")
Back to Journals » OncoTargets and Therapy » Volume 12
The tumor suppressive roles of ARHGAP25 in lung cancer cells
Received 4 March 2019
Accepted for publication 19 June 2019
Published 19 August 2019 Volume 2019:12 Pages 6699—6710
DOI https://doi.org/10.2147/OTT.S207540
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Ke Xu,1 Bin Liu,2 Yegang Ma1
1Department of Thoracic Surgery, Cancer Hospital of China Medical University, Liaoning Cancer Hospital and Institute, Shenyang, Liaoning Province, People’s Republic of China; 2Department of Medical Oncology, Cancer Hospital of China Medical University, Liaoning Cancer Hospital and Institute, Shenyang, Liaoning Province, People’s Republic of China
Correspondence: Yegang Ma
Cancer Hospital of China Medical University, No. 44 Xiaoheyan Road, Dadong District, Shenyang 110042, People’s Republic of China
Tel +86 243 191 6287
Email [email protected]
Aim: Several Rho GTPase-activating proteins (Rho GAPs) have been proved to serve as tumor suppressors in diverse human cancers. Among them, ARHGAP25 has also been found to be associated with hematopoietic cells and regulate phagocytosis. Little is known about the role of ARHGAP25 in lung cancer cells.
Methods: Quantitative real-time PCR and Western blot were used to measure the expression levels of ARHGAP25. The ability of cell growth and mobility were measured by cell proliferation and Transwell assays. Chromatin immunoprecipitation and luciferase assay were conducted to identify the transcriptional regulation.
Results: Lung cancer tissues had much lower expression level of ARHGAP25 compared to non-cancerous specimens as well as for lung cancer cells. Cell growth and mobility were strongly reduced when ARHGAP25 was overexpressed. Further, significantly negative correlation between ARHGAP25 expression and Wnt signaling pathway was observed. Overexpression of ARHGAP25 reduced the expression of β-catenin and matrix metalloproteinase-7. ARHGAP25 knockdown effect of increased abilities of cell proliferation, migration and invasion could be reversed by adding XAV939 inhibitor. The promoter site of ARHGAP25 could be bound with HOXA4. HOXA4 could regulate the transcriptional activity of ARHGAP25.
Conclusions: This study suggests that ARHGAP25 may inhibit lung cancer cell growth, migration and invasion through Wnt/β-catenin signaling pathway and its transcriptional activity can be regulated by HOXA4.
Keywords: ARHAGP25, Wnt signaling pathway, HOXA4, lung cancer, biological function
Introduction
Lung cancer is still the leading cause of cancer-related mortality worldwide, with >1.8 million newly diagnosed cases and over 1.5 million deaths annually.1,2 Non-small cell lung cancer (NSCLC) is one of the common types of lung cancer which accounts for almost 85% of the cases.3 The 5-year survival is still low mainly because initial symptoms were not prominent and highly efficient therapies were lacked in lung cancer patients with local advance or metastasis.4 Besides traditional therapies including surgery and neoadjuvant chemotherapy, targeted therapy and immunotherapy are two critical treatment strategies for lung cancer. Thus, exploring and clarifying the mechanism of lung carcinogenesis is urgent and necessary.
Rho GTPases belong to Ras superfamily which function as GTP-binding proteins. Rho GTPases are involved with multiple key biological functions such as cell cycle progression, cytoskeletal organization, and malignant transformation.5 Rho GTPases have been found to be dysregulated in tumors and play critical roles in tumor development and progression.6 Rho GTPase-activating protein 25 (ARHGAP25) belonging to Rho GTPase-activating proteins (RhoGAPs) can facilitate the intrinsic hydrolysis of GTP and negatively regulate the activity of Rho GTPases. Thuault et al first suggested that ARHGAP25 was associated with cancer.7 ARHGAP25 has also been found to be associated with hematopoietic cells and regulate phagocytosis.8,9 However, the biological function of ARHGAP25 in lung cancer has not been clarified.
Embryonic and mature lung formation has been reported to be associated with the Wnt/β-catenin signaling pathway which is relatively conservative.10 Increasing evidence indicates the importance of Wnt/β-catenin signaling pathway that is associated with proliferation, metastasis and prognosis of diverse cancers including lung cancer.11,12 Increasing investigations suggested that down-regulated β-catenin contributed to shorter survival in NSCLC implying the importance of β-catenin.13,14 Key molecules targeted by Wnt/β-catenin signaling pathway are identified including matrix metalloproteinase-2 (MMP-2), MMP-7/9, c-Myc, Axin-2.15–17 Among them, MMP-7 is found to be involved with tumor metastasis, invasion.18,19
Transcription factors encoded by the Homeobox (HOX) gene family could regulate cell differentiation and embryonic formation.20 Previous study indicated that HOXA4 was involved with the patterning of mouse lung.21 Basu et al found that HOXA4 was lowly expressed, involved with the ability of lung cell growth and mobility and suppressed the activity of Wnt/β-catenin signaling pathway.18 This indicate that HOXA4 may serve as an important transcription factor in lung cancer.
