammation, and Cardiac Injury in LPS-Treated Mice
As shown in Figure 5A–C, after LPS treatment, SOD, CAT, GSH/GSSG in the heart tissue of mice in the model group were significantly lower than those in the Sham group (P < 0.05), while SOD, CAT, GSH/GSSG in the heart tissue of the RA group were significantly higher than those in the model group (P < 0.05). The activation of inflammatory response marks one of the most essential pathology variations in sepsis-caused cardiac muscle injury. Hence, our team studied the inflammatory cell infiltration and the mRNA expression of proinflammatory cytokines in all groups. As shown in Figure 5D–F, qRT-PCR results displayed the favorable effect of RA on heart inflammatory events induced by LPS, as proven by the reduced mRNA contents of TNF-α, IL-1β and IL-6 in myocardium tissues. By virtue of the echocardiographic method, our team explored heart functions in 3 groups. RA pretreatment reinforced ejection fraction and fraction shortening in LPS-exposed animals (Figure 5G and 5H).
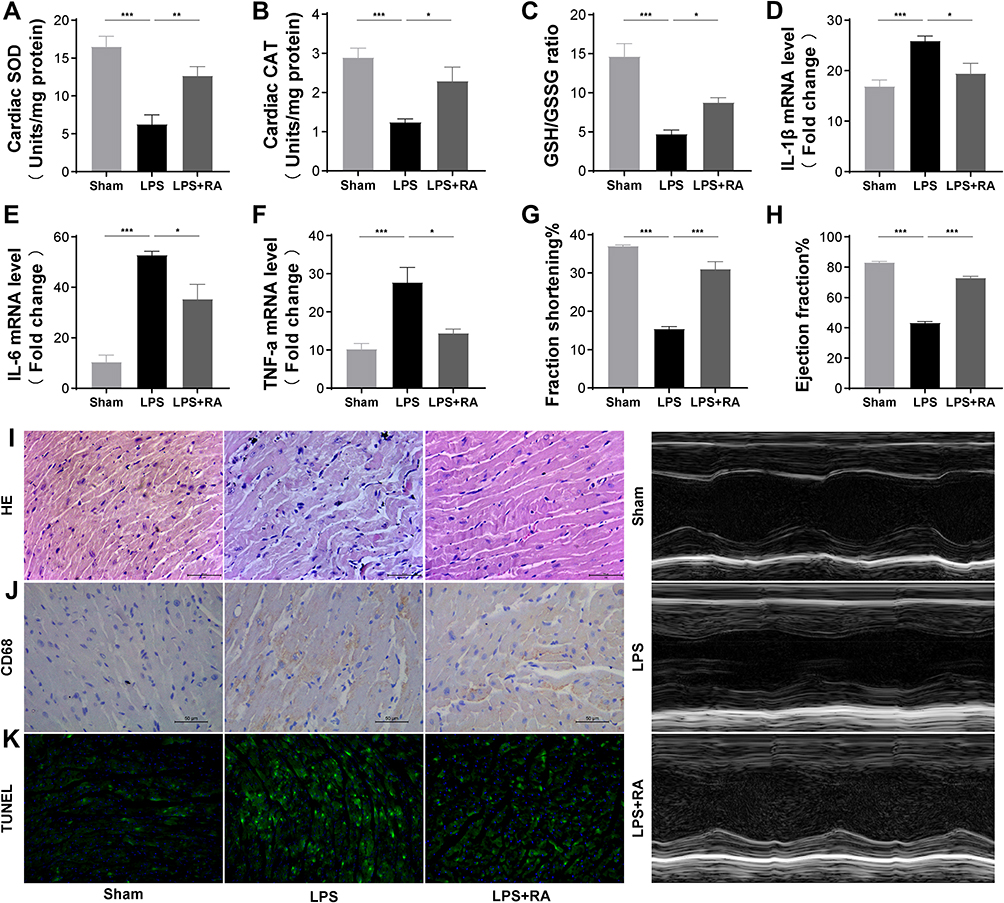 |
Figure 5 RA suppressed oxidative stress, cardiac inflammation, and cardiac injury in LPS-treated mice. (A–C) SOD, CAT, GSH/GSSG levels in myocardial tissue of each group. (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs LPS. (D–F) The mRNA levels of IL-1β, IL-6 and TNF-α in myocardial tissues of each group (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs LPS. (G and H) Effects of saline, LPS and LPS+RA fractional shortening ejection fraction (n = 6). (I–J) Representative images of the morphological analysis and inflammatory cells infiltration as reflected by the H&E staining, and immunohistochemistry staining for CD68 protein.(K) TUNEL assay was used to detect apoptosis of cardiac tissue in each group. |
The myocardium slices were dyed by H&E to evaluate the heart muscle injury and inflammatory cell infiltration. Histological features of heart damage, such as evident capillary congestion, interstitial tissue oedema, and infiltration of massive inflammation cells, were identified in the LPS group. However, in the LPS + RA group, the myocardium fibers registered obvious striation and little inflammatory infiltration was detected in heart muscle tissues (Figure 5I). IHC dyeing revealed that the infiltrative activities of CD68-labeled macrophages, which were caused by LPS, were inhibited by Retinoic acid (Figure 5J). Furthermore, the cardiac injury was evaluated in 3 groups. Tunel dyeing outcomes revealed that the LPS + RA group exhibited less programmed cell death in contrast to the LPS group (Figure 5K). Taken together, those data revealed that Retinoic acid could ameliorate oxidative stress, cardiac inflammation, cardiac injury and heart functions in septic mice.
