")
Back to Journals » Cancer Management and Research » Volume 11
The unique invasiveness of glioblastoma and possible drug targets on extracellular matrix
Authors Hatoum A , Mohammed R , Zakieh O
Received 2 September 2018
Accepted for publication 21 January 2019
Published 25 February 2019 Volume 2019:11 Pages 1843—1855
DOI https://doi.org/10.2147/CMAR.S186142
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Xueqiong Zhu
Adam Hatoum,1 Raihan Mohammed,1 Omar Zakieh2
1School of Clinical Medicine, University of Cambridge, Cambridge, UK; 2Faculty of Medicine, Imperial College London, London, UK
Abstract: Glioblastoma, or glioblastoma multiforme (GBM), is described as one of the most invasive cancer types. Although GBM is a rare disease, with a global incidence of <10 per 100,000 people, its prognosis is extremely poor. Patient survival without treatment is ~6 months, which can be extended to around 15 months with the standard treatment protocol. Given the propensity of GBM cells to show widespread local invasion, beyond the margins seen through the best current imaging techniques, tumor margins cannot be clearly defined. Recurrence is inevitable, as the highly invasive nature of GBM means complete surgical resection of the tumor is near impossible without extensive damage to healthy surrounding brain tissue. Here, we outline GBM cell invasion in the unique environment of the brain extracellular matrix (ECM), as well as a deeper exploration of the specific mechanisms upregulated in GBMs to promote the characteristic highly invasive phenotype. Among these is the secretion of proteolytic enzymes or the destruction of the ECM, as well as discussion of a novel theory of amoeboid invasion, termed the “hydrodynamic mode of invasion”. The vast heterogeneity of GBM means that there are significant redundancies in invasive pathways, which pose challenges to the development of new treatments. In the past few decades, only one major advancement has been made in GBM treatment, namely the discovery of temozolomide. Future research should look to elucidate novel strategies for the specific targeting of the invasive cells of the tumor, to reduce recurrence rates and improve patient overall survival.
Keywords: glioblastoma, glutamate, matrix metalloproteinase, uPA, hydrodynamic
Introduction
Glioblastoma, more commonly known as glioblastoma multiforme (GBM), is the most common and most aggressive type of malignant brain tumor in adults.1 Globally, it has an annual incidence of <10 per 100,000 people.2 Despite all the advances of modern medicine, it remains incurable, with an extremely poor prognosis. The Public Health England estimates the median survival as 6 months from diagnosis without treatment, close to the worst of any cancer.2 With treatment, median survival time can increase to around 15 months, although for unknown reasons, some patients can survive much longer. This treatment follows a three-pronged approach, consisting of maximal safe surgical resection, followed by concurrent temozolomide and radiotherapy, followed by temozolomide alone.3 Recurrence is inevitable, most commonly occurring within 1 cm of the surgical resection margin, owing to the highly invasive nature of GBM.
GBM is classified as a grade IV tumor (the most aggressive category according to the WHO criteria),1 and accounts for 55% of all malignant brain tumors.4 Though its cellular origins remain elusive, the astrocyte, a form of glial cell, is a principal candidate. Primary tumors represent the more aggressive de novo types, whereas the less common secondary tumors develop as a result of progression from a lower-grade glioma. The term “glioma” encompasses all brain tumors of glial cell origin, with GBM representing the most aggressive type. As such, the vast majority of glioma research studies GBM, given the dismal prognosis.
Many years of research have led to insufficient improvement in patient prognosis. In the last two decades, there has only been one major advancement, namely the discovery of temozolomide, an alkylating chemotherapy which now forms part of the standard treatment for primary GBM patients.5,6 The Stupp protocol has certainly helped increase overall survival; however, we may need to think outside the protocol to increase disease-free survival time. Currently, the only US Food and Drug Administration (FDA)-approved targeted drug for GBM treatment is the anti-VEGF antibody bevacizumab, although strong evidence of its benefits is lacking, and it may only be effective in reducing peritumoral edema. Neither increased temozolomide dosage nor bevacizumab has been shown to improve overall survival.3 There is certainly a need for new forms of GBM treatment. Consistent with most recurrence occurring within close proximity of the surgical resection margin, increasing the extent of surgical removal has been shown to increase patient survival, though this carries a greater risk of damage to eloquent or other important brain tissue. Even with the greater surgical resection, recurrence is unavoidable.
A recent landmark paper, using data from The Cancer Genome Atlas (TCGA), identified four distinct subtypes of glioblastoma: classical, proneural, neural, and mesenchymal.7 Each subtype has a unique molecular profile of protein expression and genetic mutations, with the mesenchymal subtype representing the majority of primary glioblastoma diagnoses. However, the findings of this paper have yet to translate into changes in clinical practice, and there is significant overlap between the subtypes. Traditionally, cancer research has taken a very tumor cell-centric view, typically utilizing drugs to target tumor cells. A more tumor-centric approach, focusing on the specific mechanisms utilized by invading GBM cells in the context of a complex tumor microenvironment, may yield better approaches to improve patient outcomes. This review investigates some of the mechanisms underpinning the complex interplay between tumor cells and the microenvironment to stimulate GBM cell invasion.
Glioblastoma invasion and potential cell origins
Aggressive invasiveness remains a common feature of malignant gliomas, despite high levels of tumor heterogeneity and possible divergent cells of origin.7 In vitro studies comparing central and peripheral cell samples of a GBM tumor mass showed discrepancies in levels of proliferation and invasiveness, with peripheral cells appearing markedly less proliferative but more invasive than their central counterparts.8
Although cells are inherently motile, this motility is typically restricted to specific stages of cell life such as embryonic development or, for example, immune surveillance. This process becomes dysregulated in invading cells. It is important to distinguish between migration and invasion. Migration is a normal physiological process undertaken by many cells, particularly neural stem cells (NSCs) in the brain, in which they navigate tissue boundaries. Although tissue disruption can occur in both, invasion is an unscheduled, anatomically inappropriate, and nonphysiological process.
There are clear differences between the invasiveness of other high-grade solid cancers and that of GBMs. While the former commonly intravasate into blood and lymphatic vessels, GBMs rarely intravasate into any vessels.9 Murine models have shown human glioma cells (HuN) navigating along blood vessels, in a region known as the perivascular space.10 This is reminiscent of neural progenitor cells in rodents, which also use this mechanism of vascular scaffolding, particularly in the rostral migratory stream into the olfactory bulb. While resident astrocytes do not readily migrate into central nervous system (CNS) lesion sites, NG2 cells can migrate into these areas. This is of interest since NG2 expression has been associated with an aggressive molecular signature in GBM.11
The similarities that can be drawn between these cell types improve our understanding by allowing us to draw from our knowledge of normal brain function. In addition, they provide further insight into the potential cells of origin for GBMs.
Role of glutamate signaling
GBM cells are unusual in their use of glutamate (a neurotransmitter), which acts in both an autocrine and paracrine manner as a growth factor to enhance invasion. GBM cells release significant amounts of glutamate in vitro, and glutamate is significantly increased in the microenvironment of the tumor in vivo.12 This unusual feature resembles mechanisms seen in developing neurons. While developing neurons use the NMDA receptor, GBMs utilize a specific isoform of the AMPA receptor to respond to glutamate. This particular AMPA receptor lacks the GluR2 subunit to confer Ca2+ permeability, potentially opening up the possibility of tumor-specific AMPA receptors being a therapeutic target.
Tumors with increased levels of glutamate secretion showed enhanced growth in rodent models.12 Excitotoxicity from elevated glutamate levels may act as a space-vacating agent, by killing nearby neuronal cells to facilitate invasion into surrounding parenchyma. Thus, glutamate release could also be targeted pharmacologically, although such targeting may be very toxic.
