")
Back to Journals » OncoTargets and Therapy » Volume 9
Therapeutic approaches in myelofibrosis and myelodysplastic/myeloproliferative overlap syndromes
Authors Sochacki A, Fischer M, Savona M
Received 9 November 2015
Accepted for publication 3 February 2016
Published 15 April 2016 Volume 2016:9 Pages 2273—2286
DOI https://doi.org/10.2147/OTT.S83868
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Min Li
Andrew L Sochacki,1 Melissa A Fischer,1 Michael R Savona1,2
1Department of Internal Medicine, Vanderbilt University Medical Center, 2Vanderbilt-Ingram Cancer Center, Nashville, TN, USA
Abstract: The discovery of JAK2V617F a decade ago led to optimism for a rapidly developing treatment revolution in Ph- myeloproliferative neoplasms. Unlike BCR–ABL, however, JAK2 was found to have a more heterogeneous role in carcinogenesis. Therefore, for years, development of new therapies was slow, despite standard treatment options that did not address the overwhelming symptom burden in patients with primary myelofibrosis (MF), post-essential thrombocythemia MF, post-polycythemia vera MF, and myelodysplastic syndrome (MDS)/myeloproliferative neoplasm (MPN) syndromes. JAK–STAT inhibitors have changed this, drastically ameliorating symptoms and ultimately beginning to show evidence of impact on survival. Now, the genetic foundations of myelofibrosis and MDS/MPN are rapidly being elucidated and contributing to targeted therapy development. This has been empowered through updated response criteria for MDS/MPN and refined prognostic scoring systems in these diseases. The aim of this article is to summarize concisely the current and rationally designed investigational therapeutics directed at JAK–STAT, hedgehog, PI3K–Akt, bone marrow fibrosis, telomerase, and rogue epigenetic signaling. The revolution in immunotherapy and novel treatments aimed at previously untargeted signaling pathways provides hope for considerable advancement in therapy options for those with chronic myeloid disease.
Keywords: MDS/MPN neoplasms, emerging therapy
Background
Primary myelofibrosis (MF) is a Philadelphia chromosome-negative (Ph−) myeloproliferative neoplasm (MPN) characterized by clonal megakaryocytic hyperplasia and atypia, and fibrosis within the bone marrow. Lower grade Ph− MPNs, most commonly essential thrombocythemia (ET) and polycythemia vera (PV), may progress to post-ET MF (PET-MF) and post-PV MF (PPV-MF), respectively. MF, PPV-MF, and PET-MF may have some divergent genotypes, but they have similar phenotypes and clinical course.1 This includes reduced life expectancy, endothelial dysfunction, vascular events, massive hepatosplenomegaly, significant anorexia, cachexia, and high risk of transformation to acute leukemia.2 Survival is variable, and multiple different models employ the use of specific clinical and pathologic characteristics to best assess risk.3,4
Myelodysplastic syndrome (MDS)/MPN overlap syndromes include several uncommon myeloid malignancies that have symptom burdens and proliferative features seen in MF, and are characterized by dysplastic features of MDSs. MDS/MPNs include chronic myelomonocytic leukemia (CMML), juvenile MML, atypical (BCR–ABL1−) chronic myeloid leukemia (aCML), MDS/MPN unclassifiable (MDS/MPN-U), and the World Health Organization (WHO) provisional entity: refractory anemia with ring sideroblasts and thrombocytosis (RARS-T).2,5,6 CMML is the most frequent MDS/MPN, and is marked by sustained monocytosis of at least 10% of the total white blood-cell count and equal to or greater than 1,000/μL. Juvenile MML is phenotypically similar to CMML and driven by signaling abnormalities in the RAS–RAF pathway with mutually exclusive germ-line mutations in PTPN11, KRAS, NRAS, and NF1 occurring in 90% of cases.7,8 aCML is characterized by severe granulocytic dysplasia and left-shifted leukocytosis in the absence of the BCR–ABL fusion protein. RARS-T is a provisional WHO category of disease that includes bone marrow findings similar to what is seen in RARS in the presence of sustained thrombocytosis and splenomegaly.9
Until recently, there were few available therapies to address the debilitating symptoms, morbidities, and reduced survival of MF and MDS/MPN. Development of new treatments had been limited until the past few years, and most current treatment regimens have varied effects on symptomatology without significantly impacting overall survival. However, advances in the understanding of the genetic underpinnings of MF and MDS/MPNs are now allowing for rapid development of rational therapy. Furthermore, the recent proposed response criteria for MDS/MPN and refinement of prognostic scores in MF have provided objective means to categorize objective responses.4,10,11 The aim of this article is to discuss the molecular genetics of MF and MDS/MPNs as context for current therapies and novel approaches in development.
Current pharmacologic and surgical treatments for MF and MDS/MPNs
Hydroxyurea
Hydroxyurea is an antimetabolite that inhibits ribonucleoside diphosphate reductase, resulting in G1/S arrest in the cell cycle. Hydroxyurea is typically used as a cytotoxic agent to improve leukocytosis or thrombocytosis in the proliferative phases of primary MF or MDS/MPN. Often, this is at the expense of worsening anemia, which limits its utility. Hydroxyurea has also been noted to cause a modest reduction in splenomegaly, though has little effect on symptom burden or rate of transformation to leukemia.12 There also remains some concern about the mutagenic potential of hydroxyurea with long-term use, though there remains no clear prospective evidence that this is true.13 The discovery of JAK–STAT pathway mutations and subsequent development of JAK/STAT inhibitors has reduced the use of hydroxyurea, but it remains a mainstay of therapy. Interestingly, there is some evidence that JAK2V617F status may also play a role in its effectiveness.14
Splenectomy
Palliation of symptoms from splenomegaly via splenectomy has been attempted for many decades. Though splenectomy has not been found to have an effect on survival, which averages 13–19 months postoperatively, it can provide symptomatic relief to select patients. A review of a single center over three decades found variable improvement in mechanical discomfort, thrombocytopenia, and anemia no longer requiring transfusions. However there was only a short median duration of response (12 months), with wide variability in duration of response (1–91 months).15,16 Further, the compensatory extramedullary hematopoiesis in the liver for some patients can lead to shorter postsplenectomy survival. Operative and postoperative morbidity is high, with operative mortality 5%–7% at experienced centers and rising to 25% in the perioperative window. Improvements are being made via laparoscopic splenectomy, with or without morcellation, but more data are needed for these patients.17
There are limited data that support faster postallogeneic hematopoietic stem cell transplantation (HSCT) granulocyte-count recovery in patients who have had splenectomy. However, no evidence for survival benefit in HSCT has been described.18 Splenectomy carries significant morbidity and mortality risks, with little evidence of improvement of survival, and should only be performed rarely in MF or MDS/MPN.
Allogeneic hematopoietic stem cell transplantation
The only current curative treatment for MDS/MPN and MF is HSCT.18 However, properly identifying patients that will respond to this treatment is challenging. The Dynamic International Prognostic Scoring System (DIPSS) does assist in stratifying potential HSCT candidates with MF. Patients under 65 years of age who have high-risk DIPSS scores at diagnosis have decreased mortality with HSCT compared to age-matched controls without transplant (relative risk 0.37), though the role of HSCT is less clear for intermediate- or low-risk patients.19 HSCT also appears to mitigate disease-associated severe anemia, advanced constitutional symptoms, cytogenetic aberrations, and leukoerythroblastosis.18,19 In reported case series, leukemic transformation was near universally fatal, with the majority of patients dying within 3 months of diagnosed transformation.21 Therefore, the risk of leukemic transformation is an especially important factor to consider when deciding on patient risk, and thus the impetus for HSCT. Patients with intermediate 2 and high DIPSS scoring are known to have a significantly higher risk of leukemic transformation, and thus may derive the greatest benefit from HSCT. Different mutational burdens not yet fully integrated into prognostic scores are also associated with leukemic transformation and mortality in MF and MDS/MPN.22 Decreasing the threat of leukemic transformation contributes to the benefit of HSCT in higher-risk disease.
Morbidity and mortality with HSCT remains high in MF. Reviews of HSCT for MF have found an average 45%–50% overall survival at 3 years posttransplant.23 Therefore, with low-risk disease, HSCT-associated mortality outweighs the risk of death due to MF. A retrospective review of patients who died after HSCT for MF, PPV-MF, and PET-MF found that 67% were directly related to the HSCT vs 26% from disease progression. Despite the use of reduced-intensity conditioning in the majority of patients studied, significant residual risk exists.24
Advances in transplant care have allowed for a slow but steady improvement in transplant-related mortality. Patients with MF transplanted in 2000–2010 vs 2011–2014 with similar DIPSS scores were evaluated and found to have a statistical trend with improved outcomes in the more recent cohort.25 This included a trend for improved 3-year transplant mortality, relapse rate, and actuarial survival. Incorporation of ruxolitinib pretreatment in HSCT regimens remains a pivotal and debated decision.26–29 Symptom burden is often improved at the expense of unpredictable cytopenias. There also remains a risk for cytokine-mediated discontinuation syndrome when treatment is stopped. Additional long-term prospective data are still required.
