")
Back to Journals » International Journal of Nanomedicine » Volume 12
Therapeutic effects of proteoliposomes on X-linked chronic granulomatous disease: proof of concept using macrophages differentiated from patient-specific induced pluripotent stem cells
Authors Brault J, Vaganay G, Le Roy A, Lenormand JL, Cortes S, Stasia MJ
Received 24 November 2016
Accepted for publication 7 January 2017
Published 20 March 2017 Volume 2017:12 Pages 2161—2177
DOI https://doi.org/10.2147/IJN.S128611
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Julie Brault,1,2 Guillaume Vaganay,3 Aline Le Roy,4–6 Jean-Luc Lenormand,1 Sandra Cortes,3 Marie José Stasia1,2
1UMR CNRS 5525, University of Grenoble Alpes, Grenoble, France; 2CGD Diagnosis and Research Centre, University Hospital Centre of Grenoble Alpes, Grenoble, France; 3Synthelis SAS, La Tronche, France; 4IBS, University of Grenoble Alpes, Grenoble, France; 5CNRS, IBS, University Grenoble Alpes, Grenoble, France; 6CEA, IBS, University of Grenoble Alpes, Grenoble, France
Abstract: Chronic granulomatous disease (CGD) is a rare inherited immunodeficiency due to dysfunction of the phagocytic nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex leading to severe and recurrent infections in early childhood. The main genetic form is the X-linked CGD leading to the absence of cytochrome b558 composed of NOX2 and p22phox, the membrane partners of the NADPH oxidase complex. The first cause of death of CGD patients is pulmonary infections. Recombinant proteoliposome-based therapy is an emerging and innovative approach for membrane protein delivery, which could be an alternative local, targeted treatment to fight lung infections in CGD patients. We developed an enzyme therapy using recombinant NOX2/p22phox liposomes to supply the NADPH oxidase activity in X0-linked CGD (X0-CGD) macrophages. Using an optimized prokaryotic cell-free protein synthesis system, a recombinant cytochrome b558 containing functional hemes was produced and directly inserted into the lipid bilayer of specific liposomes. The size of the NOX2/p22phox liposomes was estimated to be around 700 nm. These proteoliposomes were able to generate reactive oxygen species (ROS) in an activated reconstituted cell-free NADPH oxidase activation assay in the presence of recombinant p47phox, p67phox and Rac, the cytosolic components of the NADPH oxidase complex. Furthermore, using flow cytometry and fluorescence microscopy, we demonstrated that cytochrome b558 was successfully delivered to the plasma membrane of X0-CGD-induced pluripotent stem cell (iPSC)-derived macrophages. In addition, NADPH oxidase activity was restored in X0-CGD iPSC-derived macrophages treated with NOX2/p22phox liposomes for 8 h without any toxicity. In conclusion, we confirmed that proteoliposomes provide a new promising technology for the delivery of functional proteins to the membrane of targeted cells. This efficient liposomal enzyme replacement therapy will be useful for future treatment of pulmonary infections in CGD patients refractory to conventional anti-infectious treatments.
Keywords: protein therapy, proteoliposomes, chronic granulomatous disease, NADPH oxidase, induced pluripotent stem cells, macrophages
Introduction
Chronic granulomatous disease (CGD) is a rare inherited disorder of the innate immune system (1/200,000 births), which is caused by mutations in one of the five genes encoding the subunits of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex expressed in phagocytic cells (neutrophils, monocytes/macrophages, eosinophils).1 This enzymatic complex is composed of two membrane subunits (the redox subunit gp91phox, also called NOX2, and p22phox) both forming the flavocytochrome b558 and cytosolic components (p47phox, p67phox and p40phox). When stimulated, the cytosolic subunits translocate to the plasma membrane and associate with cytochrome b558 to form the active NADPH oxidase complex.2 Activated NADPH oxidase is responsible for the production of antimicrobial reactive oxygen species (ROS), and therefore its deficiency in CGD leads to recurrent and life-threatening infections in early childhood.3 The main genetic form of this disease is the X-linked CGD (XCGD; 70% of cases) caused by mutations in the CYBB gene leading to the absence or dysfunction of the cytochrome b558.4 Until today, bone marrow transplantation has been the only curative treatment. However, it is not devoid of risks in a context of infections and immunodeficiency, although recent progress in patient conditioning has improved the safety and efficacy of this treatment in high-risk CGD patients.5 Encouraging results were also obtained using the gene therapy approach, but the type and design of viral vector constructs used are currently under investigation to control random genome insertion and stabilization of the promoter.6,7 In addition, this therapy is highly expensive and difficult to apply to a large number of patients. The current treatment for CGD patients is antibiotic and antifungal prophylaxis throughout life. However, the leading cause of death in CGD patients is pulmonary infections of Aspergillus fumigatus that are often refractory to anti-infectious treatment, even intravenous.8 Therefore, alternative treatments to target the lungs are desperately needed to rapidly fight life-threatening pulmonary infections in CGD patients.
Protein-based therapies are a promising and safe alternative in medicine with >173 proteins approved in France for clinical use in 2014 (Biomédicaments en France: http://www.leem.org/leem-publie-l-etude-biomedicaments-en-france-etat-des-lieux-2014). However, because of their biophysical and biochemical characteristics, membrane proteins are difficult to produce in sufficient amounts for therapeutic uses using classical expression systems. The recent development of cell-free protein synthesis (CFPS) methods improved the efficiency of recombinant membrane protein production.9 In addition, their integration into liposomes to generate proteoliposomes holds great promise to vectorize therapeutic proteins.10–12 Although various strategies are currently available for the delivery of intracellular proteins,13 there is a lack of vectors for membrane proteins. Liposomes are safe nano-carriers that are ideal for the vectorization of not only chemical drugs but also a large number of biological molecules, including nucleic acids, peptides and proteins. They also provide a specific natural environment required for the insertion of functional membrane proteins. Moreover, liposomes can be chemically modified to increase their stability, to follow their biodistribution as well as to improve their targeting.14
Until now, it has been important to consider that there is no example in the literature of the use of proteoliposomes to supplement a protein deficiency in the case of genetic diseases. However, NOX2/p22phox liposomes could be an attractive delivery system for complementing NADPH oxidase activity in the ROS-deficient phagocytic cells of CGD patients. Nevertheless, it is challenging to produce both membrane subunits (NOX2 and p22phox) with heme incorporation.15 In a previous study using an optimized CFPS system, we succeeded in producing a recombinant and active truncated NOX2 protein directly incorporated into liposomes and efficiently delivered to the membrane of human carcinoma cell lines.16 More recently, an active bovine cytochrome b558 was produced in Pichia pastoris and then inserted into liposomes.17,18 However, in cellulo restoration of ROS-deficient phagocytic cells has never been reported using a human functional recombinant cytochrome b558.
In this study, we optimized the production of recombinant proteoliposomes containing an active human cytochrome b558 and we characterized their biophysical and biochemical properties. In addition, we evaluated its ability to reconstitute a functional NADPH oxidase complex in vitro. The choice of a relevant cellular model is also important for evaluating new therapeutic approaches in preclinical studies. Recently, induced pluripotent stem cells (iPSCs) have been shown to be very useful for physiopathologic modeling of diseases.19 CGD patient-specific iPSCs were successfully differentiated into neutrophils and/or macrophages recapitulating the ROS-deficient phenotype to test new therapeutic strategies.20–26 Then, using confocal microscopy and flow cytometry, we demonstrated the capability of liposomes to transport and deliver recombinant cytochrome b558 to the membrane of X0-linked CGD (X0-CGD) iPSC-derived macrophages. Finally, the therapeutic potential of the NOX2/p22phox liposomes was evidenced by the restoration of the NADPH oxidase activity of these ROS-deficient cells using the nitroblue tetrazolium (NBT) chloride test. Absence of toxicity of the proteoliposomes was also ascertained using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
Materials and methods
Chemicals and reagents
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dilinoleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and 1,2-dimyristoyl-sn-glycero-3-phosphate (DMPA) were purchased from Avanti Polar Lipids (Alabaster, AL, USA) and cholesterol from Coger (Paris, France). Chloroform, sodium dithionite (85%), flavin adenine dinucleotide (FAD; 96%), arachidonic acid (from porcine liver, >99%), dimethylsulfoxide (DMSO), Triton X-100, MTT, phorbol 12-myristate 13-acetate (PMA; >99%), NBT (98%), diphenyleneiodonium (DPI) chloride (>98%), superoxide dismutase (SOD) bovine (5,030 U/mg), anti-mouse IgG–peroxidase antibody, bovine serum albumin (BSA), Tris-buffered saline, Tween-20, magnesium acetate, potassium acetate, hemin, iron and Hoechst 33258 were purchased from Sigma-Aldrich (St Quentin Fallavier, France). NADPH (tetrasodium salt; 98% pure) was purchased from Roche Applied Science (Meylan, France). Paraformaldehyde (PFA), fetal bovine serum (FBS), goat serum, Iscove’s Modified Dulbecco’s Medium (IMDM), Alexa Fluor® 488-conjugated goat-F(ab′)2 fragment anti-mouse IgG1 (H+L), anti-NOX2 antibody (clone 54.1) and anti-p22phox antibody (clone 44.1) were from Thermo Fisher Scientific (Courtaboeuf, France). Uncoupled 7D5 monoclonal antibody specific for NOX2 (reference D162-3) was purchased from Clinisciences (Nanterre, France). Glutathione Sepharose™ 4B and PreScission protease were purchased from GE Healthcare (Velizy-Villacoublay, France) and isopropyl 1-thio-β-D-galactopyranoside (IPTG) from MP Biomedicals (Illkirch, France).
Liposome preparation
Liposomes were prepared using a 10 mg/mL lipid mixture of DOPC, DOPE, DMPA and cholesterol in chloroform. Different ratios of these lipids were prepared to obtain the liposome preparations named L1, L2, L3, L4, L5 and L6. Chloroform was evaporated using a Uniequip vacuum centrifuge (Univapo 150H). The thin lipid film was rehydrated with 50 mM HEPES pH 7.5 to obtain 30 mg/mL lipid slurry. This solution was sonicated using a tip sonicator (Branson Digital Sonifier 250) at 20% for 30 s five times and filtered (0.22 μm polyethersulfone [PES] filter).