Retinoic Acid Modulated PPARA, ITGAM, VCAM-1, IGF-1, and IL-6 in LPS-Treated Mice
To verify the network pharmacological forecast of Retinoic acid in LPS-treated mice, we performed qRT-PCR and WB observation to calculate the PPARA, ITGAM, VCAM-1, IGF-1, and IL-6 levels in healthy cardiac samples and cardiac samples from the LPS group and LPS + RA group. Posterior to the normalization with GAPDH, the expressing levels of ITGAM, VCAM-1, and IL-6 were remarkably elevated in the LPS group in contrast to the Sham group, whereas the expressing levels of those biomarkers were remarkably decreased (P< 0.05) in the RA-exposed group in contrast to the LPS group. Meanwhile, the expression levels of PPARA and IGF-1 were remarkably reduced in the LPS group in contrast to the Sham group, whereas the expressing levels of them were remarkably increased (P < 0.05) in the RA-exposed group (Figures 5E and 6A). Then, Western blot analyses verified that Retinoic acid markedly diminished the ITGAM, VCAM-1, and IL-6 protein levels compared with the LPS group, and LPS markedly decreased the protein levels of PPARA and IGF-1. In addition, RA reversed LPS-induced PPARA and IGF-1 inhibition as expected (Figure 6B–F). Therefore, those outcomes revealed that RA might suppress the stimulation of inflammation reactions and engage in the progress of SIC by means of the aforementioned molecules.
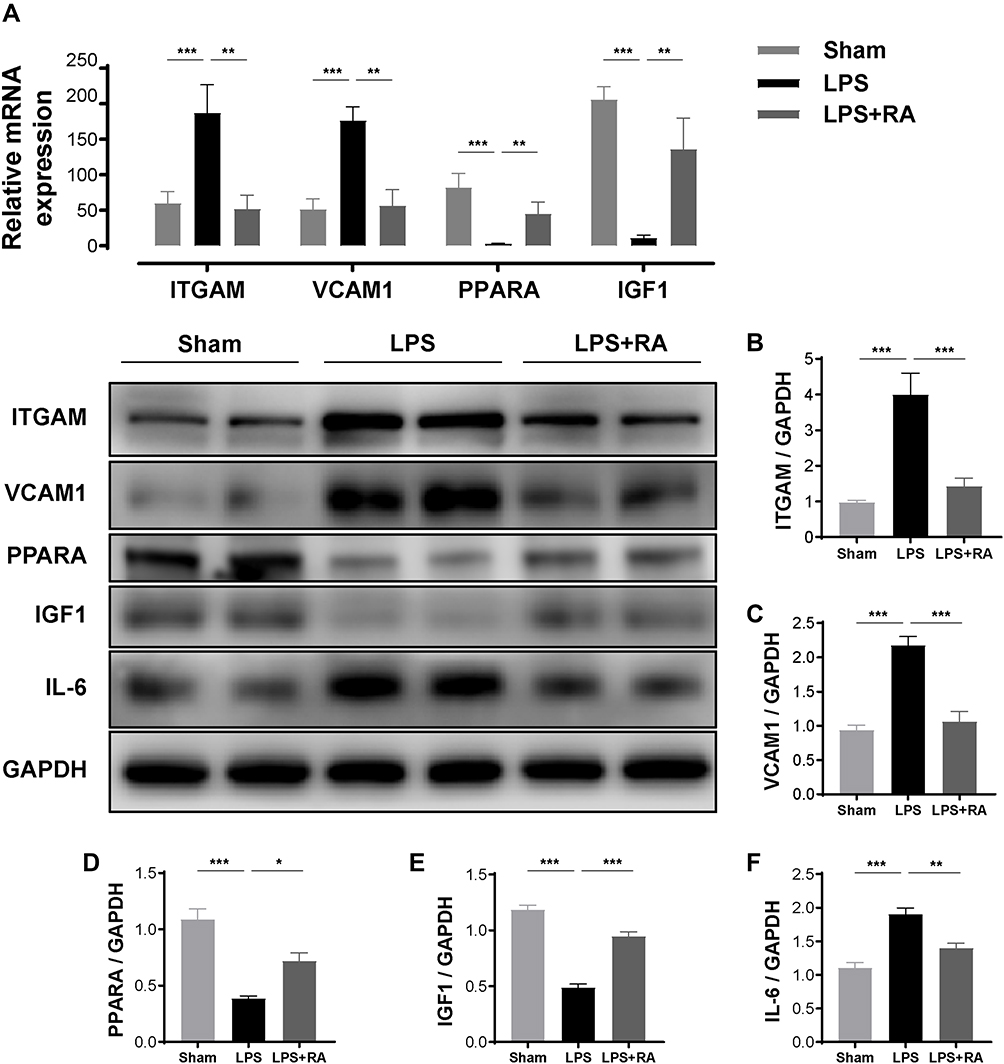 |
Figure 6 RA modulated PPARA, ITGAM, VCAM-1, IL-6 and IGF-1 in hearts of LPS-treated mice. (A) The mRNA levels of ITGAM, VCAM-1, PPARA and IGF-1 in myocardial tissues of each group (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs LPS. (B–F) The protein levels of ITGAM, VCAM-1, PPARA, IGF-1, and IL-6 in myocardial tissues of each group (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 vs LPS. |
Retinoic Acid Can Regulate the Activation of the PI3K/Akt Signaling Pathway in LPS-Treated Mice
In order to further verify the network pharmacological prediction of RA in lipopolysaccharide-induced cardiac dysfunction, Western blot analysis was performed to detect the phosphorylation level of Akt. The results showed that RA could restore the expression of P-Akt in LPS-treated mouse heart tissues (P < 0.05) (Figure 7).