Though commonly employed in refractory or high-risk MDS/MPN, HSCT data in these diseases is limited. Most experts recommend HSCT for high-risk CMML patients, as assessed by blast count. Though prognostic scoring systems do not exist for most MDS/MPNs, there are several attempts to assess risk for CMML using clinical and pathologic and more recently molecular features. This is reviewed in detail elsewhere.30
Androgens, IFNα, and thalidomide
Though wrought with toxicities, such androgens as oxymetholone, methandrostenolone, nandrolone, and fluoxymesterone have displayed improvement in anemia for some MF patients compared to transfusions alone.31 Danazol, an attenuated androgen, has also been shown to improve anemia in MF. This includes cessation of transfusions in some patients with considerably lower toxicity than the aforementioned androgens.32
IFNα alters the proliferation of fibroblasts, and has been observed clinically to reverse leukocytosis and thrombocytosis. Severe systemic (fatigue, myalgias, peripheral edema, somnolence), neurologic, and hematologic (anemia) complications have been observed with high- and low-dose regimens, which led to the development of polyethylene glycolated IFNα2a, which has become the preferred treatment modality.33 A Phase II study in 32 MF, PPV-MF, and PET-MF patients revealed improvement in cytopenias, splenomegaly, and marrow fibrosis, with approximately 50% achieving complete response OR partial response, predominantly in patients with early MF mostly consisting of low or intermediate 1 MF.34 IFNα remains underutilized, and likely has utility in some patients, particularly younger, with less advanced disease.
Heterogeneity of response is seen in MF patients treated with different thalidomide derivatives. Thalidomide is an immunomodulatory agent with known inhibition of neoangiogenesis, in addition to possible cytokine modulation. Thalidomide does have activity in MF, and has been shown in Phase II clinical trials to have a low rate of objective responses, but significant side effects of peripheral edema and neurotoxicity limit its use as a single agent.35 The addition of prednisone to thalidomide is similarly efficacious, with a reduction in toxicity.36 In Phase II clinical trials, the combination of lenalidomide plus prednisone in MF patients had similar overall response rates of 10%–35%.37–39 Pomalidomide has activity in JAK2V617F patients, but not in patients with wild-type JAK2. Limited improvement in splenomegaly, significant neuropathy, and lack of long-term survival benefits were observed.40 However, there may be benefit among some subgroups, and the effects on patients with MDS/MPN are unknown.41
Molecular genetics in MF and MDS/MPN
JAK is a protein kinase that activates STAT through phosphorylation. Janus, the two faced Roman god, is referenced with regard to the protein having a catalytic domain and a second domain that serves as an autoregulatory unit. There are four subtypes of JAKs that play a role in JAK/STAT signaling: JAK1, JAK2, JAK3, and TYK2.42 JAK1 is integral to proinflammatory signaling via cytokines TNFα, IL-1, and IL-6. JAK2 is critical for hematopoietic signaling, including thrombopoietin, erythropoietin, granulocyte-macrophage colony-stimulating factor (GM-CSF), prolactin, growth hormone, IL-3, and IL-5.43 JAK3 is implicated in B- and T-cell development, based on knockout models that develop severe combined immunodeficiency.44 TYK2, in concert with JAK2 or -3, facilitates cytokine signal transduction.45
While critical in hematopoiesis and considered important in the pathogenesis of MPNs, JAK2 was considered an inaccessible target until the last decade. Initial in vitro data in PV erythroid-progenitor cells found a lack of erythroid-colony formation in the presence of a JAK2 inhibitor and small interfering RNA.46 This initial preclinical data spurred the sequencing of DNA from PV patients, which displayed the V617F mutation in nearly all patients. Additional in vitro data showed that the presence of JAK2V617F conferred a survival benefit to cells in the absence of stimulatory cytokine signals.47 Specifically, the coexpression of the erythropoietin receptor, thrombopoietin receptor, and GM-CSF receptor with mutant JAK2V617F led to constitutive, independent activation of downstream JAK–STAT signaling.42,48–50 JAK2V617F is present in nearly all patients with PV (~95%), and is detected with considerable frequency in MF (~50%) and ET (~50%). JAK2 exon 12 mutations are present in a small fraction of MF and PV patients. MPLW515L or MPLW515K mutations, which result in constitutive activation of the TPO receptor, occur in ET and MF (5%–10%).51 Together, these mutations represent 50%–60% of MF cases. Interestingly, these mutations are also present in MDS/MPN, but their occurrence is infrequent, with the exception of JAK2V617F in RARS-T (>50% JAK2V617F-mutated).
CALR mutations have recently been shown to occur in a mutually exclusive fashion to JAK/MPL mutations in MF (25%–35%) and ET (~25%).52,53 Most JAK/MPL− MF and ET patients harbor CALR mutations (73%). Mechanistically, the effect of CALR mutations has not fully been explained. CALR-associated proliferation that is independent of JAK/STAT or cytokine signaling has been noted in vitro. Phenotypically, MF patients with CALR type I deletion appear to have longer overall survival and lower levels of disease complications, such as thrombosis. CALR type II insertions are less common and associated with poor risk, as is presence of “triple-negative” (JAK2wt/MPLwt/CALRwt) in MF.53–55 The frequencies of these mutations in MPNs are noted in Table 1.
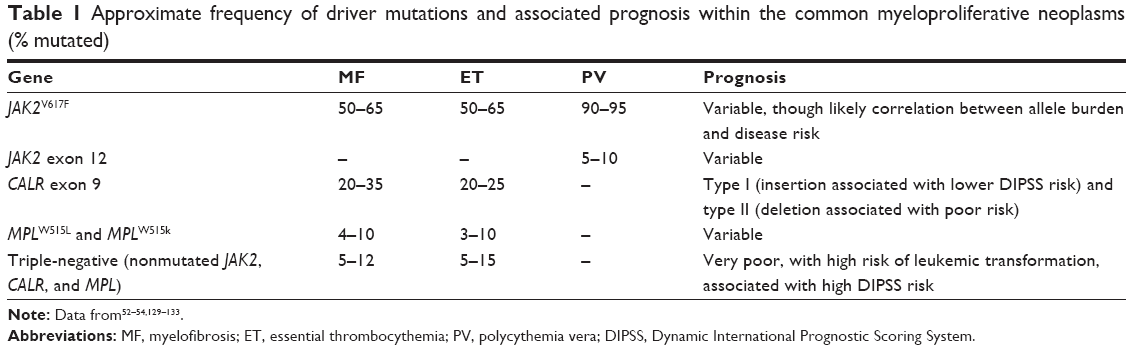 | Table 1 Approximate frequency of driver mutations and associated prognosis within the common myeloproliferative neoplasms (% mutated) |
Several other molecular aberrations have been shown to have some prognostic relevance in these diseases. ASXL, for example, is an epigenetic regulator mutated in higher-risk MF and CMML. In 500 MF patients, CALR−, ASXL1-mutated patients had a significant reduction in survival (hazard ratio 3.7).56 This mutation is seen in CMML (35%–40%), RARS-T (10%), aCML (25%), and MDS/MPN-U (14%). ASXL1-mutated CMML patients appear to have a decrease in overall survival ASXL1 can also be used in combination with age, hemoglobin, and white blood-cell count to refine prognosis further.10 Other notable mutations present in MF and MDS/MPN are displayed in Table 2.