Cell-free synthesis of recombinant NOX2/p22phox proteoliposomes and purification
The R199Q mutation was introduced into the wild-type (WT) NOX2 cDNA in pBluescript II KS (+) vector using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) and subcloned into the mammalian expression vector pEF-PGKneo as previously described.27 Then, the R199Q mutant NOX2 cDNA and p22phox cDNA sequence were subcloned by Proteogenix (Schiltigheim, France) into a specific plasmid optimized by Synthelis SAS. The two plasmids (15 μg for each plasmid) and the preformed liposome solution were added to 1 mL of the batch reaction mixture (Figure 1). The energy regeneration system (NTPs, salts, amino acids, chaperones, cofactors, etc) was developed by Synthelis SAS. Different concentrations of magnesium (0–50 mM), potassium (0–320 mM), hemin (0–20 μM) and iron (0–10 μM) were tested to optimize the protein expression. The cell-free reaction was carried out at 30°C for 16 h with gentle agitation at 400 rpm using an Escherichia coli extract.16 Liposomes prepared with identical lipid composition in the absence of the cDNA sequences encoding NOX2 and p22phox proteins were used as negative controls. To purify proteoliposomes, cell-free reactions were loaded on top of a three-step discontinuous sucrose gradient (60%, 30% and 5%) prepared in 50 mM HEPES pH 7.5 buffer. After centrifugation at 280,000 g for 1 h at 4°C, fractions were collected at each interface and analyzed using Western blotting and Coomassie blue. Proteoliposomes were stored at −80°C until use. Sterility of proteoliposome preparations was obtained after filtration of the bacterial lysate (0.22 μm), the use of sterile solutions for each component of the reaction mixture and precautions taken during the entire production process.
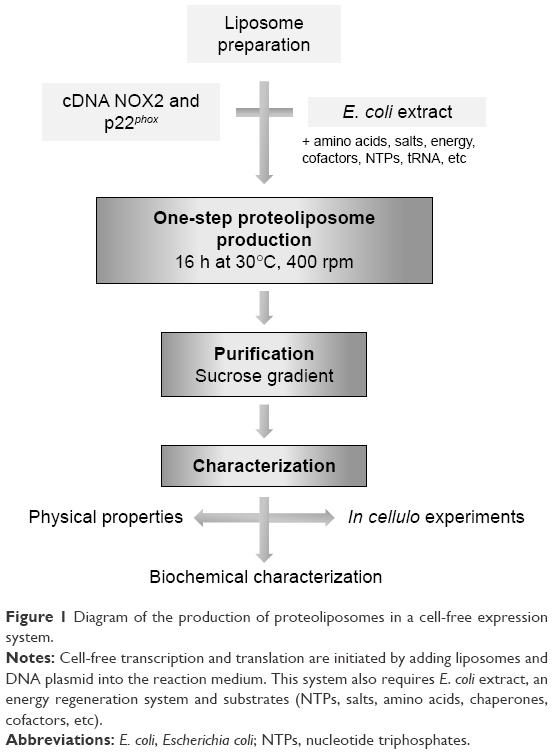 | Figure 1 Diagram of the production of proteoliposomes in a cell-free expression system. |
Proteoliposome size determination
The dynamic light scattering (DLS) technique was used to measure the average particle size using a DynaPro NanoStar instrument (Wyatt Technology Corp., Santa Barbara, CA, USA) on 4 μL samples in cyclic olefin copolymer cuvettes at room temperature. All measurements were taken in triplicate of 10 scattering readings per sample, and data analysis was performed using Dynamics V7 software (Wyatt Technology Corp.).28
Biochemical characterization of proteoliposomes
Western blotting
Samples were separated with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)29 and electrotransferred to a nitrocellulose membrane.30 The membrane was blocked by incubation for 1 h at room temperature in 1× Tris-buffered saline, 0.1% Tween-20 (TBST), 5% nonfat milk. For NOX2 and p22phox detection, the blocked membrane was incubated with an anti-NOX2 antibody (clone 54.1) or an anti-p22phox antibody (clone 44.1), respectively, diluted at 1:1,000 in TBST buffer, 5% nonfat milk. After washing, the nitrocellulose membrane was incubated with an anti-mouse IgG-peroxidase antibody diluted at 1:10,000 in TBST buffer, 5% nonfat milk. Then, the blots were developed using a chemiluminescence method (Amersham ECL; GE Healthcare). Molecular Imager Chemidoc™ XRS+ with ImageLab™ software were used for gel imaging and Western blot imaging (Bio-Rad, Marnes-la-Coquette, France).
Reduced minus-oxidized differential spectrum
The differential spectrum of purified cytochrome b558 was performed as already published.31 The reduced minus-oxidized difference spectra were recorded at room temperature with a spectrophotometer (BioTek Synergy HT, Winooski, VT, USA). Cytochrome b558 integrity was validated by the presence of two absorption bands at 426 nm (Soret) and 558 nm (α band) and the valley at ~410–411 nm. A molar absorption coefficient of ε427–411nm =200 mM−1·cm−1 was used for calculations.32
Production and purification of recombinant cytosolic subunits of the NADPH oxidase complex
P67phox, p47phox and Rac1Q61L were expressed in E. coli as a glutathione S-transferase fusion protein using a pGEX-2T vector. Briefly, protein expression was induced with IPTG (0.2 mM at 20°C for p67phox and p47phox, 0.1 mM at 37°C for Rac1) for 3 h. Rac1 and p47phox fusion proteins were purified by affinity on Glutathione Sepharose™ 4B and were cleaved directly on the matrix using thrombin33 or PreScission protease, respectively. P67phox was used without cleavage.34 The purity and cleavage of recombinant proteins were analyzed using SDS-PAGE and Coomassie blue staining. Recombinant proteins were stored at −80°C until used.
Recombinant cell-free NADPH oxidase activation assay
Cytochrome b558 (8 pmol) or negative liposomes were incubated with recombinant cytosolic subunits p67phox, p47phox and Rac1Q61L as previously described.31,34 After incubation for 10 min at room temperature with arachidonic acid, NADPH oxidase activity was measured by adding 200 μM NADPH to initiate the superoxide production in the presence of 100 μM NBT. A negative control was performed in the absence of NADPH. In some experiments, various concentrations of DPI were added 10 min before the incubation with arachidonic acid. The O2•– production was measured by following the kinetics of the NBT absorbance at 550 nm using a Mithras LB940 spectrophotometer (Berthold Technologies, Bad Wildbad, Germany). SOD-sensitive activity was determined in the presence of 0.1 mg/mL SOD. NADPH oxidase activity was expressed as moles of superoxide produced/s/mol of heme of cytochrome b558 using a molar extinction coefficient of 28.6 nM−1·cm−1 at 550 nm for NBT.35
iPSC culture and hematopoietic differentiation into mature macrophages
iPSCs were reprogrammed using a retroviral method (pMXs-based retroviruses encoding the human transcription factors Oct3/4, Sox2, Klf4 and cMyc from Addgene) from human dermal fibroblasts (hDFs) isolated from the skin biopsy of a healthy control (WT) and a X0-CGD patient as previously described.23,36 Skin biopsies were obtained after informed consent (Ethical permission from the south-east V Person Protection Committee [CPP: 09-CHUG-36, FibroCGD, N° AFSSAPS: 2009-A00944-53]). IPSCs were maintained in an undifferentiated state on irradiated mouse embryonic fibroblasts and fully characterized (Figure S1). Then, hematopoietic differentiation of iPSCs into mature macrophages was performed (Figure S2).23 Macrophages adhered on a 96-well plate for viability assay, on a 24-well plate for flow cytometry and in cellulo activity tests, and on a four-well Nunc™ Lab-Tek™ II Chamber Slide™ (Thermo Fisher Scientific, Courtaboeuf, France) for the phagocytosis experiment and confocal microscopy, as described in the following sections. Cell culture images were acquired using a Nikon Eclipse TS1000 microscope with an Elipse TS100 microscope with a digital slight DS-U3 camera (Nikon, Champigny-sur-Marne, France).
MTT assay
Cell viability was determined using an MTT assay based on the mitochondrial activity of viable cells. X0-CGD iPSC-derived macrophages (15×103 cells) were seeded in a 96-well plate and treated with NOX2/p22phox (20 pmol cytochrome b558) and negative liposomes for 8, 12 and 24 h at 37°C in IMDM. MTT was then added, and the cells were incubated for a further 3–5 h at 37°C. The absorbance was measured at 560 nm using a multiwall scanning Varioskan™ Flash spectrophotometer and SkanIt software (version 2.4.1; Thermo Fisher Scientific, Courtaboeuf, France). The percentage of cell viability was calculated as follows:
|

The viability of untreated cells was arbitrarily set at 100%. All assays were performed in triplicate. Data were expressed as mean ± standard error. A nonparametric Mann–Whitney U-test was used for statistical analysis using StatView 5.0 software (SAS Institute Inc., Cary, NC, USA). A P-value of <0.05 was considered significant.
Phagocytosis experiment
The Candida albicans strain was a clinical isolate kindly provided by the Laboratoire de Mycologie, Grenoble Alpes University Hospital, France. Fungi were prepared as previously described.37 Eight-hour liposome-treated X0-CGD macrophages were incubated with opsonized C. albicans fungi at a 3:1 multiplicity of infection (MOI) for 4 h. Phagocytosis was then analyzed with confocal microscopy as described in the following sections.
Flow cytometry
X0-CGD macrophages (1.5×105) were incubated for 8 h at 37°C with NOX2/p22phox (5 pmol cytochrome b558) or negative liposomes in IMDM and then stained with fluorescein isothiocyanate (FITC)-coupled anti-NOX2 (7D5) and anti-p22phox (clone 44.1) antibodies as previously described.23 Control staining with appropriate isotype-matched control was included to establish thresholds for positive staining. WT iPSC-derived macrophages were used as positive control. Cell fluorescence was quantified using a FACSCanto™ II flow cytometer (BD Biosciences, Le Pont de Claix, France). Data were collected and analyzed with the FACS DIVA software (BD Biosciences) and FlowJo software (Tree Star, Ashland, OR, USA).