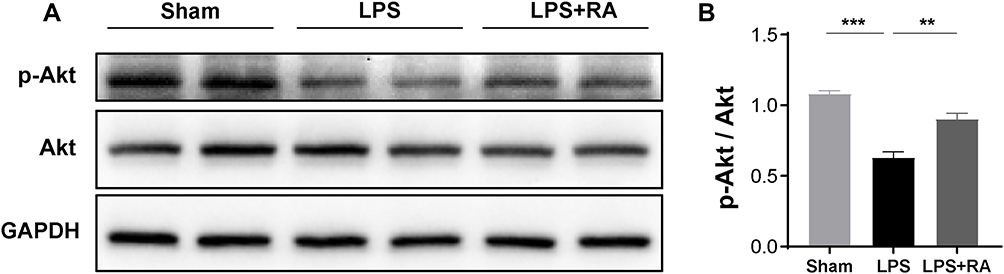 |
Figure 7 RA affects the expression of p-Akt in hearts of LPS treated mice. (A) Representative Western blot images of p-Akt, Akt. (B) Densitometric quantification analysis of the protein expression levels of p-Akt and Akt in mice. **P < 0.01, ***P < 0.001 vs LPS. |
Discussion
The definition for sepsis from the third international consensus states that “sepsis is a lethal organ function disorder induced by an aberrant reaction to infections”.39 Cytokines, especially IL‐1β, IL‐6, and TNF‐α, are major contributors in the initiation of SIC.40 It has been demonstrated that when endotoxin like LPS binds to the receptor TLR4 expressed on cardiomyocytes and macrophages, these cells can release massive inflammatory mediators, such as MCP-1, GM-CSF, IL-1α, IL-1β, IL-6, IL-7, IL-8, and IL-12.41
Retinoic acid (RA) has been reported to reduce the levels of circulation endotoxin and ameliorate survival in endotoxaemic rats.42 Furthermore, RA was a powerful derivant of vitamin A. In previous studies, administrating vitamin A to infants and minors could decrease the risks of septic diseases, immune deficiencies and inflammatory events in endemic regions with deficient vitamin.17,43,44 Eriksson et al also demonstrated that vitamin A administered before a E. coli endotoxin infusion modified the harmful events on heart-lung systems which was caused by such LPS.45 Pretreating with vitamin A counteracted the role of endotoxin in mean arterial pressure (MAP) and cardiac index (CI). Therefore, exploring the mechanism by which RA fights against SIC is quite pregnant.
Network pharmacology method is a comparatively new way to investigate the treatment potency and potential causal links of medicines on the foundation of the net of medicines and targets.46 Herein, our team utilized network pharmacology method to explore the treatment targets participating in the RA healing of SIC. We revealed that RA exposure could elevate survival rate and heart functions of LPS-induced mice while inhibiting inflammatory cytokines and oxidative stress of cardiac muscles. Furthermore, the treatment of RA reversed the production of PPARA, ITGAM, VCAM-1, IGF-1, and IL-6 in LPS-induced SIC.
PPARs are vital targets for approved and experiment medicines in substantial clinical indications, such as metabolism and inflammation illnesses.47–49 The expression of PPARA is extensive in our bodies (particularly in hearts, kidneys, and livers), as an important regulator in the heart after LPS administration.50 Described that heart PPARA expression was imperative for protecting against sepsis-triggered heart damage.51 The 3 PPAR sub-groups, PPARα, PPARγ, and PPARβ/δ generate heterodimers with their obligatory dimer partner RXR,52 and RA modulates genetic expression straightly via binding to a heterodimer of the RARs and RXRs, which are capable of binding to RAREs in the modulatory area of the targeted genes.17 These results suggest that altered RA-mediated PPARA expression might be vital for sepsis-associated end-organ damage and function disorder, particularly in cardiac tissues.