Scherer’s structures and perivascular growth
In 1938, Hans Scherer published a seminal paper, based on his work on 100 glioma patient brains, in which he described the appearance of gliomas migrating along defined brain structures. Distinct areas of growth were outlined: perineural growth, surface growth, perivascular growth, intrafascicular growth, interfibrillary growth, white and gray matter growth, and a combination of these. A notable observation was the highly adaptable nature of glioma cells, assuming the physical shape of the particular brain element they populated.9,13 The term “Scherer’s secondary structures”, coined by Scherer to describe the structures formed by gliomas around preexisting tissue, is now used to describe the structures upon which gliomas invade, as a testament to his pioneering work.
Scherer observed that a common mode of migration for GBM cells is along blood vessels. Consistent with this, in vivo studies have shown that >85% of injected tumor cells will navigate to blood vessels.14 Whether these structures are purposefully sought out or whether this process is random remains unknown, given the inherent difficulty in performing dynamic studies on invading cells. In future, refined imaging techniques may address this key question.
There is likely an array of factors involved in GBM cell attraction to blood vessels. Activation of the B2R bradykinin receptor induces cyclic changes in intracellular Ca2+ in GBM cell lines. Downstream activation of the KCa3.1 and ClC3 channels by Ca2+ is believed to mediate invasive behavior of these cells, potentially through the hydrodynamic mode of invasion. Preclinical data showing the anti-GBM effects of Icatibant, a B2R antagonist used for the treatment of hereditary angioedema, provide promise for its exploration as a potential GBM therapy.15
Other molecules have also been implicated in the induction of GBM invasion. Fibronectin, the prevalent extracellular matrix (ECM) component, is one such molecule. GBM cells exhibit increased invasion in response to stimulation with fibronectin.16 It is known to be abundant at the basement membrane, which forms part of the blood–brain barrier (BBB) around blood vessels.17 While these data are suggestive of a role in inducing perivascular invasion, it must be noted that fibronectin-induced invasion was only shown for two of the three GBM cell lines used. This illustrates the limitations of much of the research undertaken. Given the heterogeneity of GBM, this may also be indicative of multiple mechanisms existing in vivo. Identifying which of these mechanisms are active in any particular tumor remains a key challenge.
Considering this, recent studies have shown that upregulation of fibronectin, and thus the fibronectin matrix assembly (FNMA), can actually have the effect of decreasing motility and invasion.18 This is potentially by increasing cell-to-cell adhesion and cell adhesion to ECM. This process occurs via upregulation of α5 integrin and can be stimulated using dexamethasone. Interestingly, dexamethasone is already used clinically to combat cerebral edema seen in some cancers. Traditionally, dexamethasone is administered at a higher dose and tapered down, due to the number of side effects seen with steroid use. However, subsequent studies have shown that steroid doses which are 3- to 30-fold lower than those traditionally given are still capable of reducing dispersal.19 Thus, this may suggest that long-term low-dose dexamethasone should be considered for reducing recurrence in GBM patients. Also discussed is the ability of dexamethasone to decrease the ability of cells to navigate spaces in between astrocytes. This is relevant in the discussion of the hydrodynamic model of invasion discussed below.
The brain ECM provides a unique environment for invasion
Navigation of the ECM is a coordinated process, requiring a balance between cell adhesion and detachment as well as ECM destruction and remodeling. The ECM and particular cell–matrix interactions are important for determining the nature of invasion, with many of the constituents of brain ECM contributing to a pro-migratory environment for GBM cells. Although the brain ECM has a rather particular composition, studies have been undertaken to show whether alternative tissues can provide a suitable model for studying GBM invasion. Recently, an analysis of dispersal data of invading GBM cells in the brain and the mouse retina showed no significant difference.19 Therefore, mouse retina may provide a useful substrate for future study of disease progression.
The brain ECM has a unique composition; collagens, laminins, and fibronectin which are key structural ECM components in other tissues are concentrated instead in the CNS around the BBB. Likewise, certain brain ECM components are often found concentrated in NSC niches, where they are involved in regulating stemness.17 GBMs have been found to synthesize and utilize many of these molecules themselves,20 implying that they may promote stemness of GBM cells.
One such example is Tenascin C, an ECM protein which has been linked to malignancy in brain tumors.21 Tenascin C is thought to promote invasion via inhibition of RhoA leading to activation of the cell’s motility machinery. Imaging studies visualizing the distribution of Tenascin C expression showed it is produced by the tumor cells themselves rather than by resident brain cells.20 This is supported by the fact that invasive glioma cells overexpress Tenascin C in vitro. Interestingly, Tenascin C signaling is mediated by integrin-β, another ECM factor important for invasion. A recent paper showed that Tenascin C is in fact produced at the tumor margin, specifically promoting an invasive phenotype while downregulating proliferation.22
Integrins are transmembrane receptors involved in cell–cell and cell–matrix interactions. They form heterodimers consisting of 1 α and 1 β subunit (out of a possible 18 α subunits and 8 β subunits). The combination of subunits determines substrate specificity and signaling modality. In GBM, the β1 subunit is commonly upregulated, leading to enhanced invasion.23 Integrins are able to bind to extracellular ligands, such as fibronectin, predominantly via the α5 subunit,16 triggering intracellular signaling cascades which activate the motility mechanism. Mahesparan et al showed that specific integrins are highly expressed at the tumor margin.20 This may support their involvement in invasion as it adheres to the theory of pro-invasive phenotypes being most prevalent at the invasive border. However, as discussed previously, upregulation of integrins using dexamethasone may actually have a negative effect by increasing cell adhesion via FNMA to the point where invasion and migration are reduced.
Other cell-associated and cell matrix components thought to be important include SPARC, NCAM, cadherins, and connexin-43 (Cx43).21 Cx43 has recently been implicated in tumor microtubule formation, suggesting cells form a functional, therapy-resistant network capable of promoting invasion.24 However, results remain inconclusive as conflicting studies argue it in fact suppresses human GBM growth by acting as a tumor suppressor gene.25
Localization of ECM factors such as Tenascin C supports the idea that pro-invasive phenotypes are prevalent at the tumor border. The critical role of these factors could be further elucidated using novel genome editing techniques such as CRISPR/Cas9 to remove them from cell lines and study the effects on invasiveness. Furthermore, genome-wide association studies of the prevalence of GBM in individuals with mutations in any of these factors may provide further evidence of their role in GBM. These are techniques which can be applied to any identified factor thought to be important.
Role of proteases in invasion
The second key interaction with the ECM is proteolysis. An increased capacity for ECM degradation via protease secretion correlates well with increased invasion. Degradation of ECM components such as hyaluronic acid and proteoglycans, concentrated at the blood vessels, is essential to promote perivascular invasion. Quantification studies have revealed several upregulated proteases (Figure 1): uPA (a serine protease) and its receptor uPAR; cathepsin B (a cysteine protease); MMPs (zinc-dependent proteases); and ADAMs (transmembrane and secreted proteases).26,27 Many of the upregulated signaling pathways in GBM, such as EGFR, PI3K, or Wnt, appear to ultimately converge at protease expression to mediate invasiveness, particularly MMPs.