Confocal microscopy
A total of 1×105 X0-CGD macrophages were incubated at 37°C in IMDM with NOX2/p22phox (5 pmol cytochrome b558) or negative liposomes and were treated as previously described.31 Cells were stained with the 7D5 antibody (1:20) for 1 h at 4°C followed by incubation with an Alexa Fluor® 488 goat-F(ab′)2 fragment anti-mouse IgG1 (H+L) for 1 h at room temperature. Nuclei were stained with 1 μg/mL Hoechst 33258. Slides were scanned using a Zeiss LSM710 confocal laser-scanning microscope (Carl Zeiss, Jena, Germany) equipped with 488 nm argon and 405 nm diode lasers at the microscopy facility of the Institut Albert Bonniot (Grenoble, France). Image acquisition was performed by Plan-Apochromat 63x/1.4 Oil objective using ZEN software (Carl Zeiss). Images were processed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Nitroblue tetrazolium (NBT) test
X0-CGD iPSCs-derived macrophages (1.5×105) were seeded in a 24-well plate and treated for 8 h with NOX2/p22phox (5 pmol cytochrome b558) and negative liposomes. In cellulo NADPH oxidase activity was measured by the NBT reduction test as previously described.38 Briefly, macrophages were stimulated with 200 ng/mL PMA in the presence of NBT. After 1 h of incubation at 37°C with an agitation at 250 rpm, cells were washed with phosphate-buffered saline (PBS) and fixed in 4% PFA. Images were acquired using a Nikon Eclipse TS1000 microscope with a Nikon DS camera.
Results
Optimization of the production of proteoliposomes
A few years ago, we demonstrated that efficient and easy production of proteoliposomes containing a truncated form of NOX2 could be obtained in a single step with an optimized E. coli-based expression system.16 In this study, we have developed a new cell-free expression system to synthesize both entire NOX2 and p22phox proteins in a one-step reaction to produce an active proteoliposome directly. We therefore studied the impact of several parameters to optimize the yield of production and to synthesize active proteoliposomes (Figure 2).
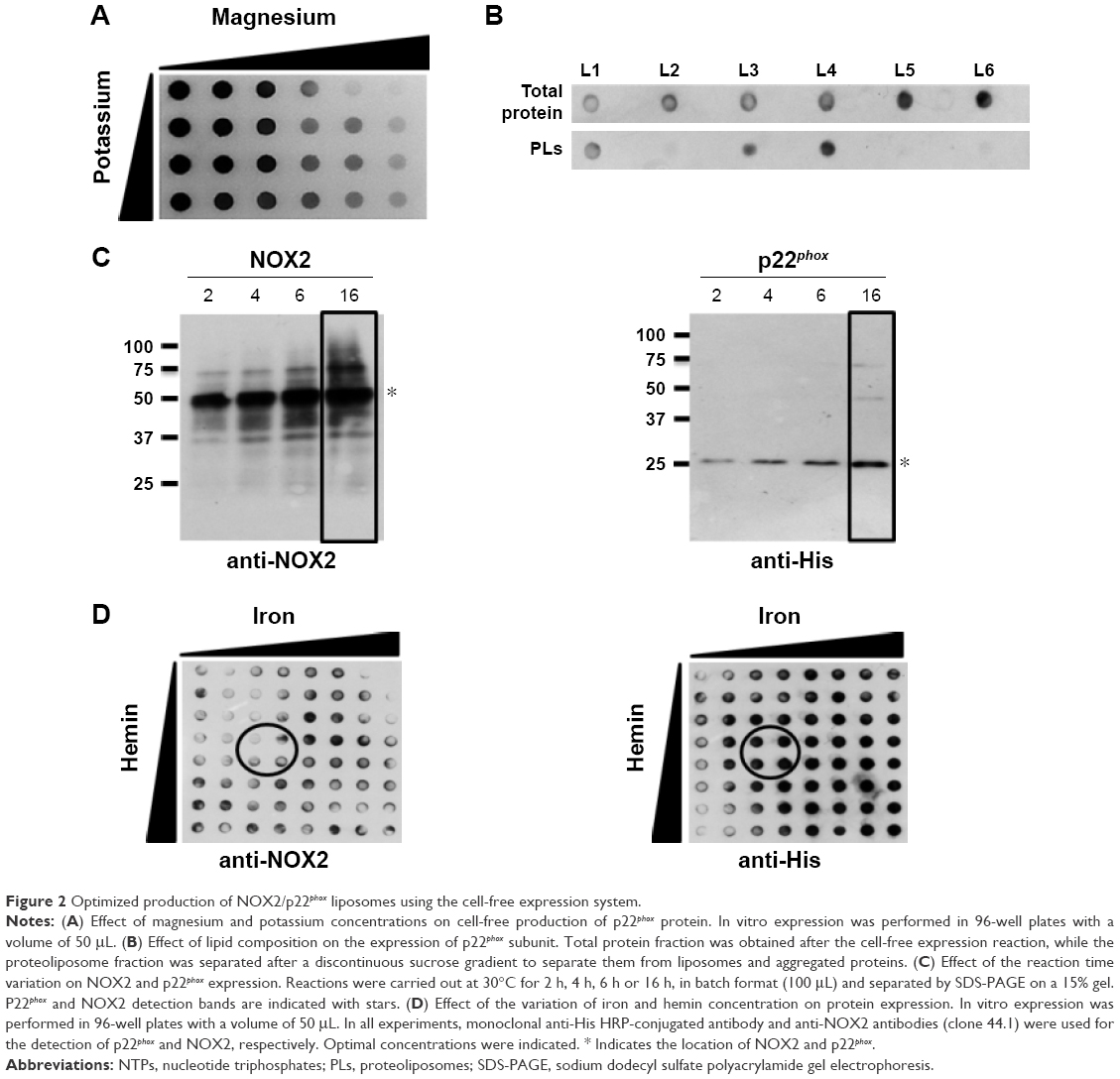 | Figure 2 Optimized production of NOX2/p22phox liposomes using the cell-free expression system. |
First, to optimize the co-expression of NOX2 and p22phox encoded by two different plasmids, several ratios of NOX2 and p22phox genetic material were tested to retain a 1:1 DNA ratio (data not shown). Optimal Mg2+ and K+ concentrations were found to be 15–20 mM and 280–320 mM, respectively (Figure 2A). The lipid composition of the lipid bilayer was also optimized using DOPE, a helper-neutral fusogenic lipid known to favor the fusion with the plasma membrane of targeted cells.39 Six compositions with different lipids, as described in the “Materials and methods” section, were compared for their ability to aid the integration of NOX2 and p22phox into liposomes. Only three of them (L1, L3 and L4) allowed the integration of both proteins to form proteoliposomes (Figure 2B). We selected the composition named “L4” for which the protein integration was the highest after purification on a sucrose gradient, as described in the “Materials and methods” section. The most adequate time of reaction for an optimal expression of both subunits was achieved in a 16 h reaction (Figure 2C). Furthermore, to reconstitute the heme groups within the NOX2 subunit, we performed a correlated screening of hemin and iron concentrations during protein expression, and the condition giving the best compromise for both proteins was chosen (Figure 2D). The reduced minus-oxidized spectra of all conditions shown in Figure 2D were tested for heme reconstitution. We chose the four conditions showing the best spectra with the two characteristic peaks of cytochrome b558 at 426 nm and 558 nm (data not shown).
In vitro characterization of NOX2/p22phox and negative liposomes
Physicochemical properties
NOX2/p22phox liposomes produced in a 16 h reaction using optimized conditions as previously determined (Figure 2) were fully characterized. The average hydrodynamic radius of NOX2/p22phox and negative liposomes was evaluated using the DLS method and was ~350±10 nm (99.6% of the population) and 427±17 nm (95.7% of the population), respectively (Figure 3A). The size of NOX2/p22phox and negative liposomes was around 700 nm and 850 nm, respectively. Then, the successful synthesis and integration of NOX2 and p22phox in the liposomes were confirmed by Coomassie brilliant blue staining and Western blotting (Figure 3B). NOX2/p22phox liposomes (and negative liposomes too) contain contaminating proteins. This is probably due to the bacterial proteins contained in the cell-free expression system. NOX2 and p22phox bands appeared only in the positive liposomes. NOX2 was detected as a non-glycosylated form at 55–58 kDa and p22phox at 20–25 kDa (Figure 3B). A band was also detected around 45 kDa with the anti-NOX2 antibody, which probably corresponds to a degraded form of NOX2.
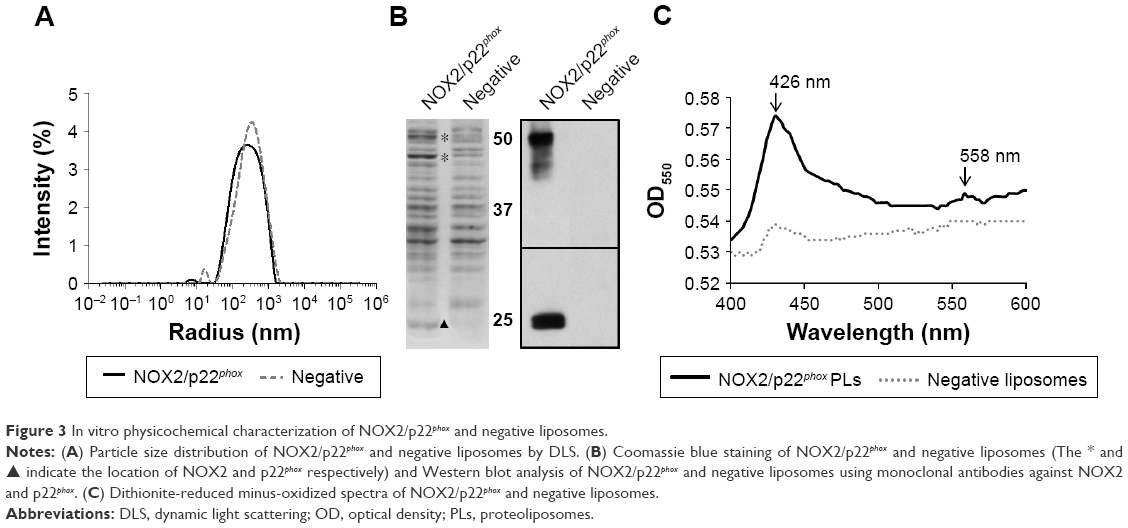 | Figure 3 In vitro physicochemical characterization of NOX2/p22phox and negative liposomes. |
The therapeutic effect of our proteoliposomes is absolutely dependent on proper integration of hemes in NOX2. Indeed, reduced minus-oxidized spectra showed the two characteristic peaks at 426 nm (Soret band) and 558 nm (α band), confirming heme incorporation within the NOX2 subunit in the NOX2/p22phox liposomes (Figure 3C). The cytochrome b558 heme concentration calculated from the absorbance at 426 nm and at 558/559 nm was 1.6 μM and 1.85 μM in NOX2/p22phox liposomes, respectively. Only a weak peak around 426 nm was detected for negative liposomes. It could be due to the presence of residual c- or b-type bacterial cytochromes in the reaction mixture.40
These results confirmed the efficiency of the optimized E. coli-based cell-free expression system to co-express both subunits of the cytochrome b558 and to integrate them into liposomes to form NOX2/p22phox liposomes with no increase in the size of proteoliposomes.