Integrin subunit alpha M (ITGAM) was discovered to be highly expressed in adults and sepsis of the newborn.53,54 A past research unraveled that ITGAM primarily facilitated septic development via fostering the nucleus, cytoplasm movement and stimulating the releasing of HMGB1.55 ITGAM block antibodies or suppressors could defend mice against the fatalness related to LPS and microbe sepsis.56 ITGAM, namely CD11b, regulates the activation, adhesion, and migration of leucocytes from blood to injury sites.57 Vitamin A and its active metabolite retinoic acid (RA) are essential for the development and function of the immune system. Recent studies have also indicated that vitamin A stimulates the development of CD11b+ dendritic cells, and affects the generation of a specific niche that drives CD11b+ dendritic cells (CD11b+ DC) differentiation.58–61 Thus, we speculated that RA might exert an anti-SIC effect via preventing the ITGAM-associated leukocyte recruitment to inflamed tissues.
The vascular cell adhesion molecule (VCAM-1), a heterodimeric molecule expressed on the surface of leukocytes, is induced in inflammatory stimulation.46,62–64 Mice deficient of endothelial selectins exhibited increased survival in an animal model of sepsis.65,66 Furthermore, increased serum content of VCAM1 was a superior predicting factor for sepsis-induced brain diseases in adult community-onset sepsis on admission.67 Recently, Moser J et al have identified that RIG-I, a new modulator of endothelium pro-inflammation stimulation functioning in parallel with TLR4, can regulate the expressing of VCAM-1 in reaction to LPS exposure.68 Gille et al reported that pretreatment with all-trans-retinoic acid prevented the TNFa-mediated VCAM-1 induction.69 In the present study, the experiment outcomes revealed the suppressive role of RA in VCAM-1 generation.
The insulin-like growth factor-1 (IGF-1), a hormone with an insulin-alike architecture, is the main mediating factor of growing hormone.70 Additionally, studies have demonstrated that oxidative stress regulates the level of IGF-1, which is reduced in the acute phase of critical patients’ blood samples,71,72 and that IGF-1 can facilitate the growth and repairment of hearts.73 Furthermore, IGF-1 may defend our hearts against sepsis-triggered myocarditis.74–76 In another study, the researchers found that RA increased the production of THREE-DIMENSIONAL human dermal equivalents (HDEs) IGF1 and IGF2.77 The WB outcome revealed that RA restored the expressing of IGF1 in LPS-induced cardiac dysfunction.
The Phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt) pathway is a classic signaling pathway. It plays an important role in regulating cell growth, proliferation, differentiation, metabolism, cytoskeletal reorganization, autophagy and apoptosis.78–80 Various growth factors and cytokines activate the PI3K/Akt signaling pathway, which ultimately phosphorylates Akt. Substantial studies have revealed that activated Akt1 has cardioprotective effects.81–83 A number of studies have found that the apoptosis of myocardial cells in Akt2 knockout mice is more serious than that in normal mice during myocardial ischemia, which reveals that Akt2 can also reduce the apoptosis of myocardial cells and protect the heart.84 Therefore, the activation of the PI3K/Akt signaling pathway can reduce cardiac injury and protect cardiac function through various ways. By analyzing the potential target of RA-treated lipopolysaccharide-induced cardiac dysfunction, we discovered that the PI3K/Akt signaling pathway was the key pathway in the RA treatment of lipopolysaccharide-induced cardiac dysfunction. After experimental verification, we found that RA could restore the activation of the PI3K/Akt signaling pathway in the heart tissues of LPS-treated mice.
Hence, our team estimate that RA might be an anti-inflammation agent in SIC. Nevertheless, there were certain deficiencies in our research. First, as the experimental subjects were merely mice, future researches have to investigate the roles of lipopolysaccharide-induced cardiac dysfunction in human. Furthermore, the effects of the 5 key targets of RA on LPS-induced cardiac dysfunction in mice require further exploration so as to unravel the corresponding mechanisms underneath.
Conclusion
In the present research, we analyzed the cellular component, biological process, molecular function, and relevant pathways of retinoic acid, as well as its molecular effects on lipopolysaccharide-induced cardiac dysfunction in mice through extensive bioinformatics analysis. Using the Cytohubba software, we successfully identified five potential key therapeutic targets. In addition, by regulating 5 survival-related key therapeutic targets and a key pathway, PI3K-Akt signaling pathway, our team confirmed that Retinoic acid could be a potential therapeutic drug for lipopolysaccharide-induced cardiac dysfunction.
Abbreviations
SIC, sepsis-induced cardiomyopathy; CC, cellular component; DEGs, differentially expressed genes; LPS, lipopolysaccharide; RA, retinoic acid; MF, molecular function; FS, fractional shortening; IHC, immunohistochemistry; TdT, terminal deoxynucleotidyl transferase; RAREs, RA response elements; MAP, Mean Arterial Pressure; PPI, protein–protein interaction; BP, biological process; CI, cardiac index; PPARs, peroxisome proliferator-activated receptors; RXR, retinoid X receptor; ATRA, all-trans retinoic acid; EF, ejection fraction; RXRs, retinoid X receptors; ITGAM, integrin subunit alpha M; HDEs, human Dermal equivalents; LN, liquid nitrogen; SMILES, simplified molecular input line entry specification; H&E, hematoxylin and eosin; WB, Western blot; RIG-I, retinoic acid inducible gene-I; RARs, RA nuclear receptors.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author and GEO database (https://www.ncbi.nlm.nih.gov/geo).