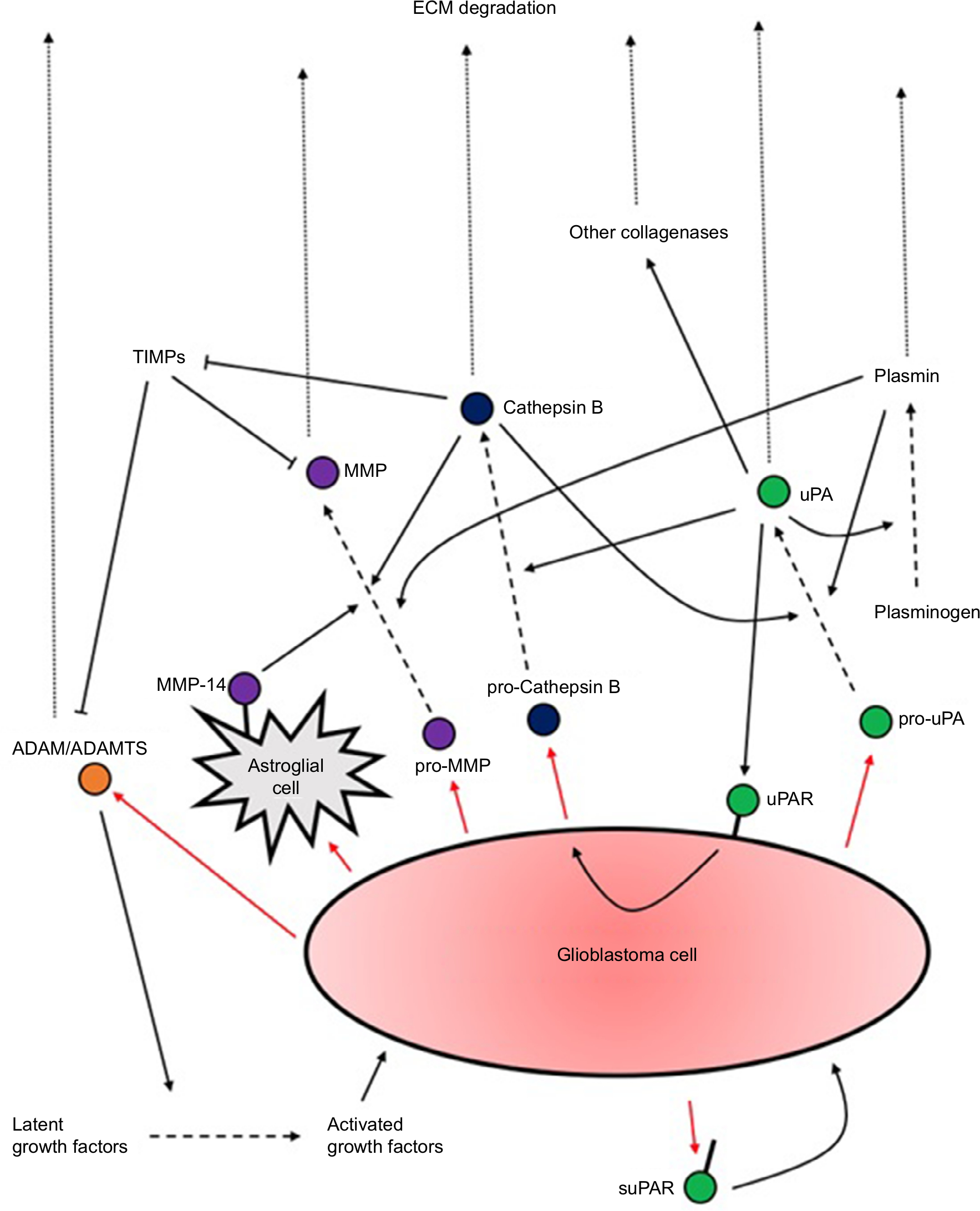 | Figure 1 A summary of proteases secretion by GBM cells and their actions upon activation. Notes: Several positive feedback loops of activation exist. What is also evident is the extensive redundancy in the activation and upregulation of the different proteases which have overlapping functions. Research should aim to identify the factors upregulated in different subtypes of GBM to allow potential tailored treatments to different patients. Abbreviations: ECM, extracellular matrix; GBM, glioblastoma multiforme; suPAR, soluble form of uPAR. |
MMPs
MMPs are thought to play a central role in invasion, owing to their ability to degrade the majority of brain ECM components. The family of zinc ion-dependent enzymes is broadly divided into six classes based on substrate specificity.28 Several MMP-knockout studies in mice have demonstrated direct evidence of the role of MMPs in tumorigenesis and cancer invasion. These mice exhibit decreased invasion of intestinal cancers, lung cancers, melanoma, and others.27 In GBM, there is a correlation between the increased MMP expression and increased invasiveness. Acting through both the degradation of the ECM and activation of pro-migratory signaling cascades, MMPs are able to promote invasive behavior. The main MMPs implicated in GBM are the gelatinases MMP-2 and MMP-9, as well as the membrane-type MT1-MMP (MMP-14).
Wang et al compared 12 samples of WHO grade IV glioblastomas to 53 lower-grade samples (grades I–III). Immunohistochemical analysis showed that MMP-2 and MMP-9 were significantly elevated in the tumor microenvironment.29 Some groups have implicated the various cells around the tumor as the main producers of MMPs, namely endothelial cells, as well as the other cells forming a significant proportion of the glioma tumor mass (infiltrating macrophages, microglia, and other infiltrating leukocytes). In contrast, Wang et al’s study showed staining was more prominent in tumor cytoplasm than in endothelial cells, suggesting the cells of the main tumor mass are the principle source of MMPs.
Wang et al also showed that MMP-2 and MMP-9 expression, as well as co-expression, increased with progressively higher tumor grade. Importantly, normal brain tissue showed no MMP-2 and MMP-9 expression, indicating that this expression is occurring de novo. Immunohistochemical data are not strictly quantitative but suggest that MMP-2 and MMP-9 are involved in the progression of GBM. A variety of qualitative studies support this now, but as highlighted by Hagemann et al,30 results are inconsistent because of the variety of different experimental approaches and cell lines used to quantify MMP expression.