In vitro NADPH oxidase activity of NOX2/p22phox liposomes
We next investigated the ability of NOX2/p22phox liposomes to produce superoxide anions in an in vitro cell-free NADPH oxidase activation assay in the presence of recombinant cytosolic proteins p47phox, p67phox and Rac1Q61L (active form of Rac1), NADPH, NBT and an adequate amount of arachidonic acid determined in preliminary assays (data not shown).31,33,34 The cytochrome b558 heme concentration given by the 426-nm peak was used for the calculation of NADPH oxidase activity. In this reconstituted system, the NADPH oxidase activity of NOX2/p22phox liposomes was 228.7 moles of superoxides O2•–/s/mol of cytochrome b558 heme (±14.7; n=3), whereas in the negative liposomes the activity was only residual (Figure 4A and B). No NADPH oxidase activity was detected in the absence of NADPH stimulation (Figure 4A). The specificity of the O2•– production was confirmed by the significant decrease in the NADPH oxidase activity in the presence of SOD (60% of SOD-sensitive NADPH oxidase activity; Figure 4B). This specific SOD-sensitive activity was estimated around 143.8±16.4 moles of superoxides O2•–/s/mol of cytochrome b558 heme. The remaining 40% of SOD-insensitive oxidase activity corresponds to the diaphorase activity of the NOX2/p22phox liposomes. The effect of various concentrations of DPI, a flavoenzyme inhibitor, was tested on the NADPH oxidase activity. We showed that 200 μM DPI was able to reduce the NADPH oxidase activity by 50% (Figure 4B).
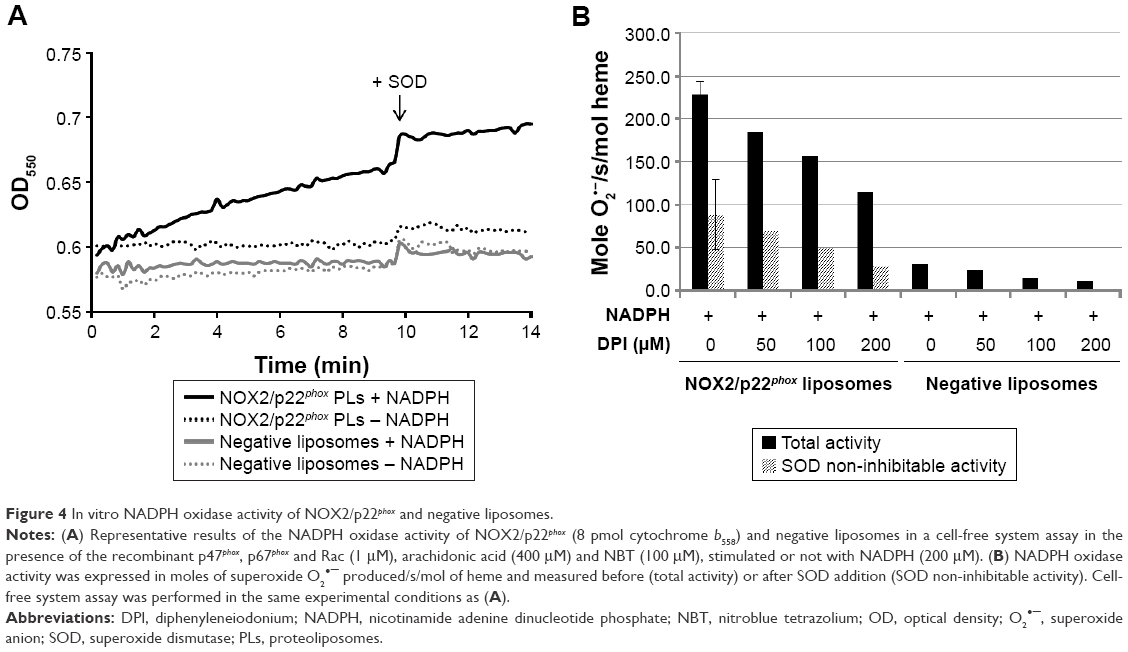 | Figure 4 In vitro NADPH oxidase activity of NOX2/p22phox and negative liposomes. |
Our results demonstrated that the NOX2/p22phox liposomes containing hemes were able to reconstitute a functional NADPH oxidase complex producing superoxide anions in vitro.
Membrane delivery of NOX2 and p22phox subunits in X0CGD iPSC-derived macrophages
One major hurdle for the use of proteoliposomes as a replacement enzyme therapy is the efficiency of delivery of the NOX2/p22phox complex at the plasma membrane of targeted cells. After 8 h of treatment of ROS-deficient X0-CGD macrophages derived from iPSCs with NOX2/p22phox or negative liposomes, the presence of the cytochrome b558 at the plasma membrane of the cells was analyzed using flow cytometry, as described in the “Materials and Methods” section (Figure 5A). The presence of both proteins was detected only in macrophages incubated with NOX2/p22phox liposomes. The mean fluorescence intensity was increased ~1.7- and 2.0-fold for NOX2 and p22phox staining, respectively, compared to negative liposome-treated X0-CGD macrophages and ~1.8- and 3.3-fold for NOX2 and p22phox staining, respectively, compared to untreated X0-CGD macrophages.
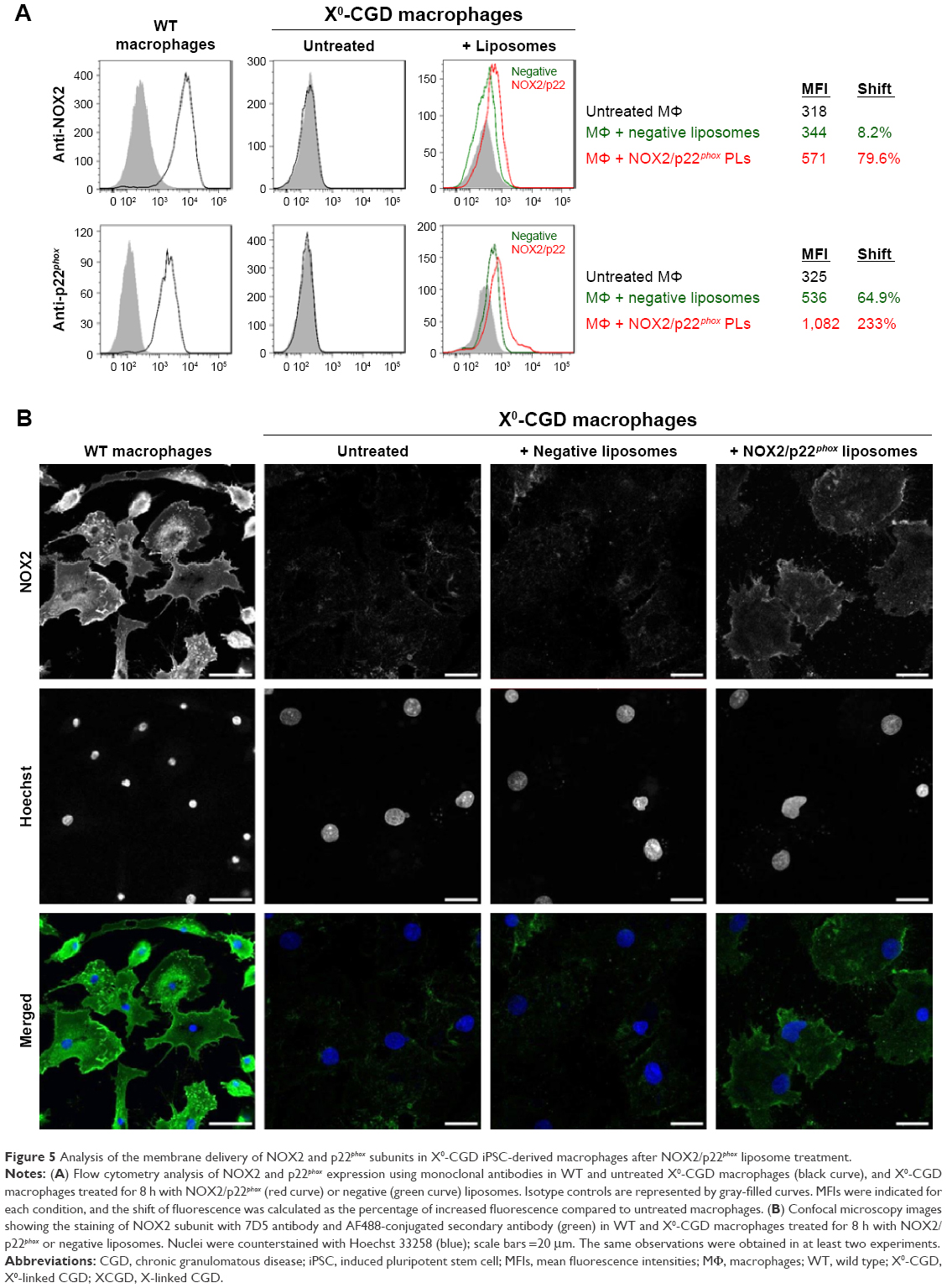 | Figure 5 Analysis of the membrane delivery of NOX2 and p22phox subunits in X0-CGD iPSC-derived macrophages after NOX2/p22phox liposome treatment. |
To further analyze the location of the delivered cytochrome b558, confocal microscopy using the 7D5 antibody targeting external epitopes of NOX2 was performed on 8 h NOX2/p22phox and negative liposome-treated X0-CGD macrophages (Figure 5B). A positive green fluorescence revealing the presence of NOX2 was detected only at the membrane of X0-CGD macrophages treated with NOX2/p22phox liposomes. Some fluorescent areas showing the presence of NOX2 were similar to the 7D5 staining of WT macrophages, suggesting a possible fusion of the NOX2/p22phox liposomes with the membrane of deficient macrophages and satisfactory delivery of cytochrome b558.