Ethics Statement
According to the Regulations and Rules of “Guidelines for Ethical Review of the Welfare in Laboratory Animals” (2018), each animal assay was accepted by the Animal Experimentation Ethics Committee of Wenzhou Medical University (Approval Ethical Inspection ID: WYYY-AEC-2021-301).
Acknowledgments
We are grateful for the experimental instruments provided by the First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China.
Author Contributions
Xi Wang and Chang Kong share first authorship. X. Wang and H. Tang conceived and designed the experiments. Ch. Kong, X. Wang and P. Liu executed the experiments and analyzed the samples. X. Wang, B. Zhou and W. Geng analyzed the data. X. Wang and Ch. Kong wrote the first version of the manuscript. All authors interpreted the data, critically revised the manuscript, and approved the final version of the manuscript. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
We acknowledge the funding received from the Natural Science Foundation of China (Grant No. 81774109 and 81973620) and Wenzhou Municipal Science and Technology Bureau (ZY2019015).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Qian M, Lou Y, Wang Y, et al. PICK1 deficiency exacerbates sepsis-associated acute lung injury and impairs glutathione synthesis via reduction of xCT. Free Radic Biol Med. 2018;1(18):23–34. doi:10.1016/j.freeradbiomed.2018.02.028
2. Fernando SM, Rochwerg B, Seely A. Clinical implications of the third international consensus definitions for sepsis and septic shock (Sepsis-3). CMAJ. 2018;36(190):E1058–9. doi:10.1503/cmaj.170149
3. Hollenberg SM, Singer M. Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol. 2021;6(18):424–434. doi:10.1038/s41569-020-00492-2
4. Weiss SL, Cvijanovich NZ, Allen GL, et al. Differential expression of the nuclear-encoded mitochondrial transcriptome in pediatric septic shock. Crit Care. 2014;6(18):623. doi:10.1186/s13054-014-0623-9
5. Meyer NJ. Finding a needle in the haystack: leveraging bioinformatics to identify a functional genetic risk factor for sepsis death. Crit Care Med. 2015;1(43):242–243. doi:10.1097/CCM.0000000000000664
6. Burnham KL, Davenport EE, Radhakrishnan J, et al. Shared and distinct aspects of the sepsis transcriptomic response to fecal peritonitis and pneumonia. Am J Respir Crit Care Med. 2017;3(196):328–339. doi:10.1164/rccm.201608-1685OC
7. Scicluna BP, van Vught LA, Zwinderman AH, et al. Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. Lancet Respir Med. 2017;10(5):816–826.
8. Jia L, Wang Y, Wang Y, et al. Heme oxygenase-1 in macrophages drives septic cardiac dysfunction via suppressing lysosomal degradation of inducible nitric oxide synthase. Circ Res. 2018;11(122):1532–1544. doi:10.1161/CIRCRESAHA.118.312910
9. Smeding L, Plotz FB, Groeneveld AB, et al. Structural changes of the heart during severe sepsis or septic shock. Shock. 2012;5(37):449–456. doi:10.1097/SHK.0b013e31824c3238
10. Vanasco V, Saez T, Magnani ND, et al. Cardiac mitochondrial biogenesis in endotoxemia is not accompanied by mitochondrial function recovery. Free Radic Biol Med. 2014;77:1–9. doi:10.1016/j.freeradbiomed.2014.08.009
11. Wagner S, Schurmann S, Hein S, et al. Septic cardiomyopathy in rat LPS-induced endotoxemia: relative contribution of cellular diastolic Ca(2+) removal pathways, myofibrillar biomechanics properties and action of the cardiotonic drug levosimendan. Basic Res Cardiol. 2015;5(110):507. doi:10.1007/s00395-015-0507-4
12. Schilling J, Lai L, Sambandam N, et al. Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism by repressing peroxisome proliferator-activated receptor gamma coactivator-1 signaling. Circ Heart Fail. 2011;4(4):474–482. doi:10.1161/CIRCHEARTFAILURE.110.959833
13. Ziolo MT, Katoh H, Bers DM. Expression of inducible nitric oxide synthase depresses beta-adrenergic-stimulated calcium release from the sarcoplasmic reticulum in intact ventricular myocytes. Circulation. 2001;24(104):2961–2966. doi:10.1161/hc4901.100379
14. Alvarez S, Vico T, Vanasco V. Cardiac dysfunction, mitochondrial architecture, energy production, and inflammatory pathways: interrelated aspects in endotoxemia and sepsis. Int J Biochem Cell Biol. 2016;81(Pt B(81)):307–314. doi:10.1016/j.biocel.2016.07.032
15. Nurrahmah QI, Madhyastha R, Madhyastha H, et al. Retinoic acid abrogates LPS-induced inflammatory response via negative regulation of NF-kappa B/miR-21 signaling. Immunopharmacol Immunotoxicol. 2021;3(43):299–308. doi:10.1080/08923973.2021.1902348