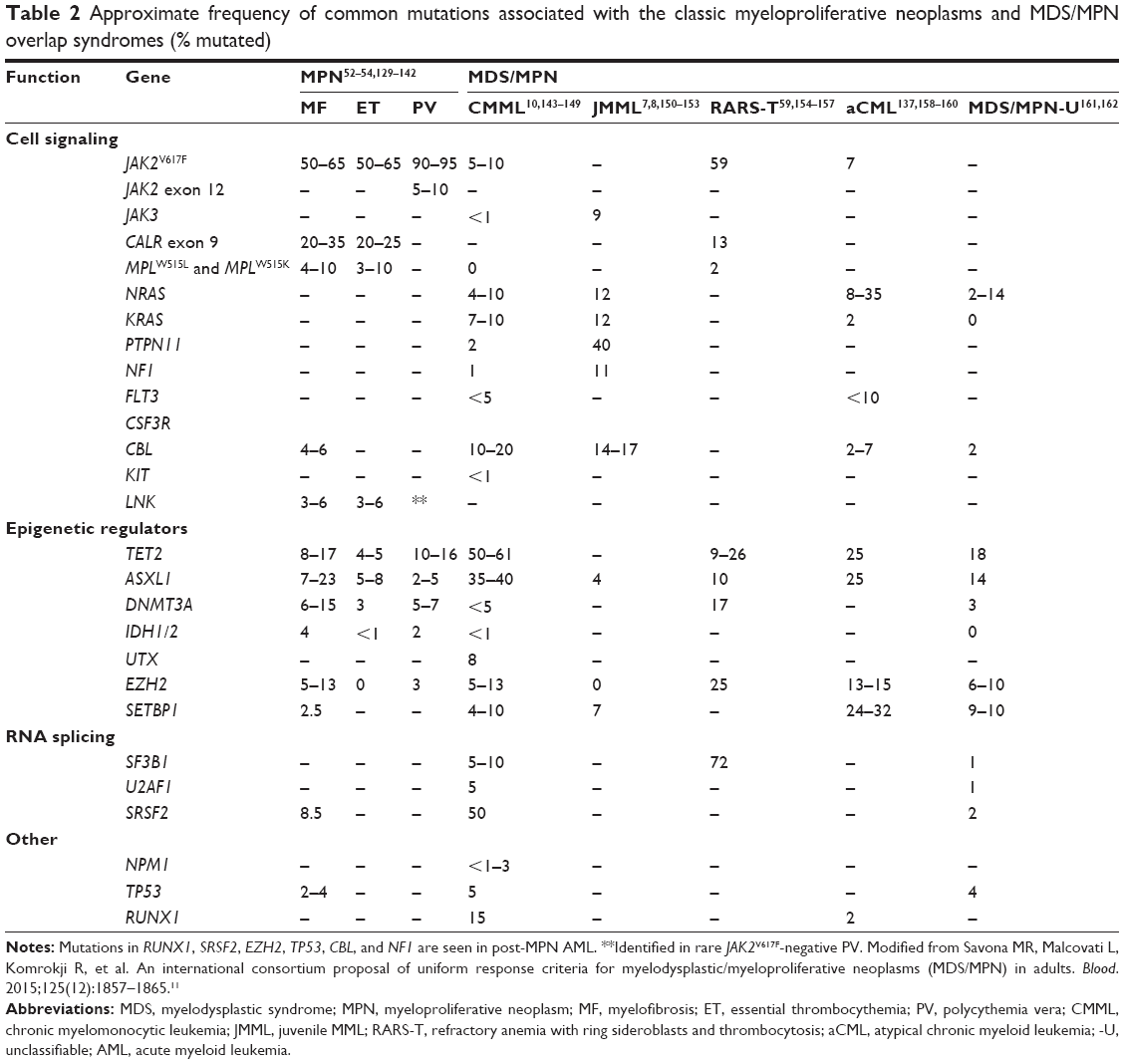 | Table 2 Approximate frequency of common mutations associated with the classic myeloproliferative neoplasms and MDS/MPN overlap syndromes (% mutated) |
Genetic hierarchy, or the order of obtaining mutations, may also impact clinical courses. For example, JAK2V617F-first compared to “TET2-mutated-first” patients had a higher rate of thrombosis in one recent analysis.57 While the correlation of genotype with phenotype is a work in progress, several molecular signatures have been recently reported, including specificity of ASXL + SRSF2 dual mutations for CMML or JAK2V617F with SF3B1 in RARS-T.58,59 Understanding these matrix molecular relationships will hopefully help with prognostication and development of rational therapy.
Emergence of JAK/STAT-directed therapy
Ruxolitinib
Ruxolitinib is a dual JAK1–JAK2 small-molecule inhibitor and ATP mimetic approved for treatment of intermediate- and high-risk MF and hydroxyurea-resistant or -intolerant PV patients. The approval of ruxolitinib for these indications was based in part on the COMFORT-I and COMFORT-II trials.60,61 COMFORT-I was a randomized, double-blind, placebo-controlled trial of 309 intermediate 2 or high-risk MF patients randomized 1:1 to oral ruxolitinib vs placebo. Patients with severe thrombocytopenia were excluded.
The primary end point of splenic volume reduction >35% at 24 weeks was robust in the ruxolitinib cohort, at 41.9%, compared to 0.7% in the placebo group (odds ratio 134.4, 95% confidence interval 18–1,004.9; P<0.001). This response was durable in 65% of patients at 48 weeks. Perhaps more telling was some degree of measured reduction in splenic volume in nearly all patients on ruxolitinib. The majority of ruxolitinib patients gained weight and had a reduction in spleen-mediated (abdominal discomfort, early satiety) and non-spleen-mediated (night sweats, pain, pruritus) symptoms. The JAK2V617F-allele burden was reduced, and critically overall survival was improved in the ruxolitinib group (hazard ratio 0.50; 95% confidence interval 0.25–0.98; P=0.04). COMFORT-II was a randomized Phase III trial of 219 MF patients assigned to ruxolitinib vs best commercially available therapy (BAT).61 BAT patients treated with any agent (67%) included hydroxyurea (47%) and glucocorticoids (16%). The primary end point of >35% splenic volume reduction was achieved in 28% of the ruxolitinib-treated patients and 0 of the BAT patients. Of note, overall survival was not statistically significant between the two groups at 48 or 61 weeks.
COMFORT-I/II suggested ruxolitinib improved the inflammatory profiles of MF patients by mediating JAK/STAT signaling independently of mutational status. The proinflammatory markers C-reactive protein, TNFα, and IL-6 were all reduced in patients treated with ruxolitinib, correlating with earlier work showing the same in vitro. There was a converse elevation of the anabolic markers leptin and erythropoietin.62,63
The COMFORT trials clearly demonstrated dose-dependent toxicities of thrombocytopenia, anemia, and to a lesser extent neutropenia, and these remain limiting factors in clinical practice. Discontinuation of ruxolitinib has been associated with acceleration of hematologic derangements, splenomegaly, and resultant MF symptoms, which in some cases can lead to severe systemic inflammatory responses. This rebound effect may occur in as many as 11% of patients who rapidly discontinue therapy.64 Taken together, these issues have inspired the development of alternative JAK/STAT regulators.
There are less MDS/MPN-specific clinical trials, due to the difficulty in diagnosis and low prevalence of disease. Recently, however, a Phase I trial evaluated 18 patients with CMML-1 treated with ruxolitinib.65 As per the 2006 International Working Group criteria, three of 15 had a hematologic improvement and one of 15 had a partial response. Splenomegaly reduction >50% was achieved in six of nine patients who exhibited baseline enlargement. Symptomatic control of B symptoms, such as night sweats, was observed in 14 of 15 patients. Grade 3 toxicities were observed in one patient with thrombocytopenia. A Phase II trial (NCT01776723) is ongoing.
Ruxolitinib in combination with lenalidomide
A total of 31 patients with MF, PPV-MF, and PET-MF were treated with ruxolitinib in combination with lenalidomide.41 International Working Group-defined response was achieved in 55% of patients, but no complete or partial responses were observed. JAK-inhibitor and lenalidomide-naïve patients appeared to have higher response rates. Despite evidence of activity, the study failed to meet predetermined efficacy or treatment success, and was discontinued prior to completion.
Pacritinib
Pacritinib is a dual JAK2 and FLT3 inhibitor. FLT3 is a tyrosine kinase with frequent aberrant signaling in hematologic malignancies, most notably acute myelogenous leukemia. Preclinical studies in MF have demonstrated that FLT3 activation of megakaryocytes results in downstream MAP-kinase activation and resultant increased inflammatory cytokines, independently of JAK2-mutational status.66 The interaction of FLT3 with PI3K/Akt and STAT signaling within MF supports its therapeutic targeting.67 While there are currently no approved FLT3 inhibitors for hematologic malignancies, sorafenib, a multikinase inhibitor, has been successfully used in FLT3-ITD-mutated AML, and several other agents with more specific activity against FLT3-ITD and FLT3-TKD are under investigation.68–71 This is reviewed expertly elsewhere.
In COMFORT-I/II, MF patients with platelet counts ≤100,000/μL were excluded. For this reason, pacritinib was first developed in patients with lower platelet counts. Interestingly, this therapy has largely been well tolerated in these thrombocytopenic patients in clinical trials. Results from a Phase II trial of pacritinib in patients with primary or secondary MF, PPV-MF, and PET-MF with baseline thrombocytopenia <100,000/μL found a 43% reduction in splenic volume after 36 weeks.72 Some patients developed similar improvements in anemia and thrombocytopenia to an additional Phase II trial.73
PERSIST-1 is a Phase III trial of pacritinib vs BAT in 327 patients with intermediate 1- or 2-risk MF, PPV-MF, and PET-MF.74 Primary outcomes are similar to the benchmark COMFORT trials, with >35% reduction in splenomegaly and >50% MF-symptom improvement at 24 weeks. Thrombocytopenia was present in a third of enrolled patients, and also improved in most cases. It is worth noting that later studies of ruxolitinib in patients with low platelets had similar findings.75 PERSIST-2 (NCT02055781) will include ruxolitinib in the BAT group and the sequential use of this second JAK–STAT active therapy.
Momelotinib
Momelotinib (CYT387) is a dual JAK1/JAK2 inhibitor with limited inhibition of TYK2.76 Initial Phase I/II clinical trials supported reduction in splenic volume (48% of patients) and a surprising improvement in anemia (70% achieved transfusion independence).77 Unfortunately, treatment-emergent peripheral neuropathy was noted in 44% of MF patients in the study.78 Onset was delayed at a median of 32 weeks, and did not resolve with dose reduction or withdrawal of momelotinib. SIMPLIFY 1 is a Phase III trial of momelotinib vs ruxolitinib in 420 patients with primary and secondary MF.79 SIMPLIFY 2 is a Phase III trial of momelotinib vs BAT in 150 primary or secondary MF, PPV-MF, and PET-MF patients, including ruxolitinib failures.80 These trials are ongoing.