Our study firstly identified down-regulation of ARHGAP25 in lung cancer tissues and cell lines, and then found that the biological functions of ARHGAP25 including cell growth, migration and invasion inhibition were mediated by the Wnt/β-catenin signaling pathway. Further experiments suggested that HOXA4 could regulate the transcription of ARHGAP25.
Materials and methods
Clinical specimens
This research was approved by the Ethics Committee of Liaoning Cancer Hospital and Institute (Shenyang, China). Written informed consent was obtained from all participants. Research in this study was performed according to the provisions of the Declaration of Helsinki of 1975. Lung cancer and adjacent noncancerous tissues were obtained from 30 patients who received surgery at Departments of Medical Oncology, Liaoning Cancer Hospital and Institute (Shenyang, China). We collected clinical data from medical records including age, sex, tumor size, smoking, and lymph node metastasis status (Table 1). All tissues were frozen in liquid nitrogen after surgical resection and stored at −80°C for further analysis.
 |
Table 1 Correlation of ARHGAP25 expression with patients’ features |
Data collection and signaling pathway analysis
We downloaded level 3 lung cancer cohort (LUAD) in the Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/). A total of 488 lung cancer specimen were included lung adenocarcinoma from TCGA dataset. We used two sample’s t-test to test the difference of ARHGAP25 expression in two groups. We used gene set enrichment analysis (GSEA) to screen out dysregulated gene sets involved with the expression of ARHGAP25. GSEA was implemented within the JAVA program (http://www.broadinstitute.org/gsea, version 3.0).22,23
Cell culture
Lung cell lines were bought from Cell Bank of Chinese Academy of Sciences (Shanghai, China). NCI-H1299, NCI-H1975, NCI-H446 cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) with 10% FBS while DMEM with 10% FBS was used culture for A549 and 293T cells. The temperature of cell incubator was set at 37°C with 5% CO2.
Total RNA extraction and qRT-PCR analysis
Total RNA was extracted using TRIzol reagent (Takara, Shanghai, China), and the PrimeScriptP RT Reagent Kit (Perfect Real Time; Takara) was used for reverse transcription. Applied Biosystems 7500 Fast Real-Time PCR System (Applied Biosystems) was applied to perform quantitative PCR. Relative expression of target genes was normalized by GAPDH. All the PCR primers involved were listed as below: ARHGAP25, 5ʹ-GAAGCAGCTCTCCATCCTTC-3ʹ and 5ʹ-CCAGGTTGTCCACACTCATC-3ʹ; β-catenin, 5ʹ-CGACACCAAGAAGCAGAGATG-3ʹ and 5ʹ-GGGACAAAGGGCAAGATTTCG-3ʹ; HOXA4 (NM_002141.4), 5ʹ-TGGATACCTGAGAAGGACTAC-3ʹ and 5ʹ-GGATGAGCTGCCTTAATG-3ʹ; MMP-7 (NM_002423.4), 5ʹ-TCTTTGGCCTACCTATAACTG-3ʹ and 5ʹ-CACTGTAATATGCGGTAAGTC-3ʹ; GAPDH (NM_001256799.2), 5ʹ-AGGCTGTTGTCATACTTC-3ʹ and 5ʹ-AATCCCATCACCATCTTC-3ʹ.
Western blot
Protein extraction buffer (Beyotime, Shanghai, China) was used to extract total protein. Proteins were quantified using BCA Protein Assay Kit (Thermo Fisher Scientific, West Palm Beach, FL, USA); 50 μg proteins per well were added to 10% or 12% SDS polyacrylamide gels and were transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA) after electrophoresis. The membranes were incubated for blocking in 5% non-fat milk at 4°C and then incubated with primary antibodies overnight. The corresponding proteins were evaluated by using the enhanced chemiluminescent substrate (Millipore, Billerica, MA, USA). Antibodies against ARHGAP25 (ab192020), MMP-7 (ab5706) and HOXA4 (ab131049), and β-catenin (#8480) and GAPDH (#5174) were used to conduct Western blot analysis.