16. Li S, Lei Y, Lei J, et al. All-trans retinoic acid promotes macrophage phagocytosis and decreases inflammation via inhibiting CD14/TLR4 in acute lung injury. Mol Med Rep. 2021;24(6). doi:10.3892/mmr.2021.12508
17. Austenaa LM, Carlsen H, Hollung K, et al. Retinoic acid dampens LPS-induced NF-kappaB activity: results from human monoblasts and in vivo imaging of NF-kappaB reporter mice. J Nutr Biochem. 2009;9(20):726–734. doi:10.1016/j.jnutbio.2008.07.002
18. Martire-Greco D, Landoni VI, Chiarella P, et al. all-trans-retinoic acid improves immunocompetence in a murine model of lipopolysaccharide-induced immunosuppression. Clin Sci. 2014;5(126):355–365. doi:10.1042/CS20130236
19. Tang M, Xie X, Yi P, et al. Integrating network pharmacology with molecular docking to unravel the active compounds and potential mechanism of simiao pill treating rheumatoid arthritis. Evid Based Complement Alternat Med. 2020;2020:5786053. doi:10.1155/2020/5786053
20. Liu Z, Huo JH, Dong WT, et al. A study based on metabolomics, network pharmacology, and experimental verification to explore the mechanism of qinbaiqingfei concentrated pills in the treatment of mycoplasma pneumonia. Front Pharmacol. 2021;12:761883. doi:10.3389/fphar.2021.761883
21. Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;11(4):682–690. doi:10.1038/nchembio.118
22. Recanatini M, Cabrelle C. Drug research meets network science: where are we? J Med Chem. 2020;16(63):8653–8666. doi:10.1021/acs.jmedchem.9b01989
23. Hopkins AL. Network pharmacology. Nat Biotechnol. 2007;10(25):1110–1111. doi:10.1038/nbt1007-1110
24. Tao W, Xu X, Wang X, et al. Network pharmacology-based prediction of the active ingredients and potential targets of Chinese herbal Radix Curcumae formula for application to cardiovascular disease. J Ethnopharmacol. 2013;1(145):1–10. doi:10.1016/j.jep.2012.09.051
25. Fang J, Liu C, Wang Q, et al. In silico polypharmacology of natural products. Brief Bioinform. 2018;6(19):1153–1171. doi:10.1093/bib/bbx045
26. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41(Database issue(41)):D991–5. doi:10.1093/nar/gks1193
27. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;7(43):e47. doi:10.1093/nar/gkv007
28. Kim S, Chen J, Cheng T, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49(D1(49)):D1388–95. doi:10.1093/nar/gkaa971
29. Szklarczyk D, Santos A, von Mering C, et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016;44(D1(44)):D380–4. doi:10.1093/nar/gkv1277
30. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;1(25):25–29. doi:10.1038/75556
31. Li S, Li L, Meng X, et al. DREAM: a database of experimentally supported protein-coding RNAs and drug associations in human cancer. Mol Cancer. 2021;1(20):148. doi:10.1186/s12943-021-01436-1
32. Altermann E, Klaenhammer TR. PathwayVoyager: pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genom. 2005;6:60. doi:10.1186/1471-2164-6-60
33. Backes C, Keller A, Kuentzer J, et al. GeneTrail–advanced gene set enrichment analysis. Nucleic Acids Res. 2007;35(Web Server issue(35)):W186–92. doi:10.1093/nar/gkm323
34. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1(47)):D607–13. doi:10.1093/nar/gky1131
35. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;11(13):2498–2504. doi:10.1101/gr.1239303
36. Liu C, He D, Zhao Q. Licoricidin improves neurological dysfunction after traumatic brain injury in mice via regulating FoxO3/Wnt/beta-catenin pathway. J Nat Med. 2020;4(74):767–776. doi:10.1007/s11418-020-01434-5
37. Chen Y, Tang J, Zhang Y, et al. Astaxanthin alleviates gestational diabetes mellitus in mice through suppression of oxidative stress. Naunyn Schmiedebergs Arch Pharmacol. 2020;393(12):2517-2527. doi:10.1007/s00210-020-01861-x
38. Deng C, Zhang B, Zhang S, et al. Low nanomolar concentrations of Cucurbitacin-I induces G2/M phase arrest and apoptosis by perturbing redox homeostasis in gastric cancer cells in vitro and in vivo. Cell Death Dis. 2016;7:e2106. doi:10.1038/cddis.2016.13
39. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;8(315):801–810. doi:10.1001/jama.2016.0287
40. Drosatos K, Lymperopoulos A, Kennel PJ, et al. Pathophysiology of sepsis-related cardiac dysfunction: driven by inflammation, energy mismanagement, or both? Curr Heart Fail Rep. 2015;2(12):130–140. doi:10.1007/s11897-014-0247-z
41. Rossol M, Heine H, Meusch U, et al. LPS-induced cytokine production in human monocytes and macrophages. Crit Rev Immunol. 2011;5(31):379–446. doi:10.1615/critrevimmunol.v31.i5.20
42. Drott PW, Meurling S, Kulander L, et al. Effects of vitamin A on endotoxaemia in rats. Eur J Surg. 1991;10(157):565–569.