Fedratinib (SAR302503)
Fedratinib is a selective JAK2 inhibitor. Early phase clinical trials found a reduction in splenomegaly and MF associated symptoms with fedratinib treatment.81,82 The JAKARTA study was a double-blind placebo-controlled study with primary end point of spleen-volume reduction similar to the COMFORT trials. Reduction in spleen volume was observed with placebo (1%), 400 mg (47%), and 500 mg (49%).83 Similar to other JAK inhibitors, diarrhea was the primary nonhematologic adverse event (AE). Ultimately, development of fedratinib was halted after several cases of Wernicke’s encephalopathy were noted. The proposed mechanism for thiamine deficiency and development of Wernicke’s involves thiamine uptake via the human THTR2.84 This toxicity appears to be directly linked to the structure of fedratinib, which competes with thiamine and has not been observed in other JAK inhibitors.
INCB039110 and INCB052793
INCB039110 is a selective JAK1 inhibitor. A Phase II clinical trial evaluated 65 MF patients treated with INCB039110 based on the total symptom score (TSS).85 Specifically, patients were evaluated for 50% reduction in symptomatic night sweats, itchiness, abdominal discomfort, pain under left ribs, early satiety, and bone pain/myalgias. At 12 weeks, 35% of patients treated with INCB039110 had a 50% reduction on the TSS, and 14% had reduction in splenic volume on computed tomography or magnetic resonance imaging. Common side effects were low-grade constipation and nausea. Anemia, grade 3 (24%), and thrombocytopenia grade 3 (24%) and grade 4 (3%) were observed. INCB052793, a more potent selective JAK1 inhibitor, is currently in a Phase I/II dose-escalation study (NCT02265510) for patients with hematologic malignancies, including MF and MDS/MPN. The more selective JAK1 inhibition lends expectation for considerable amelioration of symptoms and spleen-size reduction without dose-limiting thrombocytopenia and anemia.
Novel therapy
Hedgehog signaling and therapy
Hedgehog proteins are secreted, lipid-modified signaling proteins with final downstream antiapoptotic effects. PTCH1, SMO, SUFU, and GLI1-3 are part of the Hedgehog protein complex responsible for these signaling effects.86 Mutations or overexpression of these proteins is observed in solid malignancies, hematologic malignancies, and tissue fibrosis.87–90 Inhibition resulted in a superior chemotherapy effect and decreased tumor fibrosis within a pancreatic cancer model.91 These observations spurred exploration of Hedgehog inhibition in other diseases, specifically myeloid neoplasm with marrow fibrosis.
Further, it appears that JAK–STAT and Hedgehog pathways interact in MF. In preclinical models, granulocytes derived from MPN patients had increased Hedgehog target genes compared to controls. Mouse models treated with ruxolitinib in combination with an SMO inhibitor displayed decreases in mutant-allele burden beyond ruxolitinib or SMO inhibition alone. Bone marrow fibrosis was also reduced when used in combination, and not with ruxolitinib monotherapy.92 This led to single-agent trials and ruxolitinib combinations with SMO inhibitors in myeloid disease, specifically MF.
IPI-926 is an oral SMO inhibitor within the Hedgehog pathway. Phase I data found that IPI-926 was generally well tolerated, with limited gastrointestinal side effects and reversible elevations in hepatic transaminases.93 In a Phase II evaluation, 14 patients with MF were treated with IPI-926 for a median of 5 months, with all patients discontinuing treatment by 7.5 months, mostly due to lack of response.94 Some reduction in splenomegaly was observed, but no sustained improvements in symptoms via the MPN symptom-assessment form were recorded. IPI-926 had a heterogeneous effect on bone marrow reticulin, fibrosis, JAK2V617F-allele burden, and inflammatory cytokine profiles. Based on these results, the authors concluded that further investigation in combination with other treatments for MF may hold more promise. PF-04449913 is an oral SMO inhibitor under investigation for hematologic malignancies. In a Phase I trial in which seven of 47 patients had MF, five of seven achieved stable disease. A greater than 50% reduction in splenomegaly was found in one of seven MF patients. Side effects were limited to on-target low-grade toxicities of dysgeusia and alopecia.95
Sonidegib (LDE225) is another oral SMO inhibitor within the Hedgehog pathway. Results from a Phase I clinical trial of combination sonidegib and ruxolitinib in 23 patients with MF displayed enhanced outcomes compared to sonidegib monotherapy.96 At enrollment, most patients were at high risk on the DIPSS. Over 65% of patients had >50% reduction in splenomegaly, and over half had resolution of splenomegaly. Grade 3/4 anemia (five patients) and thrombocytopenia (one) were observed. Other serious AEs included face edema (one), hyponatremia (one), pyrexia (one), right ventricular failure (one) and creatinine-kinase elevation (two). Development of sonidegib in combination with ruxolitinib continues.
PI3K–Akt pathway and therapy
PI3K activates Akt, with resultant promotion of cellular survival and growth. PI3Kα, -β, -δ, and -γ isoforms participate in proliferation, cell differentiation, and immunity. The PI3Kδ isoform is implicated in hematopoietic proliferation and MF.97,98 TGR-1202 is a PI3Kδ-specific inhibitor that has shown promise in preliminary clinical data from a Phase I study in refractory hematologic malignancies.97,99,100 Importantly, liver toxicities were less than 5%, suggesting deviation from other PI3Kδ-toxicity profiles. Synergism between JAK and PI3Kδ inhibition is under investigation in a Phase I trial (NCT02493530) in patients with MF, PPV-MF, PET-MF, MDS/MPN, and PV resistant to hydroxyurea. This is the only ongoing trial testing the combination of ruxolitinib and a PI3Kδ-specific inhibitor.
Antifibrotics
Improvement in marrow fibrosis has been elusive in trials of antiproliferative treatments, with the exception of telomerase inhibitors. Current antifibrotic agents with efficacy in liver and lung fibrosis are being explored in MF. Novel targets include pentraxin 2, a key plasma protein that inhibits or reverses fibrosis via action on monocyte differentiation.101,102 A second target, LOXL2, is expressed in a setting of marrow hypoxia, with resultant extracellular matrix cross-linking and collagen IV assembly. PRM151 is a recombinant form of pentraxin 2. PRM151 displayed no dose-limiting toxicities in Phase I trials, and is nearing completion of a Phase II trial in MF patients with DIPSS intermediate-1, -2, or high-risk disease.102 Patients were allowed to be on ruxolitinib. Outcomes have been positive, with decreased bone marrow fibrosis (35%), improvement of anemia (40%), thrombocytopenia (40%), and symptoms via TSS (38%). Reduction in spleen size on physical exam (26%) was also noted. Patients already on ruxolitinib did not appear to have any cumulative toxicity. Simtuzumab is a monoclonal antibody against LOXL2. Simtuzumab was explored (NCT01369498) as a treatment for MF in combination with ruxolitinib, but development in myeloid disease was suspended, given failure to meet primary end point.103 Simtuzumab is also being tested in pulmonary fibrosis, nonalcoholic steatohepatitis cirrhosis, and primary sclerosing cholangitis.
Telomerase inhibition
Human cells have repetitive terminal DNA sequences that protect against loss of genetic information during replication. This sequence diminishes with age, except in cells that express telomerase. The intrinsic unlimited replication potential of malignant cells is due to the dramatic upregulation in telomerase, which makes it a potential specific target of therapy.
GRN163L (imetelstat) is an inhibitor of the RNA telomerase reverse-transcriptase template. A pilot study (NCT01731951) of imetelstat in 33 patients with mostly high-risk MF, PPV-MF, or PET-MF displayed promising molecular remission and morphologic improvement in bone marrow fibrosis.104 This striking complete response occurred in four patients. Additional patients displayed partial responses, with either molecular remission or reversal in marrow fibrosis, but not both. Improvement in cytopenias and leukocytosis was also demonstrated at a variable rate. Response was primarily seen in patients with JAK2, ASXL1, or spliceosome mutations, which holds potential promise for MDS/MPN as well, though MDS/MPN were not treated on this trial. Likewise, this is particularly important with the previously mentioned negative impact of ASXL1 mutations on survival in MF. Toxicity included myelosuppression, with dose reduction occurring in 67% of patients.
HDAC inhibitors
Access to DNA that is wrapped around histones is regulated in part by acetylation and deacetylation. Histone deacetylases (HDACs) function via interaction with the histone lysine tail and inhibit transcription. Within MF, there is elevation in HDACs compared to other MPNs that correlates with splenomegaly but not JAK2V617F status.97
Panobinostat (LBH589) is an HDAC inhibitor evaluated in a Phase I trial of 18 MF, PPV-MF, and PET-MF patients.105 Only five patients endured enough cycles to be evaluated for response. A 100% reduction in palpable splenomegaly with clinical improvement (three of five) and reduction in anemia (two of five) was observed. JAK2V617F status and previous treatment did not appear to influence response. Hematologic AEs included dose-dependent thrombocytopenia, and nonhematologic side effects were primarily gastrointestinal. A Phase II trial of higher-dose panobinostat in 35 patients with MF, PPV-MF, and PET-MF displayed unsustained reduction in splenomegaly and reduced systemic inflammation.106 However, medication toxicity resulted in a high dropout rate, with just under half of the patients completing more than two cycles. Panobinostat in combination with ruxolitinib was studied in a Phase I/II trial of 61 MF patients, with reduction in splenomegaly (79%) and similar AEs to ruxolitinib monotherapy.107 Additional single-agent and combination (ruxolitinib) trials (NCT01693601) are ongoing.