The best evidence is associated with MMP-9, initially implicated for its role in angiogenesis and neovascularization. In Wang et al’s study, samples which exhibited extensive angiogenesis showed elevated MMP-9. Direct evidence for the role of MMP-9 in invasiveness comes from knockdown studies where GBM cells transfected with antisense MMP-9 exhibited significantly impaired invasion in an in vitro Matrigel assay (Figure 2).31 In this, a cell suspension is placed in the upper chamber. After an incubation period, the most invasive cells will have passed through the pores of the Matrigel matrix into the lower chamber. These cells can then be collected, stained, and quantified. This assay can be used to test the efficacy of inhibitors which aim to reduce the invasive capacity of cells. A limitation with such studies is that Matrigel does not necessarily accurately represent the brain ECM. Nonetheless, the key role of MMP-9 is supported by the finding that 69% of primary GBM samples express active MMP-9, compared with only 17% of less aggressive secondary tumor samples.32
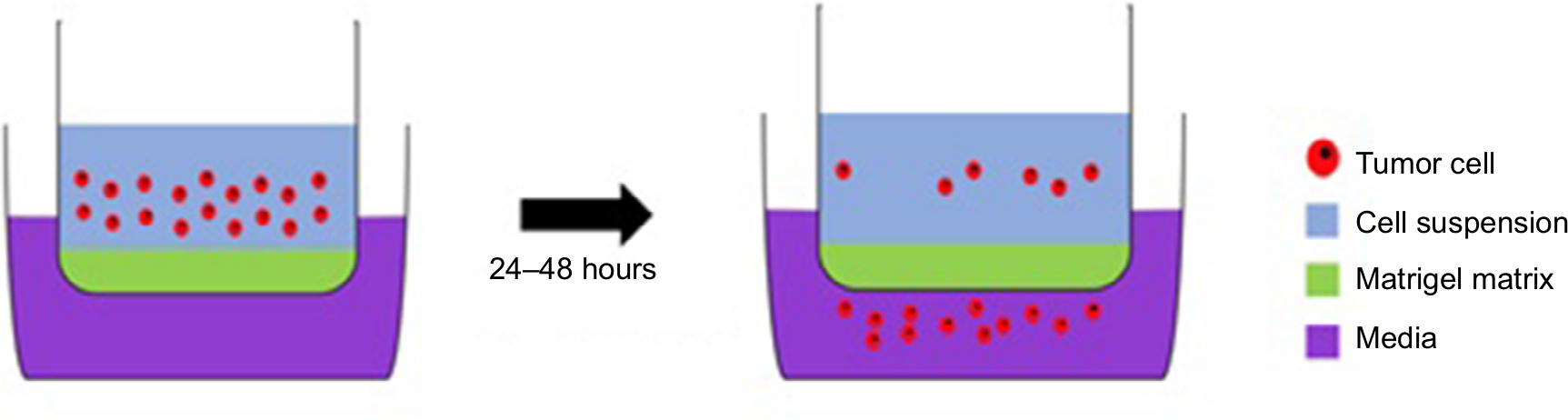 | Figure 2 A Matrigel assay: an invasion assay which allows the experimenters to assess the invasive potential of cells through the Matrigel-filled pores in a membrane separating the two compartments; this resembles invasion through the ECM. Abbreviation: ECM, extracellular matrix. |
Of the membrane-type MMPs, only MT1-MMP shows upregulation in GBM, although it differs from other MMPs discussed in that it predominantly promotes invasion via proteolytic activation of pro-MMP-2.33 Interestingly, it appears that GBM cells themselves do not express MT1-MMP, with expression rather by microglia cells which can constitute 30% of the tumor mass.34 Markovic et al showed that GBM cells induce microglial MT1-MMP expression via stimulation of microglial TLRs and intracellular signaling by MyD88 and the p38-MAPK pathway. Evidence supporting the role of MT1-MMP in invasion was shown via knockdown using shRNA. This resulted in lack of pro-MMP activation and reduced invasion. Importantly, normal brain tissue expresses little to no MT1-MMP.
Promising results in preclinical trials led to widespread excitement over MMP inhibitors (MMPIs) in the 1990s. Pharmaceutical companies accelerated their research into development of MMPIs and their testing in clinical trials. Unfortunately, targeting MMPs has shown widespread failure in all cancer types, with several potential explanations.35 As discussed, MMP-2, MMP-9, and MMP-14 are pro-invasive; however, there are other MMPs which may have beneficial antitumor roles. Broad-spectrum drugs would inhibit both these effects, culminating in a negligible improvement. Dose-escalation Phase I trials also revealed the issue of toxicity. Prolonged treatment with broad-spectrum MMPIs caused musculoskeletal pain, meaning Phase II trials were often performed with doses below the therapeutic threshold. Prinomastat, developed to specifically target MMP-2 and MMP-9 in the hope of reducing side effects, failed in Phase III trials.36 Marimastat, a broad-spectrum MMPI, showed no significant improvement of patient survival in a Phase III trial of GBM patients.37
Despite this poor performance, MMPIs remain of potential interest due to the abundance of preclinical evidence of antitumor effects. Significantly, many of the trials were in late-stage patients, and hence, early-stage trials may prove more promising. Better understanding of the side effects may also allow the administration of increased drug dosage, which could result in a greater therapeutic benefit.
uPA
uPA is a serine protease which, along with its receptor uPAR, is thought to have a role in invasion and neovascularization in gliomas. In vitro and in vivo studies of high-grade astrocytomas have shown an increased activity of uPA and uPAR which is associated with a poorer overall prognosis.27 uPA is secreted as the soluble pro-uPA form which is activated via cleavage by plasmin, a process which is more efficient if the pro-form is already bound to the receptor. Receptor-bound uPA activates plasminogen by its cleavage to plasmin, creating a positive feedback loop of activation. Plasmin, with its ability to degrade many ECM components, is able to promote invasion similar to MMPs. Furthermore, uPA is able to indirectly activate other pro-form collagenases responsible for the degradation of plasmin-resistant ECM components.
Initial studies demonstrated an association between uPA and invasiveness, as uPA/uPAR mRNA and activity were elevated in GBM cells compared to normal brain tissue and lower-grade gliomas.38,39 It was also shown that, despite heterogeneous localization within the tumor, uPA is principally expressed at the tumor margin, suggesting a role in invasion. Direct evidence was provided in the form of uPA-knockdown studies. Antisense uPA cDNA transfection resulted in a loss of invasive capacity of GBM cells, as measured by a Matrigel assay:40 42% of parental cells showed effective invasion, compared to only 12% of transfected cells. Notably, uPAR can also interact with integrins which are believed to play a role in invasion. In particular, interactions of uPAR with β1 and β3 activate distinct intracellular pathways thought to promote invasion.41 β1-uPAR binding initiates intracellular ERK signaling, one of the downstream targets of which is myosin light-chain kinase, known for its involvement in cell motility.
uPAR may also affect invasion through its association with the aggressive mesenchymal subtype.42 Using data mined from TCGA, it was found that of the top 25% of GBM samples with the highest uPAR gene signature, around 70% were of the mesenchymal subtype. uPAR expression correlated positively with expression of mesenchymal biomarkers, hence supporting its role in promoting invasiveness.
uPA binding to its receptor has other signaling capabilities which do not directly involve ECM degradation. One of these, highlighted for its role in invasion and pro-survival signaling, is the PI3K/Akt signaling pathway. An in vivo study found that stable transfection of antisense uPA cDNA resulted in a reduction of phosphorylated PI3K and Akt (the respective active forms).43 Typical anticancer therapies, such as temozolomide, induce apoptosis of cancer cells, and as such, Akt-dependent resistance to apoptosis is one of the challenges faced in treating GBM. Downregulation of uPA alone, and thus Akt, was insufficient to initiate apoptosis; however, the use of an exogenous apoptotic agent did increase the rate of apoptosis in transfected cells compared to control cells in vivo. Therefore, there is potential for the use of anti-uPA inhibitors as adjuvants alongside traditional chemo/radiotherapy.
Other studies have examined the roles of the uPAR in GBM. The soluble form of uPAR (suPAR) has been shown to act in an autocrine and paracrine manner, independently of uPA, to promote invasion and migration in the surrounding tumor cells.44 Interestingly, suPAR levels were dependent on the amount of cellular uPAR, which in turn was influenced by EGFR signaling (EGFR is typically mutated to the truncated EGFRvIII form which renders it constitutively active). Treating with exogenous EGF increased uPAR expression 30-fold, and administration of an EGF RTK inhibitor, AG1478, attenuated this response by 70%. A crucial finding was that despite decreases in uPAR expression by EGFR inhibition, cells compensated with a dramatic increase in uPA. This compensatory response could be responsible for the widespread resistance of GBM cells to anti-EGFR therapeutics. Similar mechanisms could be related to the failure of other targeted therapies.
Cathepsin B
Cathepsin B is a lysosomal cysteine protease utilized by GBM for degradation of the ECM. It is released as a pro-form which must be activated extracellularly. The protease uPA is thought to be involved in this activation, as well as influencing cathepsin B expression via uPA–uPAR interaction. Cathepsin B has a number of functions beside ECM degradation, including direct and indirect activation of pro-uPA and pro-MMPs, contributing to a complex positive feedback network utilized by invading cancers. It specifically activates MMP-2 and MMP-9 which are known to be involved in invasion, and cathepsin B expression has been associated with increased MMP activity in other cancers (eg, colon carcinoma).27 This is thought to be via inactivation of the MMPIs TIMP1 and TIMP2, leading to a shift in balance toward aberrant MMP activity.