43. Slade E, Tamber PS, Vincent JL. The surviving sepsis campaign: raising awareness to reduce mortality. Crit Care. 2003;1(7):1–2. doi:10.1186/cc1876
44. Carcillo JA. Reducing the global burden of sepsis in infants and children: a clinical practice research agenda. Pediatr Crit Care Med. 2005;3(Suppl(6)):S157–64. doi:10.1097/01.PCC.0000161574.36857.CA
45. Eriksson M, Lundkvist K, Drott P, et al. Beneficial effects of pre-treatment with vitamin A on cardiac and pulmonary functions in endotoxaemic pigs. Acta Anaesthesiol Scand. 1996;5(40):538–548. doi:10.1111/j.1399-6576.1996.tb04485.x
46. Kibble M, Saarinen N, Tang J, et al. Network pharmacology applications to map the unexplored target space and therapeutic potential of natural products. Nat Prod Rep. 2015;8(32):1249–1266. doi:10.1039/c5np00005j
47. Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;7(23):351–363. doi:10.1016/j.tem.2012.05.001
48. Choo J, Lee Y, Yan XJ, et al. A Novel Peroxisome Proliferator-activated Receptor (PPAR) gamma agonist 2-hydroxyethyl 5-chloro-4,5-didehydrojasmonate exerts anti-inflammatory effects in colitis. J Biol Chem. 2015;42(290):25609–25619. doi:10.1074/jbc.M115.673046
49. Kilu W, Merk D, Steinhilber D, et al. Heterodimer formation with retinoic acid receptor RXRalpha modulates coactivator recruitment by peroxisome proliferator-activated receptor PPARgamma. J Biol Chem. 2021;1(297):100814. doi:10.1016/j.jbc.2021.100814
50. Feingold K, Kim MS, Shigenaga J, et al. Altered expression of nuclear hormone receptors and coactivators in mouse heart during the acute-phase response. Am J Physiol Endocrinol Metab. 2004;2(286):E201–7. doi:10.1152/ajpendo.00205.2003
51. Standage SW, Waworuntu RL, Delaney MA, et al. Nonhematopoietic peroxisome proliferator-activated receptor-alpha protects against cardiac injury and enhances survival in experimental polymicrobial sepsis. Crit Care Med. 2016;8(44):e594–603. doi:10.1097/CCM.0000000000001585
52. Amoutzias GD, Pichler EE, Mian N, et al. A protein interaction atlas for the nuclear receptors: properties and quality of a hub-based dimerisation network. BMC Syst Biol. 2007;1:34. doi:10.1186/1752-0509-1-34
53. Jamsa J, Huotari V, Savolainen ER, et al. Kinetics of leukocyte CD11b and CD64 expression in severe sepsis and non-infectious critical care patients. Acta Anaesthesiol Scand. 2015;7(59):881–891. doi:10.1111/aas.12515
54. Sheneef A, Mohamed T, Boraey NF, et al. Neutrophil CD11b, CD64 and lipocalin-2: early diagnostic markers of neonatal sepsis. Egypt J Immunol. 2017;1(24):29–36.
55. Zhou H, Li Y, Gui H, et al. Antagonism of integrin CD11b affords protection against endotoxin shock and polymicrobial sepsis via attenuation of HMGB1 nucleocytoplasmic translocation and extracellular release. J Immunol. 2018;5(200):1771–1780. doi:10.4049/jimmunol.1701285
56. Hoshi M, Osawa Y, Ito H, et al. Blockade of indoleamine 2,3-dioxygenase reduces mortality from peritonitis and sepsis in mice by regulating functions of CD11b+ peritoneal cells. Infect Immun. 2014;11(82):4487–4495. doi:10.1128/IAI.02113-14
57. Sim H, Jeong D, Kim HI, et al. CD11b deficiency exacerbates methicillin-resistant staphylococcus aureus-induced sepsis by upregulating inflammatory responses of macrophages. Immune Netw. 2021;21(2):e13. doi:10.4110/in.2021.21.e13
58. Duriancik DM, Hoag KA. Vitamin A deficiency alters splenic dendritic cell subsets and increases CD8(+) Gr-1(+) memory T lymphocytes in C57BL/6J mice. Cell Immunol. 2010;2(265):156–163. doi:10.1016/j.cellimm.2010.08.006
59. Gatto D, Wood K, Caminschi I, et al. The chemotactic receptor EBI2 regulates the homeostasis, localization and immunological function of splenic dendritic cells. Nat Immunol. 2013;5(14):446–453. doi:10.1038/ni.2555
60. Beijer MR, Molenaar R, Goverse G, et al. A crucial role for retinoic acid in the development of Notch-dependent murine splenic CD8 − CD4 − and CD4 + dendritic cells. Eur J Immunol. 2013;6(43):1608–1616. doi:10.1002/eji.201343325
61. Yi T, Cyster JG. EBI2-mediated bridging channel positioning supports splenic dendritic cell homeostasis and particulate antigen capture. Elife. 2013;2:e757. doi:10.7554/eLife.00757