Vorinostat is an HDAC inhibitor evaluated in a Phase II trial of 14 patients with MF or PPV-MF. The primary outcome was a clinical response as per the International Working Group response criteria.108 Only eight of 14 patients completed the predetermined treatment period, and three of eight had concomitant hydroxyurea usage. JAK2V617F-allele burden increased in seven of eight patients, and there was no reduction in the MPN symptom-assessment form score. Additionally, abdominal pain and nausea increased in severity.
DNA methyltransferase inhibition
DNA methylation is an integral part of transcription regulation. Somatic cells use hypomethylation to increase transcriptional activity. Within the promoters of tumor-suppressor genes, hypermethylation can result in their effective inactivation. Within MF and MDS/MPN, emerging data suggests that ASXL1 is in part disrupted by alteration in methylation.109
The DNA methyltransferase inhibitor 5-azacitidine (5-Aza) is standard-of-care treatment in MDS and AML. In MF, 5-Aza has been evaluated in a Phase II trial of 34 patients.110 Eight patients displayed a response, including one partial response and seven clinical improvements. No change in bone marrow fibrosis or JAK2V617F-allele burden was observed, and this agent is less tolerated in patients with MF. Given the response seen in CMML patients in the Aza-001 trial, 5-Aza is the standard-of-care first-line therapy for patients with CMML. Interestingly, 5-Aza is less effective in more proliferative CMML cases, and this observation has informed the use of DNA methyltransferase inhibitors in MDS/MPN.111 5-Aza in combination with ruxolitinib is under investigation (NCT01787487) in MF, MDS, and MPN/MDS, and may be effective in more proliferative MDS/MPN. The other US Food and Drug Administration-approved DNA methyltransferase inhibitor, decitabine, was evaluated in 21 patients with MF.112 Seven patients demonstrated some degree of clinical response, with similar toxicity as seen with 5-Aza. Interestingly, CMML patients previously treated with 5-Aza have been treated with decitabine with modest response (overall response rate <20%) and poor overall survival.113 Combination with ruxolitinib (NCT02076191) is ongoing in patients with MPNs and postmyeloproliferative AML.
Potential future targets
Pro-viral integration site for moloney murine leukemia virus (PIM) kinases and NEDD8-activating enzymes are potential future therapies. PIM kinases are implicated in solid and hematologic tumors, and are associated with cell-cycle regulation, apoptosis, and cell growth.114 Classically, PIM stimulates the proto-oncogene MYC and inhibits its native apoptotic signals. Likewise, ABC proteins and P-glycoprotein transporters that act as efflux pumps for oncologic treatments are upregulated by PIM kinase.115 PIM-kinase inhibition appears promising in AML and multiple myeloma.116,117 In MF, STAT1 and STAT5 regulate the PIM1 kinase-promoter sequences.118 JAK/STAT signaling is directly upstream to PIM kinase, further suggesting its role in myeloproliferative signaling. INCB53914 is a pan-PIM-kinase inhibitor, and the only current PIM-kinase inhibitor entering evaluation in human trials as a potential treatment in myeloid disease.119
Pevonedistat (MLN4924) is a first in class NEDD8-activating enzyme inhibitor. This pathway is implicated in ubiquitin–proteasome protein degradation. It is currently under investigation within AML patients in combination with azacitidine.120,121
Immunotherapy
Current immunotherapies activate the patient’s immune system to recognize and attack the tumor cells. Whereas high dose IL-2 therapy and anti-CTLA4 therapy have been successful in kidney cancer and melanoma, there has been no activity with these treatments in hematologic malignancies. Allogeneic HSCT remains the gold-standard means by which host effector cells are increased and activated to combat myeloid diseases. As noted earlier, HSCT is limited to select patients, and typically is not offered to older, sicker MDS/MPN or MF patients. Further, the disease-relapse rate for patients with chronic proliferative Ph− myeloid disease (either MF or MDS/MPN) is considerable.18,122 Therefore, new therapies that increase the number and function of immune effector cells or reduce the inherent capacity of tumors to suppress immune response are being developed in myeloid malignancies.
Chimeric antigen receptor (CAR) T-cell therapy involves engineering a patient’s T-cells to express antitumor CARs. These contrived T-cells are reinfused with tumor specificity, and appear to persist in circulation while mediating their antitumor effects. Anti-CD19 CAR T-cells have been successful in treating tumors that express CD19, such as B-cell leukemias and lymphomas.123 A Phase 1 study of CAR T-cells that target NKG2D ligands is under way in patients with AML and advanced MDS–refractory anemia with excess blasts and multiple myeloma (NCT02203825). There is considerable optimism, but many scientific challenges remain ahead for CAR T-cell therapy in myeloid diseases.
Another mechanism to reactivate the immune system is to block the immune-checkpoint pathways, such as through the inhibition of the PD1 receptor. Tumor cells have been shown to express PD ligand 1, which interacts with PD1 on T-cells to decrease T-cell activity. Therefore, the blockade of PD1 allows for the reactivation of T-cell proliferation and cytotoxicity.124 Nivolumab, a monoclonal antibody against PD1,125 has been approved in the treatment of non-small-cell lung cancer and melanoma, and is currently in a Phase II study in patients with MF, PET-MF, and PPV-MF to determine if it can be used to control MF (NCT02421354).
Antitumor vaccination is also an important area of immunotherapy. Numerous studies are being performed to determine the most effective tumor antigens to target. These include individual tumor-selective antigens, such as WT1, tumor-specific neoantigens, such as the BCR–ABL fusion oncoprotein, or whole-tumor-cell (multiepitope) vaccinations, such as the GM-K562 vaccine.126 Additionally, different mechanisms of vaccine delivery are being examined, including costimulation with cytokines like GM-CSF, or the administration of the antitumor vaccines post-allo-HSCT when there is a greater chance for a graft-vs-leukemia response.127,128 Continued investigation in these areas will likely elucidate novel mechanisms to aid in the treatment of MPN and MDS/MPN.
Conclusion
Tremendous progress has been achieved in the decade since the discovery of JAK2V617F in diagnosing, prognosticating, and developing new therapeutic options for patients with MF and MDS/MPNs. Further refinement of the genetic signatures of these diseases will be key. Ideally, as therapies mature within MF, many will undergo investigation in MDS/MPNs guided by the proposed response criteria and intersecting genetic signatures now available for MDS/MPN.10,11
Significant work remains to capitalize fully on immunotherapy within myeloid disease. Combination therapy represents a large number of potential viable treatment permutations, even with agents that have limited single-agent effect. Improved targeting of proliferative pathways in MF and MDS/MPNs will potentially minimize hematologic side effects. At the same time, results are encouraging for the triad of symptom control, reduction of marrow fibrosis, and most importantly increased survival.
Disclosure
MRS has received research funding from Astex, Celgene, Incyte, and TG Therapeutics, and has advised/consulted for Baxalta, Celgene, Gilead, and Incyte. The other authors report no conflicts of interest in this work.