In addition, the efficiency of membrane delivery was estimated as the percentage of cells with positive NOX2 staining and it was around 50%–60% of the cells analyzed by flow cytometry and confocal microscopy. As expected, no NOX2 staining was detected in untreated and negative liposome-treated X0-CGD macrophages with both methods.
NOX2 location in liposome-treated X0-CGD macrophages after Candida sp. phagocytosis
Candida is a common pathogen found in CGD patients and responsible for pneumonia;1 therefore, we evaluated the location of the NOX2 subunit in liposome-treated X0-CGD macrophages in a context of fungal infection. The 7D5 staining of macrophages performed after phagocytosis of C. albicans showed a positive signal only in macrophages treated with NOX2/p22phox liposomes (Figure 6A and B). This signal was located around yeasts, indicating a phagosomal location of NOX2. In addition, no NOX2 was detected in X0-CGD macrophages treated with negative liposomes (Figure 6A).
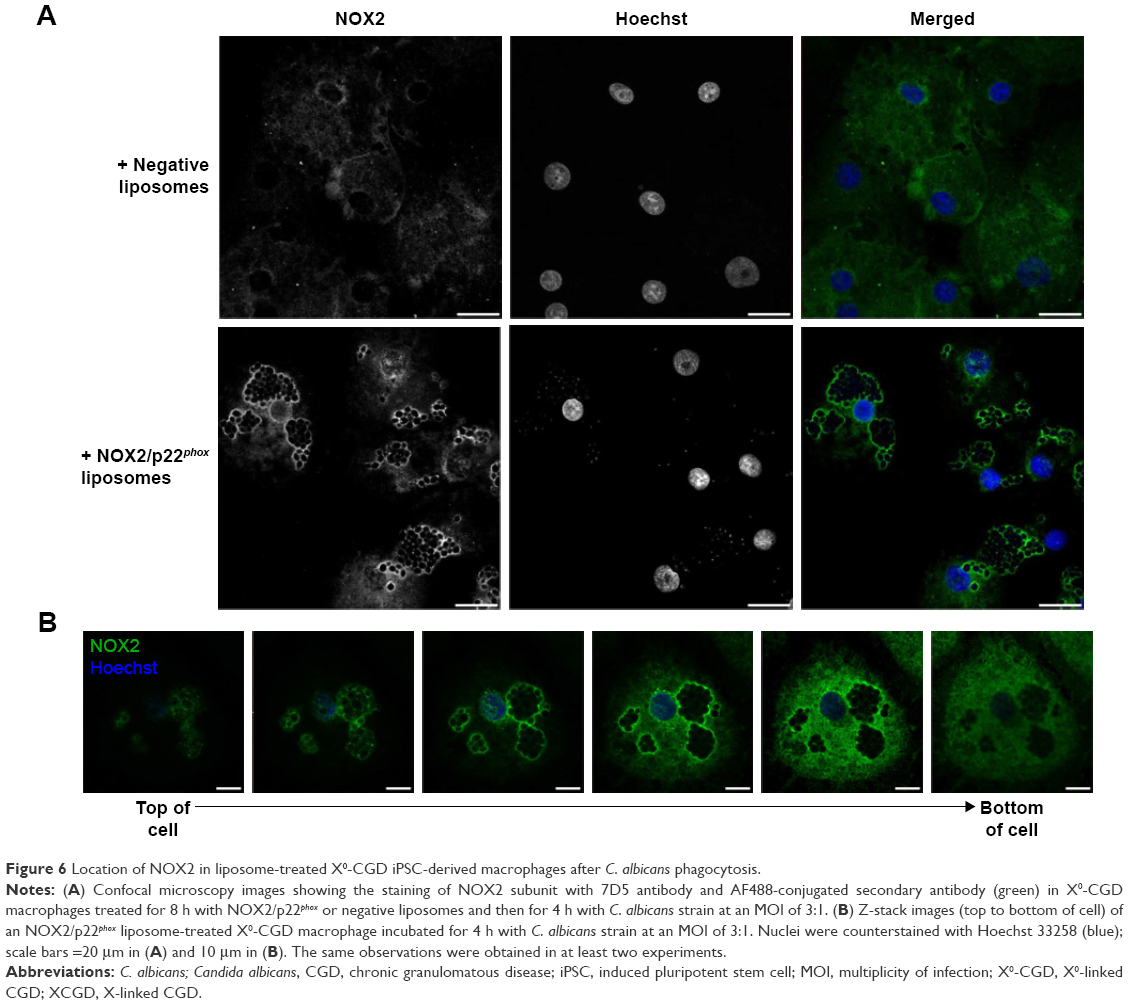 | Figure 6 Location of NOX2 in liposome-treated X0-CGD iPSC-derived macrophages after C. albicans phagocytosis. |
Safe NADPH oxidase restoration in X0-CGD iPSC-derived macrophages using NOX2/p22phox liposomes
First, we checked the viability of macrophages after exposure to a high concentration of NOX2/p22phox (20 pmol cytochrome b558 for 1.5×104 macrophages) and negative liposomes using the MTT test specific for the mitochondrial activity in living cells. No loss of viability was observed after 8 h, 12 h or 24 h treatment of X0-CGD iPSC-derived macrophages (Figure 7A). No significant morphological changes were observed between untreated and liposome-treated X0-CGD macrophages (Figure 7B). X0-CGD untreated macrophages were also incubated with IMDM alone without FBS to be sure that the absence of serum in culture medium for several hours has no influence on cell viability and morphology (Figure 7A and B).
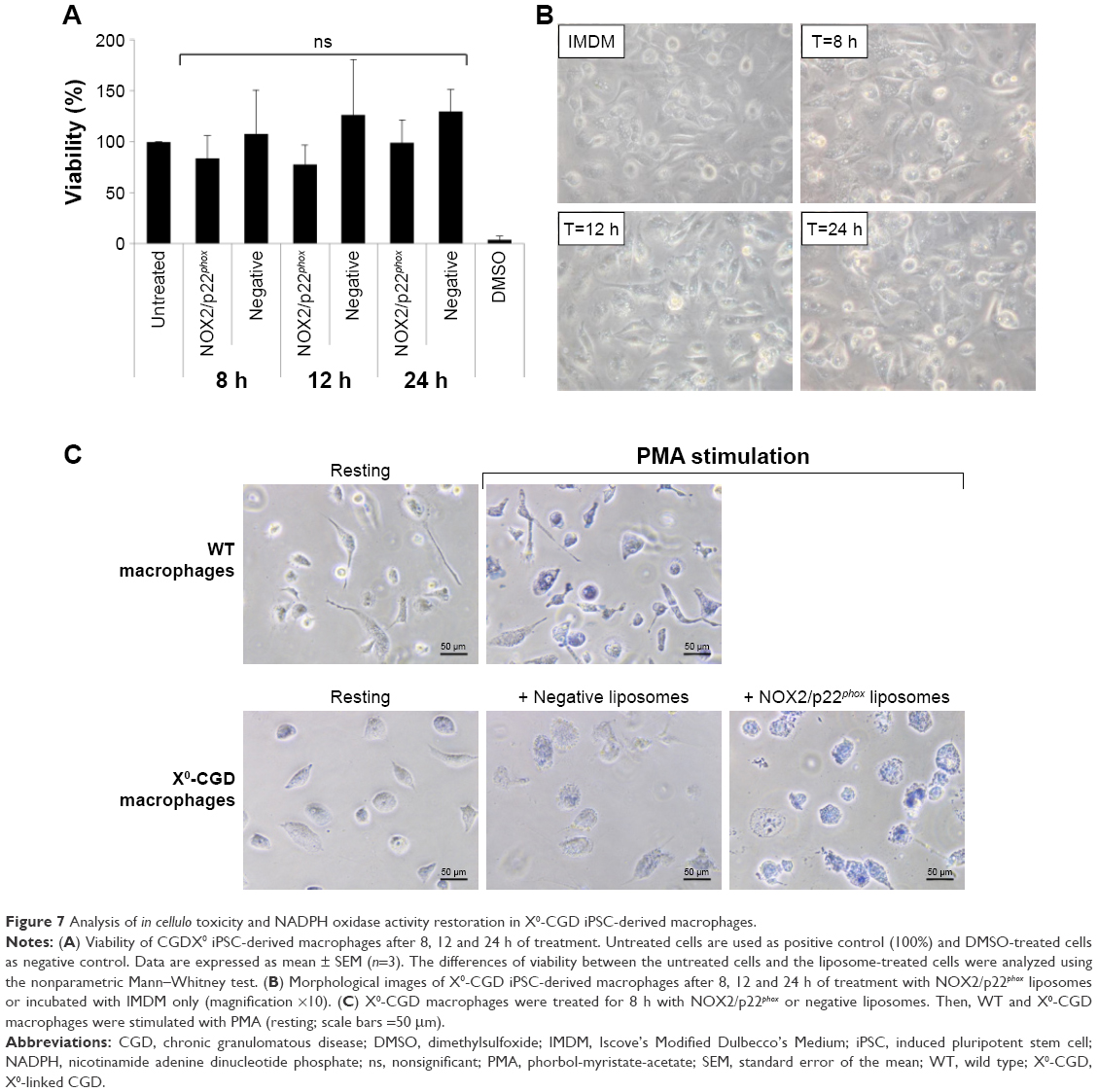 | Figure 7 Analysis of in cellulo toxicity and NADPH oxidase activity restoration in X0-CGD iPSC-derived macrophages. |
Then, we examined the ability of these proteoliposomes to restore the in cellulo NADPH oxidase activity in ROS-deficient macrophages derived from X0-CGD iPSCs. As shown in Figure 7C, the NBT test performed in X0-CGD macrophages treated with NOX2/p22phox liposomes for 8 h showed us a blue precipitate of formazan, revealing the production of ROS upon PMA stimulation. The level of this activity was quite similar to that found in WT iPSC-derived macrophages. Only residual activity was found in the X0-CGD iPSC-derived macrophages treated with negative liposomes. In conclusion, the NOX2/p22phox liposomes were functional in cellulo given that they were able to restore the NADPH activity in 8 h treated X0-CGD macrophages. This is the first time that proteoliposomes have been used for the delivery of a therapeutic membrane protein (cytochrome b558) for the purpose of enzyme replacement.