Initial in vitro studies indicated pro-cathepsin B was secreted in high levels by GBM cells despite normal brain tissue not expressing cathepsin B.45 Immunohistochemical analysis showed cathepsin B expression patterns suggestive of a role in invasion.46 Positive staining cells appeared to congregate around the vasculature, which is representative of the pattern of invasion outlined by Scherer.9,13 The same study also showed that cathepsin B mRNA transcripts were increased threefold from normal brain to low-grade glioma, and sixfold from normal brain to GBM, with increased mRNA correlating with increased activity in GBM.
As seen with uPA, knockdown experiments provide direct evidence of the importance of cathepsin B to the invasive phenotype. Downregulation using antisense cathepsin B cDNA transfection resulted in a loss of invasive capacity both in vivo and in vitro. Among the parental GBM cells, 45% showed invasion in a Matrigel assay, compared to only 15% of transfected cells. Furthermore, there was a 90% reduction in tumor formation in athymic mice injected with transfected cells compared to those injected with untreated GBM cells.47
For many years, MMPs dominated the field of protease targeting in cancer, and therefore, research into therapeutics for other proteases was somewhat neglected. Cathepsin B and cysteine proteases have only emerged in the last 10 years as viable targets with several possible strategies: small-molecule inhibitors, specific monoclonal antibodies, and upregulation of endogenous inhibitors.48
Due to the inherent nature of protease activation discussed, the expression of one protease can have a direct or indirect impact on the expression of others. For this reason, targeting a single protease alone may prove ineffective in comparison to combination targeting of several at once. In light of this, a number of studies have demonstrated this theory by showing that simultaneous targeting results in a greater attenuation of invasion. Downregulation of a combination of cathepsin B + MMP-9 or MMP-9 + uPA via shRNA resulted in a more significant reduction in the invasive potential of GBMs.49
In addition, cathepsin B and uPAR are thought to be upregulated in response to radiotherapy treatment. Similar bicistronic shRNA targeting of these two factors was able to mitigate this response and induce apoptosis in GBM xenografts.50 This indicates the potential application of combination treatment as an adjuvant to conventional therapies.
ADAMs/ADAMTSs
Despite being less widely studied than the other proteases, ADAMs and ADAMTSs are thought to be involved in the invasive process by virtue of their ability to degrade the ECM. The disintegrin domain within their structure allows their interaction with integrins, and therefore, they can influence cell adhesion. They also function as “sheddases”, cleaving specific extracellular fragments of proteins to release soluble ectodomains. Many ECM-bound growth factors are activated by ADAMs in this way. Often, the receptors are also substrates, and thus, cell signaling can be directly activated.26 ADAMTSs are similar to ADAMs in their function but only exist in the secreted form due to the lack of a transmembrane domain. Like MMPs, they are endogenously regulated by TIMPs, although in the case of ADAMTSs, TIMPs appear to have a higher level of selectivity, with the number being inhibited by TIMP-3 alone.
Specifically, ADAMTS-4 and ADAMTS-5 have been found to be elevated in surgical glioma samples, leading to deeper research into their expression.51 Various glioma samples express these ADAMTSs when grown in situ, with GBM samples showing the greatest expression of both, implicating these factors in malignancy. Solid GBM tumor samples appear to have levels of ADAMTS-4 and ADAMTS-5 elevated far above those seen in cultured cell samples, prompting uncertainty over which cells within the tumor microenvironment are actually producing them. Immunohistochemical double-labeling has confirmed that expression is restricted to GBM and astroglial cells. The discrepancies in levels of ADAMTSs between sample sources are attributable to the nature of their regulation in vivo. Their expression is inducible by glioma-derived cytokines such as TGF-β and IL-1β, affecting ADAMTS-4 and ADAMTS-5, respectively. The role of ADAMTS-5 in invasion has been established to be via cleavage of brevican, a proteoglycan in the brain ECM which is also a target of MMP-2.26
Likewise, it has been established that ADAM-17 is able to promote an invasive phenotype in GBM cells. It acts as a primary sheddase for the activation of EGFR ligands such as TGF-α and TNF-α. Upon receptor binding, these ligands activate downstream signaling pathways, including the PI3K/Akt pathway, known to promote invasion.52 Inhibition of ADAM-17 is able to attenuate invasion (as measured in a Matrigel assay) via disruption of this pathway activation. Intriguingly, isolated GBM stem cells (GSCs), identified using the positive stem cell marker CD133, expressed higher levels of ADAM-17 than their non-stem cell counterparts.53 GSCs are known to have a relatively greater invasive potential, with ADAM-17 involved in their maintenance and invasion via the pathways described. ADAMs and ADAMTSs are yet to be explored as potential GBM therapeutics; however, their emergence as potential therapeutics in other conditions holds promise for GBM.
There is a considerable array of evidence for the upregulation of proteases in gliomas and particularly GBM. There was much interest in this area 20–30 years ago, but the failure of many developed drugs, such as the MMPI Marimastat, has discouraged further studies and trials.37 Nonetheless, protease inhibitors remain potential therapeutics and elucidating novel targets may yield more desirable results.
Hydrodynamic model of invasion
The failure of therapeutics targeting proteases has stimulated exploration of novel ideas regarding mechanism of GBM invasiveness. An emerging model of this is the hydrodynamic model of invasion, a form of amoeboid invasion which differs from the typical mesenchymal type of invasion (Figure 3). Whereas mesenchymal invasion involves the enzymatic disruption of the ECM by proteases to clear paths for the migration of invading cells, amoeboid invasion describes a more intricate method of navigating the extracellular space. This theory arose through the analysis of electron micrograph images depicting the structural changes undergone by invading GBM cells. From Scherer, it is understood the brain parenchyma consists of many tortuous extracellular spaces which invading cells must navigate. During such challenges, cells appeared to be elongated and slender wedge shaped, a characteristic feature of extreme volume reduction. Many studies have implicated K+ and Cl− channels for their role in volume-dependent cell shape changes. The use of selective K+ and Cl− channel inhibitors demonstrated the dependence of cell volume reduction on these channels.54 What was not clear was whether this process of volume reduction was necessary for, and temporally linked to, active invasion of GBM cells.