References
Ma Z, DeBusk L, Chandra P, et al. Array-Based Comparative Genomic Hybridization (aCGH) In Myelofibrosis Reveals Divergent Genetic Copy Number Alterations Among Patients With Post-Essential Thrombocythemia-Myelofibrosis/Post-Polycythemia Rubra Vera-Myelofibrosis (PET-MF and PPV-MF) and Pat. Blood. 2013;122(21):5266. | ||
Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007;110(4):1092–1097. | ||
Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. | ||
Passamonti F, Cervantes F, Vannucchi AM, et al. Dynamic International Prognostic Scoring System (DIPSS) predicts progression to acute myeloid leukemia in primary myelofibrosis. Blood. 2010;116(15):2857–2858. | ||
Padron E, Garcia-Manero G, Patnaik MM, et al. An international data set for CMML validates prognostic scoring systems and demonstrates a need for novel prognostication strategies. Blood Cancer J. 2015;5:e333. | ||
Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. | ||
Niemeyer CM, Kratz CP. Paediatric myelodysplastic syndromes and juvenile myelomonocytic leukaemia: molecular classification and treatment options. Br J Haematol. 2008;140(6):610–624. | ||
Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29(4):465–468. | ||
Foucar K. Myelodysplastic/myeloproliferative neoplasms. Am J Clin Pathol. 2009;132(2):281–289. | ||
Itzykson R, Kosmider O, Renneville A, et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2428–2436. | ||
Savona MR, Malcovati L, Komrokji R, et al. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood. 2015;125(12):1857–1865. | ||
Martínez-Trillos A, Gaya A, Maffioli M, et al. Efficacy and tolerability of hydroxyurea in the treatment of the hyperproliferative manifestations of myelofibrosis: results in 40 patients. Ann Hematol. 2010;89(12):1233–1237. | ||
Hanft VN, Fruchtman SR, Pickens CV, Rosse WF, Howard TA, Ware RE. Acquired DNA mutations associated with in vivo hydroxyurea exposure. Blood. 2000;95(11):3589–3593. | ||
Sirhan S, Lasho TL, Hanson CA, Mesa RA, Pardanani A, Tefferi A. The presence of JAK2V617F in primary myelofibrosis or its allele burden in polycythemia vera predicts chemosensitivity to hydroxyurea. Am J Hematol. 2008;83(5):363–365. | ||
Mesa RA, Nagorney DS, Schwager S, Allred J, Tefferi A. Palliative goals, patient selection, and perioperative platelet management: outcomes and lessons from 3 decades of splenectomy for myelofibrosis with myeloid metaplasia at the Mayo Clinic. Cancer. 2006;107(2):361–370. | ||
Santos FP, Tam CS, Kantarjian H, et al. Splenectomy in patients with myeloproliferative neoplasms: efficacy, complications and impact on survival and transformation. Leuk Lymphoma. 2014;55(1):121–127. | ||
Somasundaram SK, Massey L, Gooch D, Reed J, Menzies D. Laparoscopic splenectomy is emerging ‘gold standard’ treatment even for massive spleens. Ann R Coll Surg Engl. 2015;97(5):345–348. | ||
Deeg HJ, Gooley TA, Flowers ME, et al. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Blood. 2003;102(12):3912–3918. | ||
Kroger N, Giorgino T, Scott BL, et al. Impact of allogeneic stem cell transplantation on survival of patients less than 65 years of age with primary myelofibrosis. Blood. 2015;125(21):3347–3350; quiz 3364. | ||
Cervantes F, Barosi G, Demory JL, et al. Myelofibrosis with myeloid metaplasia in young individuals: disease characteristics, prognostic factors and identification of risk groups. Br J Haematol. 1998;102(3):684–690. | ||
Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single-institution experience with 91 cases. Blood. 2005;105(3):973–977. | ||
Patel KP, Newberry KJ, Luthra R, et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood. 2015;126(6):790–797. | ||
Gupta V, Hari P, Hoffman R. Allogeneic hematopoietic cell transplantation for myelofibrosis in the era of JAK inhibitors. Blood. 2012;120(7):1367–1379. | ||
Robin M, Tabrizi R, Mohty M, et al. Allogeneic haematopoietic stem cell transplantation for myelofibrosis: a report of the Société Française de Greffe de Moelle et de Thérapie Cellulaire (SFGM-TC). Br J Haematol. 2011;152(3):331–339. | ||
Bregante S, Dominietto A, Ghiso A, et al. Improved outcome of alternative donor transplantations in patients with myelofibrosis: from unrelated to haploidentical family donors. Biol Blood Marrow Transplant. 2016;22(2):324–329. | ||
Francois S, Huynh A, Cassinat B, et al. Ruxolitinib before allogeneic hematopoietic stem cell transplantation (HSCT) in patients with myelofibrosis: a preliminary descriptive report of the JAK ALLO study, a phase II trial sponsored by Goelams-FIM in collaboration with the SFGMTC. Blood. 2013;122(21):306. | ||
Jaekel N, Behre G, Behning A, et al. Allogeneic hematopoietic cell transplantation for myelofibrosis in patients pretreated with the JAK1 and JAK2 inhibitor ruxolitinib. Bone Marrow Transplant. 2014;49(2):179–184. | ||
Stübig T, Alchalby H, Ditschkowski M, et al. JAK inhibition with ruxolitinib as pretreatment for allogeneic stem cell transplantation in primary or post-ET/PV myelofibrosis. Leukemia. 2014;28(8):1736–1738. | ||
Rubio MT, Legrand F, Kiladjian J-J, et al. Ruxolitinib for patients with primary or secondary myelofibrosis before allogeneic hematopoietic stem cell transplantation (allo-HSCT): a retrospective study of the Société Française de Greffe de Moelle et de Thérapie Cellulaire (SFGM-TC). Blood. 2013;122(21):2111. | ||
Mughal TI, Cross NC, Padron E, et al. An International MDS/MPN working group’s perspective and recommendations on molecular pathogenesis, diagnosis and clinical characterization of myelodysplastic/myeloproliferative neoplasms. Haematologica. 2015;100(9):1117–1130. | ||
Brubaker LH, Brière J, Laszlo J, et al. Treatment of anemia in myeloproliferative disorders: a randomized study of fluoxymesterone v transfusions only. Arch Intern Med. 1982;142(8):1533–1537. | ||
Cervantes F, Alvarez-Larrán A, Domingo A, Arellano-Rodrigo E, Montserrat E. Efficacy and tolerability of danazol as a treatment for the anaemia of myelofibrosis with myeloid metaplasia: long-term results in 30 patients. Br J Haematol. 2005;129(6):771–775. | ||
Tefferi A, Elliot MA, Yoon SY, et al. Clinical and bone marrow effects of interferon alfa therapy in myelofibrosis with myeloid metaplasia. Blood. 2001;97(6):1896. | ||
Silver RT, Feldman EJ, Ritchie E, et al. Recombinant interferon alpha (rIFN) may retard progression of early myelofibrosis by reducing splenomegaly and by decreasing marrow fibrosis. Blood. 2013;122(21):4053. | ||
Thomas DA, Giles FJ, Albitar M, et al. Thalidomide therapy for myelofibrosis with myeloid metaplasia. Cancer. 2006;106(9):1974–1984. | ||
Mesa RA, Steensma DP, Pardanani A, et al. A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood. 2003;101(7):2534–2541. | ||
Mesa RA, Yao X, Cripe LD, et al. Lenalidomide and prednisone for myelofibrosis: Eastern Cooperative Oncology Group (ECOG) phase 2 trial E4903. Blood. 2010;116(22):4436–4438. | ||
Quintás-Cardama A, Kantarjian HM, Manshouri T, et al. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol. 2009;27(28):4760–4766. | ||
Thapaliya P, Tefferi A, Pardanani A, et al. International working group for myelofibrosis research and treatment response assessment and long-term follow-up of 50 myelofibrosis patients treated with thalidomide-prednisone based regimens. Am J Hematol. 2011;86(1):96–98. | ||
Begna KH, Pardanani A, Mesa R, et al. Long-term outcome of pomalidomide therapy in myelofibrosis. Am J Hematol. 2012;87(1):66–68. | ||
Daver N, Cortes J, Newberry K, et al. Ruxolitinib in combination with lenalidomide as therapy for patients with myelofibrosis. Haematologica. 2015;100(8):1058–1063. | ||
Vainchenker W, Constantinescu SN. A unique activating mutation in JAK2 (V617F) is at the origin of polycythemia vera and allows a new classification of myeloproliferative diseases. Hematology Am Soc Hematol Educ Program. 2005:195–200. | ||
Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene. 2007;26(47):6724–6737. | ||
Nosaka T, van Deursen JM, Tripp RA, et al. Defective lymphoid development in mice lacking Jak3. Science. 1995;270(5237):800–802. | ||
Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Semin Cell Dev Biol. 2008;19(4):385–393. | ||
James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. | ||
Kralovics R, Teo SS, Buser AS, et al. Altered gene expression in myeloproliferative disorders correlates with activation of signaling by the V617F mutation of Jak2. Blood. 2005;106(10):3374–3376. | ||
Lu X, Levine R, Tong W, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci U S A. 2005;102(52):18962–18967. | ||
Steensma DP, Dewald GW, Lasho TL, et al. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and myelodysplastic syndromes. Blood. 2005;106(4):1207–1209. | ||
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. | ||
Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112(1):141–149. | ||
Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. | ||
Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. | ||