Discussion
The first production of a recombinant cytochrome b558 was performed in 1993 using a baculovirus/insect cell expression system.41 However, >20 years were required to produce this cytochrome again. Recombinant production of cytochrome b558 is indeed highly challenging because it is a complex of two membrane proteins, with six transmembrane domains for NOX2 and two to four transmembrane domains for p22phox that are intimately linked to be functional.42 In X0-CGD only NOX2 is mutated, but the absence of its expression in phagocytes leads to the absence of p22phox as well. Conversely in AR220-CGD characterized by the absence of mutated p22phox, NOX2 is also not expressed.43 This underlines that the synthesis of cytochrome b558 involved the concomitant synthesis of both proteins. In addition, some sequences of NOX2 and p22phox were demonstrated to be in interaction to stabilize the complex.44,45
The first advantage of using a cell-free expression system is that it is a completely open system in which each parameter of the reaction can be modified depending on the protein to be produced, and this format is also particularly suitable for the co-expression of membrane proteins. Several ratios of DNA templates expressing NOX2 and p22phox separately were tested to optimize the concomitant expression of both proteins. Optimal magnesium and potassium concentrations (known to be important for optimizing in vitro cell-free transcription and translation46) and the synthesis reaction time were also determined.
Then, to obtain a fully functional and stabilized cytochrome b558, two hemes need to be incorporated in NOX2. Heme incorporation is required for heterodimerization of NOX2 and p22phox, stable expression of both subunits at the plasma membrane and therefore for the NADPH oxidase activity.47,48 In the literature, synthesis of other heme proteins using the E. coli in vitro coupled transcription/translation system required the addition of hemin at a concentration around 5–20 μM.49 In the case of cytochrome b558 production using baculovirus/insect cells or yeasts, the heme precursor δ-aminolevulinic acid or hemin was used.17,18,41 However, hemin must only be added to our system because of the absence of mitochondria in the bacterial lysate that is responsible for heme generation. Therefore, screening of the optimal hemin and iron concentrations was essential to ascertain satisfactory heme production and cytochrome b558 expression. This was evidenced by its characteristic reduced minus-oxidized spectrum. It is also well established that heme incorporation is essential for the electron transfer within the cytochrome b558 from the NADPH to the FAD sites and through the hemes to reduce molecular oxygen leading to the generation of the superoxide anion.50 The in vitro recombinant cell-free NADPH oxidase activation assay measured a total production of superoxide anions of 228.7 moles/s/mol of cytochrome b558 heme with NOX2/p22phox liposomes. This is equivalent to the NADPH oxidase activity of the bovine cytochrome b558 produced in yeasts.17 Our specific SOD-sensitive NADPH oxidase activity of 143.8 moles of superoxides O2•–/s/mol of cytochrome b558 heme is also within the same range of value as the activity of the detergent-solubilized recombinant human cytochrome b558 produced in baculovirus/insect cells.41 The maximum turnover measured in an amphiphile-activated cell-free system using neutrophil membrane is generally 100 mol O2•–/s/mol of cytochrome b558 heme,51 and it decreases when cytochrome b558 is reconstituted in liposomes.52 The high turnover obtained with our NOX2/p22phox liposomes could be explained by the fact that we introduced the R199Q mutation in NOX2 that improves ROS production after activation four- to eightfold.27,33 The DPI concentration used to inhibit the NADPH oxidase activity of the reconstituted complex was higher than the concentration usually used in phagocytes (200 μM versus 2.5–5 μM in whole cells). However, a similar observation was made for the bovine NOX2 expressed in yeast.17 Probably, the active site of the enzyme was less accessible when incorporated into liposomes than in the plasma membrane of phagocytes.
The molecular mass of NOX2 and p22phox produced was 55–58 kDa and 21–25 kDa, respectively. This suggests that NOX2 was in a non-glycosylated form (as is always the case in an E. coli-producing system) but was still active, as for murine NOX2.53 In human phagocytes, the mature form of NOX2 is glycosylated and detected at around 91 kDa. But it has been demonstrated that glycosylation is not required for NOX2-p22phox heterodimer formation, heme incorporation and superoxide anion generation.41,54–56 The glycosylated form of NOX2 was produced using a yeast system, whereas a weakly or non-glycosylated form of NOX2 was obtained when cell-free systems based on rabbit reticulocyte lysates or a baculovirus/insect cell system were used.17,18,41,57–59
Our E. coli-based cell-free expression system is able to produce 100 times and 20 times more recombinant human cytochrome b558 than previous reports showing the synthesis of a bovine cytochrome b558 from 1 L of a P. pastoris or baculovirus/insect cell system, respectively.18 Our production is also 1,000 times greater than the first attempt using 109 baculovirus/insect cells.41 The second advantage of this E. coli cell-free expression system was that cytochrome b558 is directly incorporated into liposomes in <24 h, contrary to cell-based expression systems. In addition, these cell-based expression systems require supplementary steps of cytochrome b558 purification and incorporation into liposomes after detergent removal, which could be a critical step leading to heterogeneous preparations and the protein’s loss of function.60 Furthermore, the process is scalable and applicable for a therapeutic approach. Several previous studies have demonstrated the ability to scale the cell-free expression system linearly to the liter scale for soluble protein production.61
The presence of liposomes in the reaction mixture is essential for the solubilization of the newly synthesized NOX2 and p22phox subunits. Lipids are the normal environment of membrane proteins, so liposomes are perfectly relevant vectors to obtain a proper folding of a protein and to allow its vectorization. However, the lipid composition is an important parameter to consider because of its influence on the stability of liposomes as well as the activity of the protein inserted. A mixture of DOPC and DOPE is commonly used to form liposomes.62 Therefore, cholesterol content, the choice of synthetic lipids and the proportion of the fusogenic lipid DOPE were determined on the basis of our previous results16,63 and the current study. The choice of the lipid composition for optimal NOX2/p22phox production while maintaining their functionality was also compatible with efficient vectorization of both proteins in the plasma membrane of X0-CGD iPSC-derived macrophages, as evidenced by confocal analysis. Stable expression of NOX2 and p22phox subunits at the plasma membrane is possible only after NOX2-p22phox heterodimer formation. Thus, this membrane incorporation confirmed that the heterodimerization of both proteins occurred during their synthesis in the cell-free system. In addition, we demonstrated a phagosomal membrane location of NOX2 after Candida sp. phagocytosis. In normal macrophages, cytochrome b558 is first located in the plasma membrane and then in the phagosomal membrane after phagocytosis.64 In this study, we focused on macrophages because of their easy production from iPSCs23 but first of all because of their long lifetime in the body (several months in some tissues), making them an advantageous target for protein therapy. The in vitro macrophages used for the experiments were nondividing cells, like their counterparts in the blood circulation.
Then, satisfactory delivery of both subunits was definitively confirmed by the restoration of the NADPH oxidase activity in ROS-deficient macrophages after NOX2/p22phox liposome treatment and PMA stimulation. The NBT slide test is a robust test that is easy to perform and that requires a small number of cells. For these reasons, it is commonly used for routine diagnosis of CGD patients.65 It is known that if 5%–10% of circulating phagocytes are efficient, individuals are protected against infections. This means that a partial correction of the cells with proteoliposomes can be sufficient to prevent or fight infections. Using the NBT test, we proved that NOX2/p22phox liposomes were able to reconstitute a functional NADPH oxidase complex in X0-CGD macrophages. This is the first time that a membrane protein has been delivered by liposomes to restore a deficient enzyme activity. In addition, MTT assay results demonstrated that this approach is not toxic for cells. However, further studies will also be needed to investigate the interaction between proteoliposomes and phagocytes in greater detail. In the context of antitumor therapy, liposomes containing the pro-apoptotic Bak protein designed by our team have already demonstrated powerful activity to induce apoptosis in cellulo in human colorectal carcinoma cell lines and in vivo in a mouse model of glioblastoma.11,12 It was the first demonstration of the efficacy of a proteoliposome-based therapy in a preclinical study.
NOX2 is mainly expressed in neutrophils and alveolar and interstitial macrophages in the lungs, which are the first line of defense against Aspergillus invasion.66 As a consequence, in a further step, we intend to evaluate the biodistribution, safety and efficacy of NOX2/p22phox liposomes in a CYBB knockout mouse model of pulmonary infection. We hope that these preclinical studies will confirm the therapeutic potential of NOX2/p22phox liposomes and then lead to an experimental clinical trial in XCGD patients. Indeed, the first cause of death of CGD patients is lung infections mainly due to Aspergillus sp.8 Nebulization is the most relevant administration route for a local treatment with liposomes because 75% of the administered dose will accumulate in the lungs, preferentially in the small airways and alveoli for the particles with a diameter <1–5 μm.67 Therefore, a local and targeted therapy against acute infections refractory to conventional treatments would be extremely useful in CGD patients.