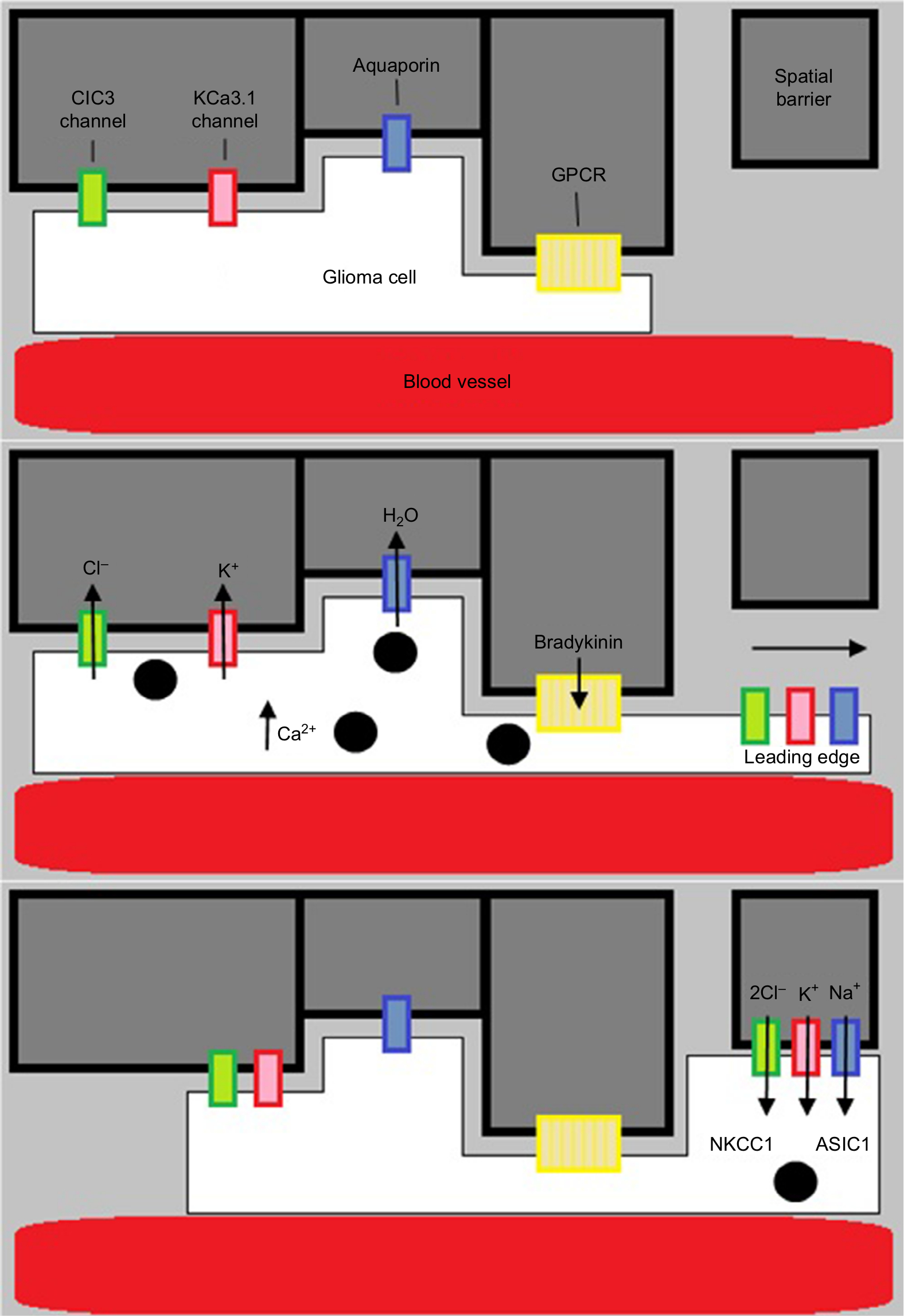 | Figure 3 Diagram showing the hydrodynamic model of invasion. Notes: Activation of GPCR (eg, by bradykinin binding to B2R) causes intracellular Ca2+ to rise, leading to Ca2+-dependent K+ and Cl− efflux through CIC3 and KCa3.1 channels. Water then follows out of the cell through aquaporins down its osmotic gradient. This decreases cellular volume, allowing the glioma cells to migrate. They then increase volume via ion influx through NKCC1 and ASIC1 channels. |
A series of in vitro and in vivo experiments using quantitative three-dimensional multiphoton and confocal time-lapse microscopy indeed confirmed that invading glioma cells reduced their volume by 30%–35% upon encountering a tortuous space. This was shown to be irrespective of the starting volume or the width of the barrier.55 These results were indeed consistent with the values obtained from hyperosmotic dehydration experiments, reflecting the maximal possible volume reduction for a cell. In this condition, the invading cells have essentially extruded all free, unbound water from the cytoplasm to allow infiltration of tortuous spaces. To confirm the involvement of Cl− and K+ channels, Watkins and Sontheimer performed the same experiments in the presence of the respective inhibitors. As predicted, transwell migration was significantly blunted in the presence of the inhibitors.
The hydrodynamic model suggests that cells repurpose Cl− and K+ currents to shift their osmotic potential.10 Invading GBM cells encounter spatial barriers, such as other cells, as they migrate. GPCR stimulation results in an increase in intracellular calcium levels, leading to ClC3 and KCa3.1/1.1 channel opening and a hypoosmotic intracellular environment. The efflux of Cl− and K+ out of the cell forces water to follow through aquaporins and a subsequent reduction in cell volume. Upon passing the barrier, GBM cells can regain volume by ion influx through the NKCC transporter or the ASIC1 ion channel. Knowledge of the specific channels involved allows their evaluation as potential therapeutic targets.
Concerning the specific channels implicated, the outward flowing chloride current, which is absent in normal astrocytes, is thought to be the main driver of this mechanism.54 Ca2+ entry into the cell and subsequent depolarization activates voltage-gated ClC3 channels.15 Despite the voltage-gated nature of the ClC family, calcium entry alone is capable of mediating activation via calcium-calmodulin-dependent phosphorylation. Chloride channel inhibitors have been explored for their potential use in reducing GBM migration, but currently no efficacious agents have been found. However, chlorotoxin, a poison extracted from the Israeli desert scorpion, has been shown to be tolerated in Phase I trials, and thus, Phase II was indicated.56 No major advancements have since been made. Incidentally, it was also discovered that chlorotoxin exclusively binds to glioma tissue, leading to its testing as a tumor tissue-delineating agent in surgery.
As discussed, normal astrocytes do not carry outward flowing chloride currents. At physiological levels and resting potential, chloride is at equilibrium; hence, there is no chloride current upon channel opening. Upregulated and overactive NKCC channels in GBMs are responsible for the elevated intracellular concentration of chloride ions that is needed to drive the volume changes. NKCC transporters use the inward Na+ gradient to accumulate chloride. The isoform NKCC1 has been shown to be constitutively expressed in gliomas, and this expression positively correlates with invasiveness.57 NKCC knockdown showed markedly reduced cell invasion when tested in vitro. Furthermore, such channels have been implicated for their role in influencing cytoskeletal dynamics to accelerate invasion.58 Hence, these observations raise NKCC as a viable therapeutic target, particularly considering the availability of an FDA-approved inhibitor, Bumetanide (a diuretic drug).
Potassium channels are responsible for maintaining electro-neutrality within the cell by balancing the anionic shift out of the cell with an equal cation shift. GBM cells express two functional isoforms of the KCa family (calcium-dependent potassium channels): KCa3.1 and a spliced KCa1.1 isoform specific to gliomas which exhibits enhanced calcium sensitivity.10
The mechanism of action of many factors involved in stimulating invasion centers around this model of volume fluctuation. The known chemoattractant bradykinin binds to the GPCR B2R, leading to an IP3R-dependent calcium increase, which activates the chloride and potassium channels.15 Interestingly, EGFR is involved in regulating NKCC activity via downstream signaling through PI3K and Akt.57 EGFR also exerts a direct role in increasing calcium concentrations via an IP3R-mediated process. This is significant as the majority of primary GBMs carry some form of activating mutation for EGFR.7
Overall, this theory provides a novel perspective on potential modes of GBM invasion. It presents several potential new therapeutic targets, namely the ion channels involved, to be explored in future research. What remains unclear is whether invasive mechanisms such as this are mutually exclusive to the secretion of proteases and destruction of the ECM within a single tumor population. The vast heterogeneity of GBM, as well as intratumor heterogeneity, suggests that multiple mechanisms may be activated simultaneously, and hence, combination therapies are further implicated.