Tefferi A, Lasho T, Finke C, et al. Type 1 vs type 2 calreticulin mutations in primary myelofibrosis: differences in phenotype and prognostic impact. Leukemia. 2014;28(7):1568–1570. | ||
Tefferi A, Lasho TL, Tischer A, et al. The prognostic advantage of calreticulin mutations in myelofibrosis might be confined to type 1 or type 1-like CALR variants. Blood. 2014;124(15):2465–2466. | ||
Tefferi A, Guglielmelli P, Lasho TL, et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: an international study of 570 patients. Leukemia. 2014;28(7):1494–1500. | ||
Ortmann CA, Kent DG, Nangalia J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med. 2015;372(7):601–612. | ||
Meggendorfer M, Roller A, Haferlach T, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood. 2012;120(15):3080–3088. | ||
Broseus J, Alpermann T, Wulfert M, et al. Age, JAK2V617F and SF3B1 mutations are the main predicting factors for survival in refractory anaemia with ring sideroblasts and marked thrombocytosis. Leukemia. 2013;27(9):1826–1831. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. | ||
Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. | ||
Quintás-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–3117. | ||
Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127. | ||
Tefferi A, Pardanani A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin Proc. 2011;86(12):1188–1191. | ||
Padron E, Dezern AE, Vaddi K, et al. A multi-institution phase I trial of ruxolitinib in chronic myelomonocytic leukemia (CMML). Clin Cancer Res. Epub 2016 Feb 8. | ||
Desterke C, Bilhou-Nabera C, Guerton B, et al. FLT3-mediated p38-MAPK activation participates in the control of megakaryopoiesis in primary myelofibrosis. Cancer Res. 2011;71(8):2901–2915. | ||
Kiyoi H. Flt3 inhibitors: recent advances and problems for clinical application. Nagoya J Med Sci. 2015;77(1–2):7–17. | ||
Zhang W, Konopleva M, Shi YX, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008;100(3):184–198. | ||
Ravandi F, Cortes JE, Jones D, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28(11):1856–1862. | ||
Tallman MS, Schiller G, Trone D, et al. Results of a phase 2 randomized, open-label, study of lower doses of quizartinib (AC220;ASP2689) in subjects with FLT3-ITD positive relapsed or refractory acute myeloid leukemia (AML). Blood. 2013;122(21):494. | ||
Stone R, Mandrekar S, Sanford B. The multi-kinase inhibitor midostaurin prolongs survival compared with placebo in combination with daunorubicin/cytarabine induction, high-dose consolidation, and as maintenance therapy in newly diagnosed acute myeloid leukemia patients age 18-60 with FLT3 mutations: an international prospective randomized placebo-controlled double-blind trial (CALGB 10603/RATIFY [Alliance]). Poster presented at: 57th ASH Annual Meeting and Exposition; December 5–8, 2015; Orlando, FL. | ||
Dean JP, Cernohous P, Komrokji RS, et al. Pacritinib, a dual JAK2/FLT3 inhibitor: an integrated efficacy and safety analysis of phase II trial data in patients with primary and secondary myelofibrosis (MF) and platelet counts ≤100,000/μl. Blood. 2013;122(21):395. | ||
Komrokji RS, Seymour JF, Roberts AW, et al. Results of a phase 2 study of pacritinib (SB1518), a JAK2/JAK2(V617F) inhibitor, in patients with myelofibrosis. Blood. 2015;125(17):2649–2655. | ||
Mesa RA, Egyed M, Szoke A, et al. Results of the PERSIST-1 phase III study of pacritinib (PAC) versus best available therapy (BAT) in primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF), or post-essential thrombocythemia-myelofibrosis (PET-MF). J Clin Oncol. 2015;33 Suppl:LBA7006. | ||
Talpaz M, Paquette R, Afrin L, et al. Interim analysis of safety and efficacy of ruxolitinib in patients with myelofibrosis and low platelet counts. J Hematol Oncol. 2013;6(1):81. | ||
Tyner JW, Bumm TG, Deininger J, et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood. 2010;115(25):5232–5240. | ||
Pardanani A, Laborde RR, Lasho TL, et al. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia. 2013;27(6):1322–1327. | ||
Abdelrahman RA, Begna KH, Al-Kali A, et al. Momelotinib treatment-emergent neuropathy: prevalence, risk factors and outcome in 100 patients with myelofibrosis. Br J Haematol. 2015;169(1):77–80. | ||
Winton EF, Devos T, Oh ST, et al. Phase III randomized, double-blind, active-controlled study of momelotinib versus ruxolitinib in patients with myelofibrosis. J Clin Oncol. 2015;33 Suppl:TPS7101. | ||
Gupta V, Baer MR, Oh ST, et al. Phase III randomized, open-label, active-controlled study of momelotinib versus best available therapy in ruxolitinib-treated patients with myelofibrosis. J Clin Oncol. 2015;33 Suppl:TPS7102. | ||
Pardanani A, Tefferi A, Jamieson C, et al. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J. 2015;5:e335. | ||
Pardanani A, Gotlib JR, Jamieson C, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29(7):789–796. | ||
Harrison CN, Cortes JE, Cervantes F, et al. Results of a randomized, double-blind, placebo-controlled phase iii study (JAKARTA) of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis (MF). Blood. 2013;122(21):393. | ||
Zhang Q, Zhang Y, Diamond S, et al. The Janus kinase 2 inhibitor fedratinib inhibits thiamine uptake: a putative mechanism for the onset of Wernicke’s encephalopathy. Drug Metab Dispos. 2014;42(10):1656–1662. | ||
Mascarenhas JO, Talpaz M, Gupta V, et al. Primary analysis results from an open-label phase II study of INCB039110, a selective JAK1 inhibitor, in patients with myelofibrosis. Blood. 2014;124(21):714. | ||
Low JA, de Sauvage FJ. Clinical experience with Hedgehog pathway inhibitors. J Clin Oncol. 2010;28(36):5321–5326. | ||
Irvine DA, Copland M. Targeting hedgehog in hematologic malignancy. Blood. 2012;119(10):2196–2204. | ||
Greenbaum LE. Hedgehog signaling in biliary fibrosis. J Clin Invest. 2008;118(10):3263–3265. | ||
McGowan SE, McCoy DM. Platelet-derived growth factor-A and sonic hedgehog signaling direct lung fibroblast precursors during alveolar septal formation. Am J Physiol Lung Cell Mol Physiol. 2013;305(3):L229–L239. | ||
Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer. 2003;3(12):903–911. | ||
Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. | ||
Keller MD, Rampal RK, Shank K, et al. Improved efficacy of combination of JAK2 and hedgehog inhibitors in myelofibrosis. Blood. 2013;122(21):666. | ||
Jimeno A, Weiss GJ, Miller WH Jr, et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin Cancer Res. 2013;19(10):2766–2774. | ||
Sasaki K, Gotlib JR, Mesa RA, et al. Phase II evaluation of IPI-926, an oral Hedgehog inhibitor, in patients with myelofibrosis. Leuk Lymphoma. 2015;56(7):2092–2097. | ||
Martinelli G, Oehler VG, Papayannidis C, et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: a phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015;2(8):e339–e346. | ||
Gupta V, Koschmieder S, Harrison CN, et al. Phase 1B dose-escalation study of sonidegib (LDE225) in combination with ruxolitinib (INC424) in patients with myelofibrosis. Poster presented at: 56th ASH Annual Meeting and Exposition; December 6–9, 2014; San Francisco, CA. | ||
Wang JC, Chen C, Dumlao T, et al. Enhanced histone deacetylase enzyme activity in primary myelofibrosis. Leuk Lymphoma. 2008;49(12):2321–2327. | ||
Meadows SA, Nguyen HM, Queva C, et al. PI3Kδ inhibitor idelalisib inhibits AKT signaling in myelofibrosis patients on chronic JAK inhibitor therapy. Blood. 2013;122(21):4065. | ||
Burris HA, Patel MR, Lanasa MC, et al. Activity of TGR-1202, a novel once-daily PI3Kδ inhibitor, in patients with relapsed or refractory hematologic malignancies. J Clin Oncol. 2014;32(15 Suppl):2513. | ||
Gutierrez M, Lanasa M, Deng C, et al. A phase I dose escalation study of TGR-1202, a novel PI3K-δ inhibitor, for patients with relapsed or refractory hematologic malignancies. Blood. 2013;122(21):4373. | ||
Nanthakumar CB, Hatley RJ, Lemma S, Gauldie J, Marshall RP, Macdonald SJ. Dissecting fibrosis: therapeutic insights from the small-molecule toolbox. Nat Rev Drug Discov. 2015;14(10):693–720. | ||
Verstovsek S, Mesa RA, Foltz LM, et al. Phase 2 trial of PRM-151, an anti-fibrotic agent, in patients with myelofibrosis: stage 1 results. Blood. 2014;124(21):713. | ||
Verstovsek S, Savona MR, Mesa RA, et al. A phase 2 study to evaluate the efficacy and safety of simtuzumab in adult subjects with primary, post polycythemia vera (PV) or post essential thrombocythemia (ET) myelofibrosis. Poster presented at: 57th ASH Annual Meeting and Exposition; December 5–8, 2015; Orlando, FL. | ||
Tefferi A, Lasho TL, Begna KH, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015;373(10):908–919. | ||
Mascarenhas J, Lu M, Li T, et al. A phase I study of panobinostat (LBH589) in patients with primary myelofibrosis (PMF) and post-polycythaemia vera/essential thrombocythaemia myelofibrosis (post-PV/ET MF). Br J Haematol. 2013;161(1):68–75. | ||
DeAngelo DJ, Mesa RA, Fiskus W, et al. Phase II trial of panobinostat, an oral pan-deacetylase inhibitor in patients with primary myelofibrosis, post-essential thrombocythaemia, and post-polycythaemia vera myelofibrosis. Br J Haematol. 2013;162(3):326–335. | ||