Conclusion
For the first time, we have described the successful production of recombinant therapeutic proteoliposomes that demonstrate their efficiency in treating a membrane enzyme deficiency. In a single step using an optimized E. coli-based expression system, we were able to generate liposomes containing a functional cytochrome b558 into the lipid bilayer of liposomes with heme incorporation. These NOX2/p22phox liposomes successfully reconstituted an NADPH oxidase complex in vitro, as shown by the production of superoxide anions after stimulation. Furthermore, membrane delivery of NOX2 and p22phox was confirmed by the presence of both subunits at the plasma membrane of macrophages and also at the phagosome membrane for NOX2. In cellulo experiments with X0-CGD iPSC-derived macrophages revealed no toxicity of the proteoliposomes at any incubation time. In addition, NOX2/p22phox liposome treatment of these cells restored NADPH oxidase activity.
In the future, this promising new liposomal enzyme replacement therapy could become a local curative alternative against life-threatening lung infections refractory to conventional antibiotic and antifungal therapy in CGD patients.
Acknowledgments
The authors thank the University Grenoble Alpes (AGIR program 2014) and the French-Swiss Interregional program (Interreg – Feder, 2013–2015) for funding this work. They also thank Sylvain Beaumel, Cécile Martel and Michèle Mollin for their helpful and valuable work at the Centre Diagnostic et Recherche sur la CGD, and Pascal Gabard for his advice on the miniaturized recombinant cell-free system. They acknowledge the platforms of the Grenoble Instruct Center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) supported by the French Infrastructure for Integrated Structural Biology Initiative FRISBI (ANR-10-INSB-05-02) and by the Grenoble Alliance for Integrated Structural Cell Biology GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). They also thank Mrs Linda Northrup for editing the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
van den Berg JM, van Koppen E, Ahlin A, et al. Chronic granulomatous disease: the European experience. PLoS One. 2009;4(4):e5234. | ||
Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine. 2000;79(3):170–200. | ||
Holland SM. Chronic granulomatous disease. Hematol Oncol Clin North Am. 2013;27(1):89–99. | ||
Roos D, Kuhns DB, Maddalena A, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis. 2010;45(3):246–265. | ||
Gungor T, Teira P, Slatter M, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;383(9915):436–448. | ||
Kaufmann KB, Chiriaco M, Siler U, et al. Gene therapy for chronic granulomatous disease: current status and future perspectives. Curr Gene Ther. 2014;14(6):447–460. | ||
Touzot F, Hacein-Bey-Abina S, Fischer A, Cavazzana M. Gene therapy for inherited immunodeficiency. Expert Opin Biol Ther. 2014;14(6):789–798. | ||
Winkelstein JA, Marino MC, Johnston RB Jr, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine. 2000;79(3):155–169. | ||
Sachse R, Dondapati SK, Fenz SF, Schmidt T, Kubick S. Membrane protein synthesis in cell-free systems: from bio-mimetic systems to bio-membranes. FEBS Lett. 2014;588(17):2774–2781. | ||
Liguori L, Lenormand JL. Production of recombinant proteoliposomes for therapeutic uses. Methods Enzymol. 2009;465:209–223. | ||
Liguori L, Marques B, Villegas-Mendez A, Rothe R, Lenormand JL. Liposomes-mediated delivery of pro-apoptotic therapeutic membrane proteins. J Control Release. 2008;126(3):217–227. | ||
Liguori L, Pastorino F, Rousset X, et al. Anti-tumor effects of bak-proteoliposomes against glioblastoma. Molecules. 2015;20(9):15893–15909. | ||
Stewart MP, Sharei A, Ding X, Sahay G, Langer R, Jensen KF. In vitro and ex vivo strategies for intracellular delivery. Nature. 2016;538(7624):183–192. | ||
Torchilin VP. Multifunctional nanocarriers. Adv Drug Deliv Rev. 2006; 58(14):1532–1555. | ||
Cross AR, Rae J, Curnutte JT. Cytochrome b-245 of the neutrophil superoxide-generating system contains two nonidentical hemes. Potentiometric studies of a mutant form of gp91phox. J Biol Chem. 1995;270(29):17075–17077. | ||
Marques B, Liguori L, Paclet MH, et al. Liposome-mediated cellular delivery of active gp91(phox). PLoS One. 2007;2(9):e856. | ||
Ezzine A, Souabni H, Bizouarn T, Baciou L. Recombinant form of mammalian gp91(phox) is active in the absence of p22(phox). Biochem J. 2014;462(2):337–345. | ||
Ostuni MA, Lamanuzzi LB, Bizouarn T, Dagher MC, Baciou L. Expression of functional mammal flavocytochrome b(558) in yeast: comparison with improved insect cell system. Biochim Biophys Acta. 2010;1798(6):1179–1188. | ||
Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481(7381):295–305. | ||
Merling RK, Sweeney CL, Chu J, et al. An AAVS1-targeted minigene platform for correction of iPSCs from all five types of chronic granulomatous disease. Mol Ther. 2015;23(1):147–157. | ||
Zou J, Sweeney CL, Chou BK, et al. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood. 2011;117(21):5561–5572. | ||
Jiang Y, Cowley SA, Siler U, et al. Derivation and functional analysis of patient-specific induced pluripotent stem cells as an in vitro model of chronic granulomatous disease. Stem Cells. 2012;30(4):599–611. | ||
Brault J, Goutagny E, Telugu N, et al. Optimized generation of functional neutrophils and macrophages from patient-specific induced pluripotent stem cells: ex vivo models of X(0)-linked, AR22(0)- and AR47(0)-chronic granulomatous diseases. Biores Open Access. 2014;3(6):311–326. | ||
Flynn R, Grundmann A, Renz P, et al. CRISPR-mediated genotypic and phenotypic correction of a chronic granulomatous disease mutation in human iPS cells. Exp Hematol. 2015;43(10):838–848.e3. | ||
Dreyer AK, Hoffmann D, Lachmann N, et al. TALEN-mediated functional correction of X-linked chronic granulomatous disease in patient-derived induced pluripotent stem cells. Biomaterials. 2015;69:191–200. | ||
Laugsch M, Rostovskaya M, Velychko S, et al. Functional restoration of gp91phox-oxidase activity by BAC transgenesis and gene targeting in X-linked chronic granulomatous disease iPSCs. Mol Ther. 2016;24(4):812–822. | ||
Li XJ, Grunwald D, Mathieu J, Morel F, Stasia MJ. Crucial role of two potential cytosolic regions of Nox2, 191TSSTKTIRRS200 and 484DESQANHFAVHHDEEKD500, on NADPH oxidase activation. J Biol Chem. 2005;280(15):14962–14973. | ||
Sutkeviciute I, Thepaut M, Sattin S, et al. Unique DC-SIGN clustering activity of a small glycomimetic: a lesson for ligand design. ACS Chem Biol. 2014;9(6):1377–1385. | ||
Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. | ||
Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76(9):4350–4354. | ||
Picciocchi A, Debeurme F, Beaumel S, et al. Role of putative second transmembrane region of Nox2 protein in the structural stability and electron transfer of the phagocytic NADPH oxidase. J Biol Chem. 2011;286(32):28357–28369. | ||
Light DR, Walsh C, O’Callaghan AM, Goetzl EJ, Tauber AI. Characteristics of the cofactor requirements for the superoxide-generating NADPH oxidase of human polymorphonuclear leukocytes. Biochemistry. 1981;20(6):1468–1476. | ||