62. Lush CW, Cepinskas G, Kvietys PR. LPS tolerance in human endothelial cells: reduced PMN adhesion, E-selectin expression, and NF-kappaB mobilization. Am J Physiol Heart Circ Physiol. 2000;3(278):H853–61. doi:10.1152/ajpheart.2000.278.3.H853
63. Ulbrich H, Eriksson EE, Lindbom L. Leukocyte and endothelial cell adhesion molecules as targets for therapeutic interventions in inflammatory disease. Trends Pharmacol Sci. 2003;12(24):640–647. doi:10.1016/j.tips.2003.10.004
64. van Meurs M, Wulfert FM, Knol AJ, et al. Early organ-specific endothelial activation during hemorrhagic shock and resuscitation. Shock. 2008;2(29):291–299. doi:10.1097/SHK.0b013e318145a7c1
65. Matsukawa A, Lukacs NW, Hogaboam CM, et al. Mice genetically lacking endothelial selectins are resistant to the lethality in septic peritonitis. Exp Mol Pathol. 2002;1(72):68–76. doi:10.1006/exmp.2001.2416
66. Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Lab Invest. 2006;1(86):9–22. doi:10.1038/labinvest.3700366
67. Su CM, Cheng HH, Tsai TC, et al. Elevated serum vascular cell adhesion molecule-1 is associated with septic encephalopathy in adult community-onset severe sepsis patients. Biomed Res Int. 2014;2014:598762. doi:10.1155/2014/598762
68. Moser J, Heeringa P, Jongman RM, et al. Intracellular RIG-I signaling regulates TLR4-independent endothelial inflammatory responses to endotoxin. J Immunol. 2016;11(196):4681–4691. doi:10.4049/jimmunol.1501819
69. Gille J, Paxton LL, Lawley TJ, et al. Retinoic acid inhibits the regulated expression of vascular cell adhesion molecule-1 by cultured dermal microvascular endothelial cells. J Clin Invest. 1997;3(99):492–500. doi:10.1172/JCI119184
70. Lembo G, Rockman HA, Hunter JJ, et al. Elevated blood pressure and enhanced myocardial contractility in mice with severe IGF-1 deficiency. J Clin Invest. 1996;11(98):2648–2655. doi:10.1172/JCI119086
71. Ahasic AM, Tejera P, Wei Y, et al. Predictors of circulating insulin-like growth factor-1 and insulin-like growth factor-binding protein-3 in critical illness. Crit Care Med. 2015;12(43):2651–2659. doi:10.1097/CCM.0000000000001314
72. Barabutis N. Growth hormone releasing hormone in endothelial barrier function. Trends Endocrinol Metab. 2021;6(32):338–340. doi:10.1016/j.tem.2021.03.001
73. Ungvari Z, Csiszar A. The emerging role of IGF-1 deficiency in cardiovascular aging: recent advances. J Gerontol a Biol Sci Med Sci. 2012;6(67):599–610. doi:10.1093/gerona/gls072
74. Wahlander H, Isgaard J, Jennische E, et al. Left ventricular insulin-like growth factor I increases in early renal hypertension. Hypertension. 1992;1(19):25–32. doi:10.1161/01.hyp.19.1.25
75. He H, Chang X, Gao J, et al. Salidroside mitigates sepsis-induced myocarditis in rats by regulating IGF-1/PI3K/Akt/GSK-3beta signaling. Inflammation. 2015;6(38):2178–2184. doi:10.1007/s10753-015-0200-7
76. Donohue TJ, Dworkin LD, Lango MN, et al. Induction of myocardial insulin-like growth factor-I gene expression in left ventricular hypertrophy. Circulation. 1994;2(89):799–809. doi:10.1161/01.cir.89.2.799
77. Shim JH, Shin DW, Lee TR, et al. The retinoic acid-induced up-regulation of insulin-like growth factor 1 and 2 is associated with prolidase-dependent collagen synthesis in UVA-irradiated human dermal equivalents. J Dermatol Sci. 2012;1(66):51–59. doi:10.1016/j.jdermsci.2011.12.008
78. Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;3(13):195–203. doi:10.1038/nrm3290
79. Hamzehzadeh L, Atkin SL, Majeed M, et al. The versatile role of curcumin in cancer prevention and treatment: a focus on PI3K/AKT pathway. J Cell Physiol. 2018;10(233):6530–6537. doi:10.1002/jcp.26620
80. Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;4(88):435–437. doi:10.1016/s0092-8674(00)81883-8
81. Matsui T, Tao J, Del MF, et al. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;3(104):330–335. doi:10.1161/01.cir.104.3.330
82. Matsui T, Li L, Wu JC, et al. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem. 2002;25(277):22896–22901. doi:10.1074/jbc.M200347200
83. Debosch B, Sambandam N, Weinheimer C, et al. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J Biol Chem. 2006;43(281):32841–32851. doi:10.1074/jbc.M513087200
84. Roberts DJ, Tan-Sah VP, Smith JM, et al. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem. 2013;33(288):23798–23806. doi:10.1074/jbc.M113.482026
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.