Kiladjian JJ, Heidel FH, Vannucchi AM, et al. Efficacy, safety, and confirmation of the recommended phase 2 dose of ruxolitinib plus panobinostat in patients with intermediate or high-risk myelofibrosis. Blood. 2014;124(21):711. | ||
Andersen CL, Mortensen NB, Klausen TW, Vestergaard H, Bjerrum OW, Hasselbalch HC. A phase II study of vorinostat (MK-0683) in patients with primary myelofibrosis and post-polycythemia vera myelofibrosis. Haematologica. 2014;99(1):e5–e7. | ||
Nischal S, Bhattacharyya S, Christopeit M, et al. Methylome profiling reveals distinct alterations in phenotypic and mutational subgroups of myeloproliferative neoplasms. Cancer Res. 2013;73(3):1076–1085. | ||
Quintas-Cardama A, Tong W, Kantarjian H, et al. A phase II study of 5-azacitidine for patients with primary and post-essential thrombocythemia/polycythemia vera myelofibrosis. Leukemia. 2008;22(5):965–970. | ||
Wolfromm A, Dreyfus F, Vey N, et al. Treatment of advanced CMML by azacitidine (AZA) in a compassionate program: the GFM experience in 38 patients (pts). Poster presented at: 53rd ASH Annual Meeting and Exposition; December 10–13, 2011; San Diego, CA. | ||
Odenike OM, Godwin JE, Van Besien K, et al. Phase II trial of low dose, subcutaneous decitabine in myelofibrosis. Poster presented at: 50th ASH Annual Meeting and Exposition; December 6–9, 2008; San Francisco, CA. | ||
Harel S, Cherait A, Berthon C, et al. Outcome of patients with high risk myelodysplastic syndrome (MDS) and advanced chronic myelomonocytic leukemia (CMML) treated with decitabine after azacitidine failure. Leuk Res. 2015;39(5):501–504. | ||
Warfel NA, Kraft AS. PIM kinase (and Akt) biology and signaling in tumors. Pharmacol Ther. 2015;151:41–49. | ||
Mahadevan D, Shirahatti N. Strategies for targeting the multidrug resistance-1 (MDR1)/P-gp transporter in human malignancies. Curr Cancer Drug Targets. 2005;5(6):445–455. | ||
Keane N, Reidy M, Natoni A, Raab M, O’Dwyer M. Targeting the Pim kinases in multiple myeloma. Blood Cancer J. 2015;5(7):e325. | ||
Koblish H, Shin N, Hall L, et al. Activity of the pan-PIM kinase inhibitor INCB053914 in models of acute myelogenous leukemia. Cancer Res. 2015;75(15 Suppl):5416. | ||
Radin AI, Kim HT, Grant BW, et al. Phase II study of α2 interferon in the treatment of the chronic myeloproliferative disorders (E5487): a trial of the Eastern Cooperative Oncology Group. Cancer. 2003;98(1):100–109. | ||
Shin N, Covington M, Wynn R, et al. Characterization of INCB053914, a novel pan-PIM kinase inhibitor. Cancer Res. 2015;75(15 Suppl):5397. | ||
Swords RT, Erba HP, DeAngelo DJ, et al. Pevonedistat (MLN4924), a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol. 2015;169(4):534–543. | ||
Swords RT, Savona MR, Maris MB, et al. Pevonedistat (MLN4924), an Investigational, first-in-class NAE inhibitor, in combination with azacitidine in elderly patients with acute myeloid leukemia (AML) considered unfit for conventional chemotherapy: updated results from the phase 1 C15009 trial. Blood. 2014;124(21):2313. | ||
Kröger N, Holler E, Kobbe G, et al. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2009;114(26):5264–5270. | ||
Libutti S. Genetically engineered lymphocytes and adoptive cell therapy: cancer immunotherapy’s smart bombs. Cancer Gene Ther. 2015;22(2):63. | ||
Armand P. Immune checkpoint blockade in hematologic malignancies. Blood. 2015;125(22):3393–3400. | ||
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | ||
Smith BD, Kasamon YL, Kowalski J, et al. K562/GM-CSF immunotherapy reduces tumor burden in chronic myeloid leukemia patients with residual disease on imatinib mesylate. Clin Cancer Res. 2010;16(1):338–347. | ||
Bachireddy P, Burkhardt UE, Rajasagi M, Wu CJ. Haematological malignancies: at the forefront of immunotherapeutic innovation. Nat Rev Cancer. 2015;15(4):201–215. | ||
Ho VT, Vanneman M, Kim H, et al. Biologic activity of irradiated, autologous, GM-CSF-secreting leukemia cell vaccines early after allogeneic stem cell transplantation. Proc Natl Acad Sci U S A. 2009;106(37):15825–15830. | ||
Cazzola M, Kralovics R. From Janus kinase 2 to calreticulin: the clinically relevant genomic landscape of myeloproliferative neoplasms. Blood. 2014;123(24):3714–3719. | ||
Abdel-Wahab O, Levine RL. Genetics of the myeloproliferative neoplasms. In: Verstovsek S, Tefferi A, editors. Myeloproliferative Neoplasms. Heidelberg: Springer; 2011:39–68. | ||
Rumi E, Pietra D, Pascutto C, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood. 2014;124(7):1062–1069. | ||
Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–1869. | ||
Chang TY, Dvorak CC, Loh ML. Bedside to bench in juvenile myelomonocytic leukemia: insights into leukemogenesis from a rare pediatric leukemia. Blood. 2014;124(16):2487–2497. | ||
Kim E, Abdel-Wahab O. Focus on the epigenome in the myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. 2013;2013:538–544. | ||
Stegelmann F, Bullinger L, Schlenk R, et al. DNMT3A mutations in myeloproliferative neoplasms. Leukemia. 2011;25(7):1217–1219. | ||
Laborde R, Patnaik M, Lasho T, et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia. 2013;27(10):2100–2102. | ||
Piazza R, Valletta S, Winkelmann N, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013;45(1): 18–24. | ||
Greaves WO, Verma S, Bisrat T, et al. TP53 mutation is rare in primary myelofibrosis. Leuk Lymphoma. 2013;54(7):1552. | ||
Raza S, Viswanatha D, Frederick L, et al. TP53 mutations and polymorphisms in primary myelofibrosis. Am J Hematol. 2012;87(2):204–206. | ||
Passamonti F, Maffioli M, Caramazza D, Cazzola M. Myeloproliferative neoplasms: from JAK2 mutations discovery to JAK2 inhibitor therapies. Oncotarget. 2011;2(6):485–490. | ||
Soriano G, Heaney M. Polycythemia vera and essential thrombocythemia: new developments in biology with therapeutic implications. Curr Opin Hematol. 2013;20(2):169–175. | ||
Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol. 2011;29(5):573–582. | ||
Such E, Germing U, Malcovati L, et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood. 2013;121(15):3005–3015. | ||
Jankowska AM, Makishima H, Tiu RV, et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood. 2011;118(14):3932–3941. | ||
Makishima H, Yoshida K, Nguyen N, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45(8):942–946. | ||
Patnaik M, Padron E, LaBorde R, et al. Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. Leukemia. 2013;27(7):1504–1510. | ||
Score J, Hidalgo-Curtis C, Jones AV, et al. Inactivation of polycomb repressive complex 2 components in myeloproliferative and myelodysplastic/myeloproliferative neoplasms. Blood. 2012;119(5):1208–1213. | ||
Jankowska AM, Szpurka H, Tiu RV, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009;113(25):6403–6410. | ||
Kar SA, Jankowska A, Makishima H, et al. Spliceosomal gene mutations are frequent events in the diverse mutational spectrum of chronic myelomonocytic leukemia but largely absent in juvenile myelomonocytic leukemia. Haematologica. 2013;98(1):107–113. | ||
Loh ML, Sakai DS, Flotho C, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009;114(9): 1859–1863. | ||
Muramatsu H, Makishima H, Jankowska AM, et al. Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood. 2010;115(10):1969–1975. | ||
Sakaguchi H, Okuno Y, Muramatsu H, et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat Genet. 2013;45(8):937–941. | ||
Sugimoto Y, Muramatsu H, Makishima H, et al. Spectrum of molecular defects in juvenile myelomonocytic leukaemia includes ASXL1 mutations. Br J Haematol. 2010;150(1):83–87. | ||
Visconte V, Makishima H, Jankowska A, et al. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia. 2012;26(3):542–545. | ||
Eder C, Weissmann S, Meggendorfer M, et al. RARS-T patients harbor SF3B1 mutations in 90.2% and can be characterized by mutations in ASXL1 and other spliceosome genes in most of the remaining cases. Blood. 2013;122(21):2764. | ||
Szpurka H, Jankowska AM, Makishima H, et al. Spectrum of mutations in RARS-T patients includes TET2 and ASXL1 mutations. Leuk Res. 2010;34(8):969–973. | ||
Szpurka H, Tiu R, Murugesan G, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood. 2006;108(7):2173–2181. | ||
Tiu RV, Gondek LP, O’Keefe CL, et al. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood. 2011;117(17):4552–4560. | ||
Gotlib J, Maxson JE, George TI, Tyner JW. The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment. Blood. 2013;122(10):1707–1711. | ||
Makishima H, Jankowska A, Tiu R, et al. Novel homo-and hemizygous mutations in EZH2 in myeloid malignancies. Leukemia. 2010;24(10):1799–1804. | ||
Liu Y, Tabarroki A, Visconte V, et al. A prognostic scoring system for unclassifiable MDS and MDS/MPN. Poster presented at: 54th ASH Annual Meeting and Exposition; December 8–11, 2012; Atlanta, GA. | ||
Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42(8):722–726. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.