Carrichon L, Picciocchi A, Debeurme F, et al. Characterization of superoxide overproduction by the D-Loop(Nox4)-Nox2 cytochrome b(558) in phagocytes-differential sensitivity to calcium and phosphorylation events. Biochim Biophys Acta. 2011;1808(1):78–90. | ||
Debeurme F, Picciocchi A, Dagher MC, et al. Regulation of NADPH oxidase activity in phagocytes: relationship between FAD/NADPH binding and oxidase complex assembly. J Biol Chem. 2010;285(43):33197–33208. | ||
Roerig DL, Mascaro L Jr, Aust SD. Microsomal electron transport: tetrazolium reduction by rat liver microsomal NADPH-cytochrome c reductase. Arch Biochem Biophys. 1972;153(2):475–479. | ||
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. | ||
Defendi F, Decleva E, Martel C, Dri P, Stasia MJ. A novel point mutation in the CYBB gene promoter leading to a rare X minus chronic granulomatous disease variant – impact on the microbicidal activity of neutrophils. Biochim Biophys Acta. 2009;1792(3):201–210. | ||
Segal AW, Levi AJ. The mechanism of the NBT (nitroblue tetrazolium) test. Clin Sci. 1973;44(6):26. | ||
Lonez C, Lensink MF, Kleiren E, Vanderwinden JM, Ruysschaert JM, Vandenbranden M. Fusogenic activity of cationic lipids and lipid shape distribution. Cell Mol Life Sci. 2010;67(3):483–494. | ||
Scott RI, Poole RK. A re-examination of the cytochromes of Escherichia coli using fourth-order finite difference analysis: their characterization under different growth conditions and accumulation during the cell cycle. J Gen Microbiol. 1982;128(8):1685–1696. | ||
Rotrosen D, Yeung CL, Katkin JP. Production of recombinant cytochrome b558 allows reconstitution of the phagocyte NADPH oxidase solely from recombinant proteins. J Biol Chem. 1993;268(19):14256–14260. | ||
Stasia MJ. CYBA encoding p22(phox), the cytochrome b558 alpha polypeptide: gene structure, expression, role and physiopathology. Gene. 2016;586(1):27–35. | ||
Stasia MJ, Li XJ. Genetics and immunopathology of chronic granulomatous disease. Semin Immunopathol. 2008;30(3):209–235. | ||
Beaumel S, Grunwald D, Fieschi F, Stasia MJ. Identification of NOX2 regions for normal biosynthesis of cytochrome b558 in phagocytes highlighting essential residues for p22phox binding. Biochem J. 2014;464(3):425–437. | ||
von Lohneysen K, Noack D, Wood MR, Friedman JS, Knaus UG. Structural insights into Nox4 and Nox2: motifs involved in function and cellular localization. Mol Cell Biol. 2010;30(4):961–975. | ||
Craig D, Howell MT, Gibbs CL, Hunt T, Jackson RJ. Plasmid cDNA-directed protein synthesis in a coupled eukaryotic in vitro transcription-translation system. Nucleic Acids Res. 1992;20(19):4987–4995. | ||
Yu L, Zhen L, Dinauer MC. Biosynthesis of the phagocyte NADPH oxidase cytochrome b558. Role of heme incorporation and heterodimer formation in maturation and stability of gp91phox and p22phox subunits. J Biol Chem. 1997;272(18):27288–27294. | ||
DeLeo FR, Burritt JB, Yu L, Jesaitis AJ, Dinauer MC, Nauseef WM. Processing and maturation of flavocytochrome b558 include incorporation of heme as a prerequisite for heterodimer assembly. J Biol Chem. 2000;275(18):13986–13993. | ||
Miyazaki-Imamura C, Oohira K, Kitagawa R, Nakano H, Yamane T, Takahashi H. Improvement of H2O2 stability of manganese peroxidase by combinatorial mutagenesis and high-throughput screening using in vitro expression with protein disulfide isomerase. Protein Eng. 2003; 16(6):423–428. | ||
Yu L, Quinn MT, Cross AR, Dinauer MC. Gp91(phox) is the heme binding subunit of the superoxide-generating NADPH oxidase. Proc Natl Acad Sci U S A. 1998;95(14):7993–7998. | ||
Pick E. Cell-free NADPH oxidase activation assays: “in vitro veritas”. Methods Mol Biol. 2014;1124:339–403. | ||
Taylor RM, Lord CI, Riesselman MH, et al. Characterization of surface structure and p47phox SH3 domain-mediated conformational changes for human neutrophil flavocytochrome b. Biochemistry. 2007;46(49):14291–14304. | ||
Björgvinsdóttir H, Zhen L, Dinauer MC. Cloning of murine gp91phox cDNA and functional expression in a human X-linked chronic granulomatous disease cell line. Blood. 1996;87(5):2005–2010. | ||
Kleinberg ME, Rotrosen D, Malech HL. Asparagine-linked glycosylation of cytochrome b558 large subunit varies in different human phagocytic cells. J Immunol. 1989;143(12):4152–4157. | ||
Paclet MH, Coleman AW, Burritt J, Morel F. NADPH oxidase of Epstein-Barr-virus immortalized B lymphocytes. Effect of cytochrome b(558) glycosylation. Eur J Biochem. 2001;268(19):5197–5208. | ||
Harper AM, Chaplin MF, Segal AW. Cytochrome b-245 from human neutrophils is a glycoprotein. Biochem J. 1985;227(3):783–788. | ||
Parkos CA, Allen RA, Cochrane CG, Jesaitis AJ. Purified cytochrome b from human granulocyte plasma membrane is comprised of two polypeptides with relative molecular weights of 91,000 and 22,000. J Clin Invest. 1987;80(3):732–742. | ||
Wallach TM, Segal AW. Analysis of glycosylation sites on gp91phox, the flavocytochrome of the NADPH oxidase, by site-directed mutagenesis and translation in vitro. Biochem J. 1997;321(pt 3):583–585. | ||
Yu L, DeLeo FR, Biberstine-Kinkade KJ, Renee J, Nauseef WM, Dinauer MC. Biosynthesis of flavocytochrome b558. gp91(phox) is synthesized as a 65-kDa precursor (p65) in the endoplasmic reticulum. J Biol Chem. 1999;274(7):4364–4369. | ||
Rigaud JL, Pitard B, Levy D. Reconstitution of membrane proteins into liposomes: application to energy-transducing membrane proteins. Biochim Biophys Acta. 1995;1231(3):223–246. | ||
Zawada JF, Yin G, Steiner AR, et al. Microscale to manufacturing scale-up of cell-free cytokine production – a new approach for shortening protein production development timelines. Biotechnol Bioeng. 2011;108(7):1570–1578. | ||
Seddon AM, Curnow P, Booth PJ. Membrane proteins, lipids and detergents: not just a soap opera. Biochim Biophys Acta. 2004;1666(1–2):105–117. | ||
Liguori L, Blesneac I, Madern D, Vivaudou M, Lenormand JL. Single-step production of functional OEP24 proteoliposomes. Protein Expr Purif. 2010;69(1):106–111. | ||
Casbon AJ, Allen LA, Dunn KW, Dinauer MC. Macrophage NADPH oxidase flavocytochrome B localizes to the plasma membrane and Rab11-positive recycling endosomes. J Immunol. 2009;182(4):2325–2339. | ||
Ochs HD, Igo RP. The NBT slide test: a simple screening method for detecting chronic granulomatous disease and female carriers. J Pediatr. 1973;83(1):77–82. | ||
Trocme C, Deffert C, Cachat J, et al. Macrophage-specific NOX2 contributes to the development of lung emphysema through modulation of SIRT1/MMP-9 pathways. J Pathol. 2015;235(1):65–78. | ||
Kuzmov A, Minko T. Nanotechnology approaches for inhalation treatment of lung diseases. J Control Release. 2015;219:500–518. |
Supplementary materials
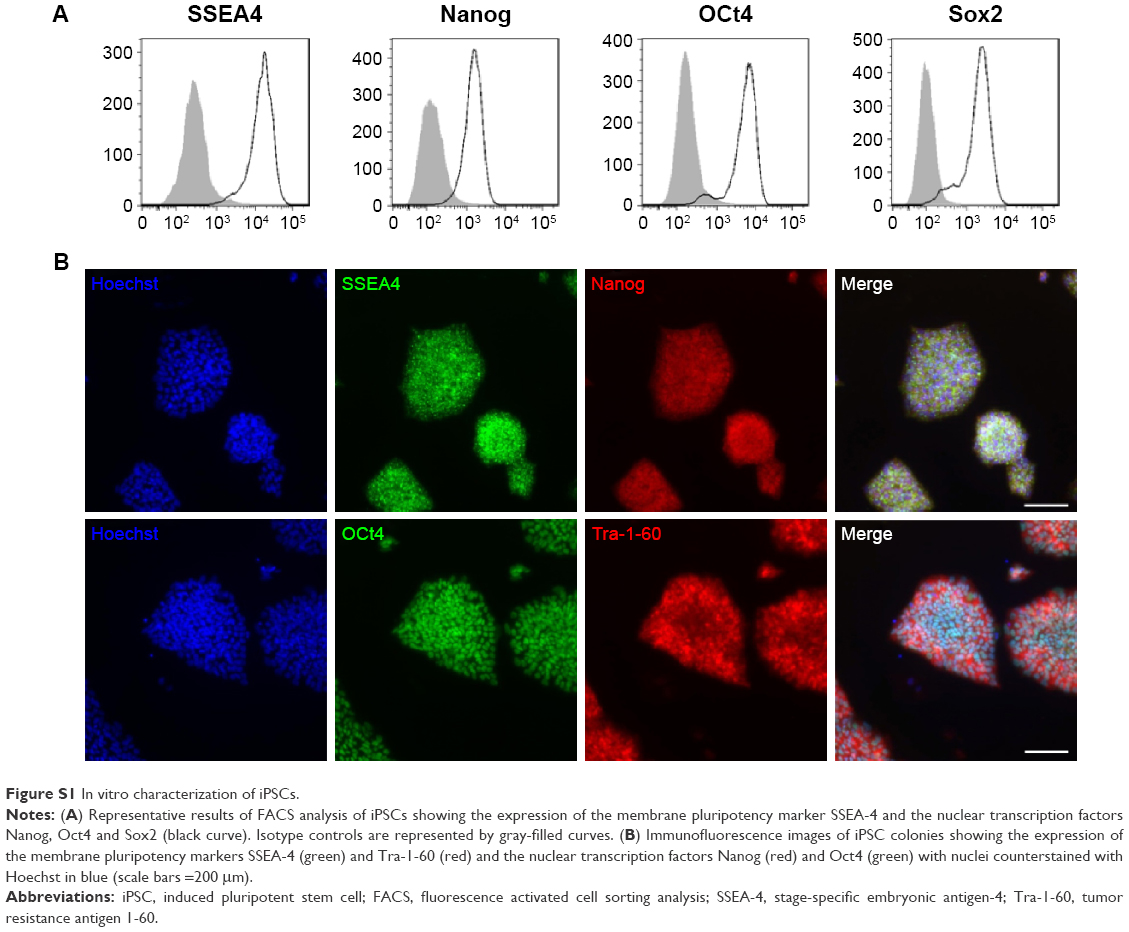 | Figure S1 In vitro characterization of iPSCs. |
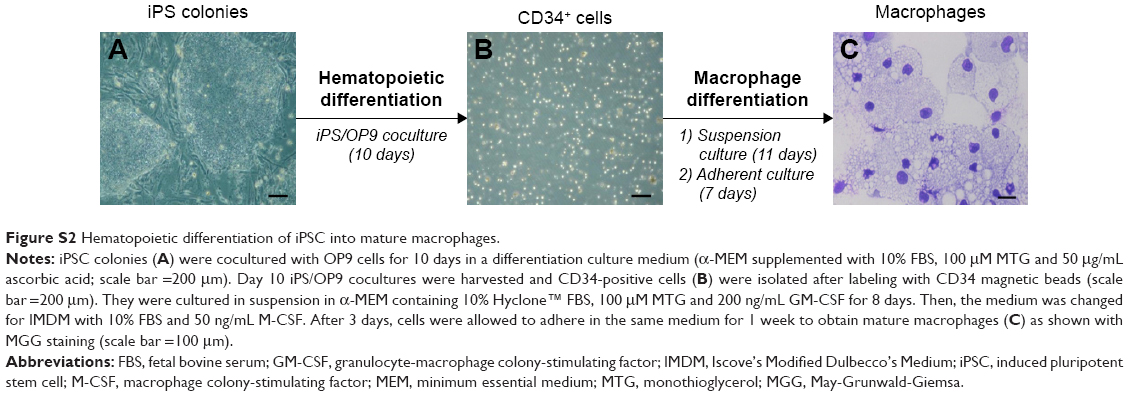 | Figure S2 Hematopoietic differentiation of iPSC into mature macrophages. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.