Conclusion
Despite important advances in our knowledge of GBM, patient outcomes remain poor. Tumor recurrence is an inevitable fate, owing to the highly invasive cells which spread well beyond the boundaries of primary tumors visible through modern imaging techniques and thus persist after surgery. Consequently, there is certainly a need for novel therapeutics and new treatment strategies in the targeting of GBM. Table 1 shows a summary of the current ongoing Phase III and Phase IV studies into drug options for glioblastoma treatment. Unchecked proliferation is a prerequisite for cancer progression; however, targeting solely the proliferative mechanisms of GBMs is unlikely to improve patient outcomes compared with temozolomide. Such treatments fail to address the unique property of GBM to extensively invade into surrounding brain tissue.
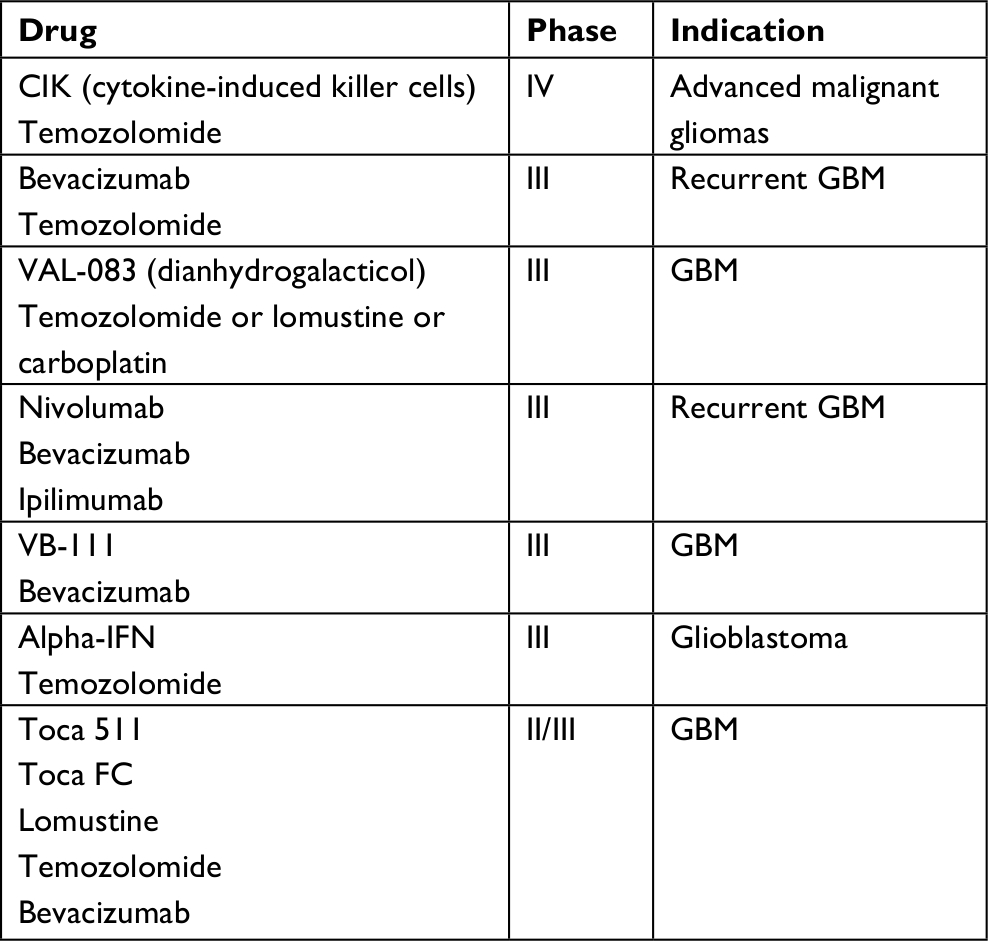 | Table 1 A summary of the current ongoing Phase III and Phase IV trials Abbreviation: GBM, glioblastoma multiforme. |
Common problems with other areas of cancer research are also relevant in GBM research. Primary cell cultures used for xenografts are grown in vitro and effectively consist of a monoculture which is not representative of the heterogenous nature of tumor cell populations. On top of this, cell culture conditions which do not resemble stem cell niches are unable to promote the growth of tumor stem cells appropriately, despite these likely playing a major role in tumor progression and invasiveness. Furthermore, the physiological constraints and tissue structure and composition faced by the tumor cell in humans and mice are very different.
Although it is important to develop new treatments alongside basic research, a deeper knowledge of GBM pathogenesis is likely the most effective way to create paradigmatic shifts in treatment strategies. Lessons must be learned from the previous failings of clinical trials, such as those of MMPIs. Deep tumor analysis, including DNA and RNA sequencing of patient tumor samples, may allow careful selection of participants for future clinical trials, to ensure the targeted pathways are indeed upregulated in their tumors. These techniques may also allow the development of tailored treatments to patients. Future research should look to elucidate these specific treatments for patients with different GBM subtypes which utilize distinct invasive mechanisms.
The significant heterogeneity of GBMs is becoming increasingly appreciated and understood and is likely compounding the difficulties encountered when developing novel therapies. Many of the invasive mechanisms described exhibit extensive redundancy. Redundancies in these pathways may also explain the exceptionally high resistance of GBMs to monotherapies. Therefore, there is indeed an indication for future research into novel approaches exploring the potential benefits of combination therapies. For example, simultaneous targeting of two or more secreted proteases may reduce the negating effects of redundancies in their functions. Targeting several known invasive mechanisms at once may result in significant improvements in patient overall survival rates.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005;109(1):93–108. | ||
Brodbelt A, Greenberg D, Winters T, et al. Glioblastoma in England: 2007–2011. Eur J Cancer. 2015;51(4):533–542. | ||
Weller M, Wick W, Aldape K, et al. Glioma. Nat Rev Dis Primers. 2015;39:15017. | ||
Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. | ||
Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. | ||
Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. | ||
Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. | ||
Molina JR, Hayashi Y, Stephens C, Georgescu M-M. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia. 2010;12(6):453–IN5. | ||
Scherer HJ. Structural development in gliomas. Am J Cancer. 1938;34(3):333–351. | ||
Cuddapah VA, Robel S, Watkins S, Sontheimer H. A neurocentric perspective on glioma invasion. Nat Rev Neurosci. 2014;15(7):455–465. | ||
Al-Mayhani MTF, Grenfell R, Narita M, et al. NG2 expression in glioblastoma identifies an actively proliferating population with an aggressive molecular signature. Neuro Oncol. 2011;13(8):830–845. | ||
Venkatesh H, Monje M. Neuronal activity in ontogeny and oncology. Trends Cancer. 2017;3(2):89–112. | ||
Scherer HJ. A critical review: the pathology of cerebral gliomas. J Neurol Neurosurg Psychiatry. 1940;3(2):147–177. | ||
Montana V, Sontheimer H. Bradykinin promotes the chemotactic invasion of primary brain tumors. J Neurosci. 2011;31(13):4858–4867. | ||
Cuddapah VA, Turner KL, Seifert S, Sontheimer H. Bradykinin-induced chemotaxis of human gliomas requires the activation of KCa3.1 and ClC-3. J Neurosci. 2013;33(4):1427–1440. | ||
Ohnishi T, Arita N, Hiraga S, et al. Fibronectin-mediated cell migration promotes glioma cell invasion through chemokinetic activity. Clin Exp Metastasis. 1997;15(5):538–546. | ||
Quail DF, Joyce JA. The microenvironmental landscape of brain tumors. Cancer Cell. 2017;31(3):326–341. | ||
Shannon S, Vaca C, Jia D, et al. Dexamethasone-mediated activation of fibronectin matrix assembly reduces dispersal of primary human glioblastoma cells. Plos One. 2015;10(8):e0135951. | ||
Meleis AM, Mahtabfar A, Danish S, Foty RA. Dexamethasone-mediated inhibition of glioblastoma neurosphere dispersal in an ex vivo organotypic neural assay. Plos One. 2017;12(10):e0186483. | ||
Mahesparan R, Read TA, Lund-Johansen M, Skaftnesmo KO, Bjerkvig R, Engebraaten O. Expression of extracellular matrix components in a highly infiltrative in vivo glioma model. Acta Neuropathol. 2003;105(1):49–57. | ||
Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004;70(2):217–228. | ||
Xia S, Lal B, Tung B, Wang S, Goodwin CR, Laterra J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro Oncol. 2016;18(4):507–517. | ||
Kwiatkowska a, Symons M. Signaling determinants of glioma cell invasion. Glioma Signaling. 2013:121–141. | ||