Cells transfection
The coding regions of ARHGAP25 and HOXA4 were amplified using corresponding primers as follows and integrated with lentivirus vector: ARHGAP25: 5ʹ-CGGAATTCATGTCCCTAAAATTGCCAAGG -3ʹ (EcoRI); 5ʹ-CGGGATCCTTAAGCCTCGGTCTTG-3ʹ (BamHI). HOXA4: forward 5ʹ- CCCAAGCTTATGACCATGAGCTCGTTTTTG-3ʹ (EcoRI); 5ʹ-CGGAATTCTTATATGGAGGAGGGAACG-3ʹ (BamHI). Sequences underlined represented the restriction sites. ShRNAs targeting ARHGAP25 were constructed and integrated with pLKO.1 plasmids. Sites for targeting were listed as below: shARHGAP25#1 (sites 1255–1273), CCATCCTTCCTCGTGACAA; shARHGAP25#2 (sites 1738–1756), GGACTCAAACACTCCCTAA; shARHGAP25#3 (sites 1895–1913), CCAACTTTCTGACTCCCAA. Lipofectamine 2000 was used to facilitate the transfection of these constructs together with lentivirus plasmids. We collected lentiviruses after transfection for 48–72 hrs and then used them to transfect cells.
Tumor cell growth
Cell growth rate was evaluated using Cell Counting Kit-8 (CCK-8). Briefly, a density of 1500 cells/well was cultured in 96-well plates using 100 µL medium where these cells were transfected with corresponding lentivirus or vectors. ARHGAP25 shRNAs and XAV939 (Selleck Chemicals, Houston, TX, USA) were transfected into NCI-H1299 cells. Cell culture medium was replaced with CCK-8 solution at 0, 24, 48 and 72 hrs, respectively, and cultured for 1 hr at 37°C. A microplate reader (Bio-Rad) was used to evaluate OD at 450 nm.
Cell migration and invasion assay
Cells were collected after transfected for 48 hrs and then 2×105 cells were seeded into the upper chamber of the transwell chambers. The lower chamber was filled with 600 μL cultured medium attenuated by 20% FBS. Cells which have passed through the membranes were fixed after incubation at 37°C with 5% CO2, followed by crystal violet staining and cell counting.
Transwell invasion was conducted as transwell migration while the membranes were appended with Matrigel (BD Bioscience, Franklin Lakes, NJ, USA).
Luciferase assay
To evaluate transfection efficiency, designated combinations of pGL3-HOXA4 wild type (WT), pGL3-HOXA4 overexpression, pGL3 with siRNA and plasmids were transiently transfected in triplicate with lipo 3000 transfection reagent (Invitrogen) according to the manufacturer’s protocol. Luciferase activity was evaluated after transfection for 24 hrs using the Dual-Luciferase Reporter Assay System (Promega, Fitchburg, WI, USA). Luciferase activity of the construct was divided by the corresponding renilla luciferase activity to normalize the variation of transfection efficiency.
Chromatin immunoprecipitation (ChIP)
A cChIP assay kit (Upstate, Millipore) was used to conduct ChIP assay in NCI-H1299 cells based on the manufacturer’s instruction. ChIP analysis was performed as previously mentioned.24 Immunoprecipitated DNA was amplified with primers targeting ARHGAP25 promoter: 5ʹ-AGTTTGAACGCTGACAGGAAGG-3ʹ (forward), 5ʹ-GGGAGGGTTTTGATGTATTTGG-3ʹ (reverse) and nonspecific rabbit IgG was applied as negative control. Agarose gel electrophoresis (2%) was used to resolve PCR products followed by PCR which was performed in duplicate in a reaction system (10 mL) containing eluted immunoprecipitated DNA (3 mL). The amount of DNA combining with the corresponding antibody was evaluated by comparing with the total input DNA in each assay.
Statistical analysis
All statistical analyses were carried out using GraphPad Prism (version 6.0; GraphPad Software, Inc., La Jolla, CA, USA). Statistical test of mRNA expression of ARHGAP25 in our own cohort was performed with Student’s t-test. Correlation between ARHGAP25 expression and clinical parameters was tested by chi-squared test or Fisher’s exact test. All the in vitro experiments were repeated at least three times. ANOVA test was used to examine the differences in vitro experiments. Differences were considered significant when P-value was <0.05.