Osswald M, Jung E, Sahm F, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;303(7580):93–98. | ||
Huang R, Lin Y, Wang CC, et al. Connexin 43 suppresses human glioblastoma cell growth by down-regulation of monocyte chemotactic protein 1, as discovered using protein array technology. Cancer Res. 2002;62(10):2806–2812. | ||
Mentlein R, Hattermann K, Held-Feindt J. Lost in disruption: role of proteases in glioma invasion and progression. Biochim Biophys Acta. 2012;1825(2):178–185. | ||
Rao JS. Molecular mechanisms of glioma invasiveness: the role of proteases. Nat Rev Cancer. 2003;3(7):489–501. | ||
Nakada M, Okada Y, Yamashita J. The role of matrix metalloproteinases in glioma invasion. Front Biosci. 2003;8(5):e261–e269. | ||
Wang M, Wang T, Liu S, Yoshida D, Teramoto A. The expression of matrix metalloproteinase-2 and-9 in human gliomas of different pathological grades. Brain Tumor Pathol. 2003;20(2):65–72. | ||
Hagemann C, Anacker J, Ernestus R-I, Vince GH. A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J Clin Oncol. 2012;3(5):67–79. | ||
Kondraganti S, Mohanam S, Chintala SK, et al. Selective suppression of matrix metalloproteinase-9 in human glioblastoma cells by antisense gene transfer impairs glioblastoma cell invasion. Cancer Res. 2000;60(24):6851–6855. | ||
Choe G, Park JK, Jouben-Steele L, et al. Active matrix metalloproteinase 9 expression is associated with primary glioblastoma subtype. Clin Cancer Res. 2002;8(9):2894–2901. | ||
Fillmore HL, VanMeter TE, Broaddus WC. Membrane-type matrix metalloproteinases (MT-MMPs): expression and function during glioma invasion. J Neurooncol. 2001;53(2):187–202. | ||
Markovic DS, Vinnakota K, Chirasani S, et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A. 2009;106(30):12530–12535. | ||
Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–2392. | ||
Bissett D, O’Byrne KJ, von Pawel J, et al. Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J Clin Oncol. 2005;23(4):842–849. | ||
Levin VA, Phuphanich S, Alfred Yung WK, et al. Randomized, double-blind, placebo-controlled trial of marimastat in glioblastoma multiforme patients following surgery and irradiation. J Neurooncol. 2006;78(3):295–302. | ||
Yamamoto M, Sawaya R, Mohanam S, et al. Expression and localization of urokinase-type plasminogen activator in human astrocytomas in vivo. Cancer Res. 1994;54(14):3656–3661. | ||
Yamamoto M, Sawaya R, Mohanam S, et al. Expression and localization of urokinase-type plasminogen activator receptor in human gliomas. Cancer Res. 1994;54(18):5016–5020. | ||
Mohanam S, Jasti SL, Kondraganti SR, et al. Stable transfection of urokinase-type plasminogen activator antisense construct modulates invasion of human glioblastoma cells. Clin Cancer Res. 2001;7(8):2519–2526. | ||
Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11(1):23–36. | ||
Gilder AS, Natali L, van Dyk DM, et al. The urokinase receptor induces a mesenchymal gene expression signature in glioblastoma cells and promotes tumor cell survival in neurospheres. Sci Rep. 2018;8(1):2982. | ||
Chandrasekar N, Mohanam S, Gujrati M, Olivero WC, Dinh DH, Rao JS. Downregulation of uPA inhibits migration and PI3K/Akt signaling in glioblastoma cells. Oncogene. 2003;22(3):392–400. | ||
Gilder AS, Jones KA, Hu J, et al. Soluble urokinase receptor is released selectively by glioblastoma cells that express epidermal growth factor receptor variant III and promotes tumor cell migration and invasion. J Biol Chem. 2015;290(24):14798–14809. | ||
McCormick D. Secretion of cathepsin B by human gliomas in vitro. Neuropathol Appl Neurobiol. 1993;19(2):146–151. | ||
Rempel SA, Rosenblum ML, Mikkelsen T, et al. Cathepsin B expression and localization in glioma progression and invasion. Cancer Res. 1994;54(23):6027–6031. | ||
Mohanam S, Jasti SL, Kondraganti SR, et al. Down-regulation of cathepsin B expression impairs the invasive and tumorigenic potential of human glioblastoma cells. Oncogene. 2001;20(28):3665–3673. | ||
Palermo C, Joyce JA. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharmacol Sci. 2008;29(1):22–28. | ||
Veeravalli KK, Chetty C, Ponnala S, et al. MMP-9, uPAR and cathepsin B silencing downregulate integrins in human glioma xenograft cells in vitro and in vivo in nude mice. PLoS One. 2010;5(7):e11583. | ||
Malla RR, Gopinath S, Alapati K, Gorantla B, Gondi CS, Rao JS. uPAR and cathepsin B inhibition enhanced radiation-induced apoptosis in gliomainitiating cells. Neuro Oncol. 2012;14(6):745–760. | ||
Held-Feindt J, Paredes EB, Blömer U, et al. Matrix-degrading proteases ADAMTS4 and ADAMTS5 (disintegrins and metalloproteinases with thrombospondin motifs 4 and 5) are expressed in human glioblastomas. Int J Cancer. 2006;118(1):55–61. | ||
Zheng X, Jiang F, Katakowski M, Lu Y, Chopp M. ADAM17 promotes glioma cell malignant phenotype. Mol Carcinog. 2012;51(2):150–164. | ||
Chen X, Chen L, Chen J, et al. ADAM17 promotes U87 glioblastoma stem cell migration and invasion. Brain Res. 2013;1538:151–158. | ||
Soroceanu L, Manning TJ, Sontheimer H. Modulation of glioma cell migration and invasion using Cl− and K+ ion channel blockers. J Neurosci. 1999;19(14):5942–5954. | ||
Watkins S, Sontheimer H. Hydrodynamic cellular volume changes enable glioma cell invasion. J Neurosci. 2011;31(47):17250–17259. | ||
Mamelak AN, Rosenfeld S, Bucholz R, et al. Phase I single-dose study of intracavitary-administered iodine-131-TM-601 in adults with recurrent high-grade glioma. J Clin Oncol. 2006;24(22):3644–3650. | ||
Garzon-Muvdi T, Schiapparelli P, ap Rhys C, et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 2012;10(5):e1001320. | ||
Schiapparelli P, Guerrero-Cazares H, Magaña-Maldonado R, et al. NKCC1 regulates migration ability of glioblastoma cells by modulation of actin dynamics and interacting with cofilin. EBioMedicine. 2017;21:94–103. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.