Results
ARHGAP25 is down-regulated in lung cancer tissues and cell lines
We initially analyzed ARHGAP25 expression on lung cancer cohort derived from TCGA. The expression of ARHGAP30 was significantly down-regulated in cancer tissues compared with adjacent normal lung tissues (Figure 1A, P<0.0001). For confirmation, we collected 30 paired samples to measure the expression of ARHGAP25. Consistently, tumor tissues had lower expression of ARHGAP25 than paired normal tissues (Figure 1B, P<0.001). We then divided these 30 cases into low- or high-expression group using the median mRNA level as the cut-off value. ARHGAP25 expression was correlated with tumor size and lymph node metastasis (Table 1). In addition, we evaluated the protein level of ARHGAP25 on seven paired samples and found that ARHGAP25 was also decreased at protein level in tumor tissues (Figure 1C, Figure S1). Immunohistochemical assay also indicated that ARHGAP25 level was much lower in lung cancer tissues compared to paired normal tissues (Figure S2).
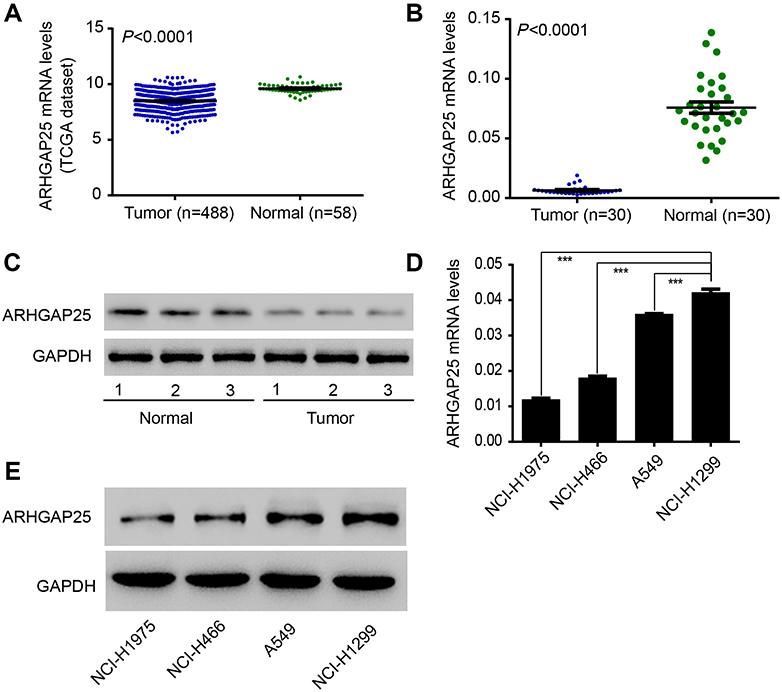 |
Figure 1 The expression of ARHGAP25 in lung cancer was measured. The difference of ARGFAP25 expression in cancerous and non-cancerous samples was compared in two cohorts (A) n=546; (B) n=60, respectively. (C) Western blot was conducted between paired cancerous and non-cancerous samples (n=6). (D and E) Western blot and qRT-PCR were conducted in four lung cancer cell lines. ***P<0.001. |
Also, the expression of ARHGAP25 was evaluated in four lung cancer cell lines using qRT-PCR and Western blot (Figure 1D and E). Among these cell lines, NCI-H1299 cell showed higher expression levels while NCI-H1975 and NCI-H446 cells displayed lower expression levels. Therefore, these three cell lines were used for further experiments. Altogether, these results implied that the expression of ARHGAP25 was remarkably reduced in lung cancer.
ARHGAP25 inhibited tumor growth
To explore roles of ARHGAP25, we initially overexpressed ARHGAP25 by transfecting lentivirus into two cell lines (NCI-H1975 and NCI-H446). Our results suggested that ARHGAP25 was significantly increased at both mRNA and protein levels in these two cell lines, while vector group had no influence on ARHGAP25 expression (Figure 2A and B). Also, after transfected for 48 and 72 hrs, cell growth rate was much lower in ARHGAP25-overexpressed group than that in vector group while the difference between vector group and WT group was not significant (Figure 2C). These results were consistent with that in NCI-H446 cells (Figure 2D). These results implied that cell growth could be inhibited by ARHGAP25 in lung cancer.
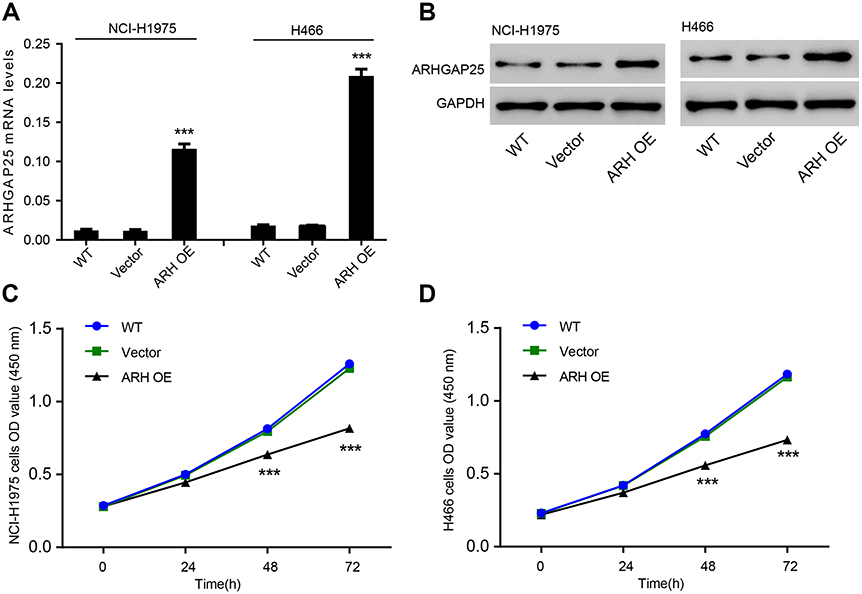 |
Figure 2 ARHGAP25 inhibited cell growth. ARHGAP25 expression was evaluated by using RT-PCR (A) and Western blot (B) in NCI-H446 and NCI-H1975 cells in which ARHGAP25 was overexpressed. Wild type group was a control group. Cell growth rate was compared in NCI-H446 and NCI-H1975 cells, respectively (C and D). ***P<0.001. |
ARHGAP25 inhibited tumor migration and invasion
We performed transwell assays to test the influence of ARHGAP25 on cell mobility. Our results indicated that the migration and invasive capabilities of ARHGAP25 overexpressed group were significantly reduced compared with vector group in both NCI-H1975 (Figure 3A) and NCI-H446 (Figure 3B). Our finding indicated that ARHGAP25 might repress the mobility of lung cancer.
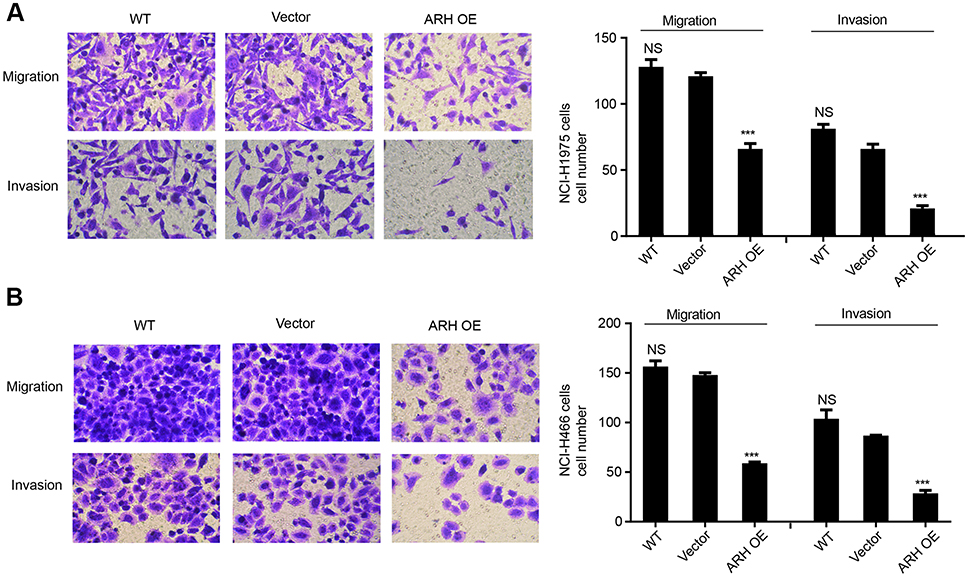 |
Figure 3 ARHFAP25 inhibited lung cancer cell, migration and invasion. Transwell invasion assay was performed in NCI-H1299 (A) and NCI-H446 (B) cells in which ARHGAP25 was overexpressed. Wild type group was used as control group. ***P<0.001. |
ARHGAP25 inhibited the Wnt/β-catenin signaling pathway
By using GSEA in lung cancer cohort from TCGA, we observed negative correlation between ARHGAP25 expression and the Wnt/β-catenin pathway (Figure 4A). To further our analysis, the expression of β-catenin and MMP-7 which were downstream effectors of Wnt/β-catenin pathway was further evaluated. We found that the activity of the Wnt/β-catenin signaling pathway could be inhibited by ARHGAP25 (Figure 4B–D).
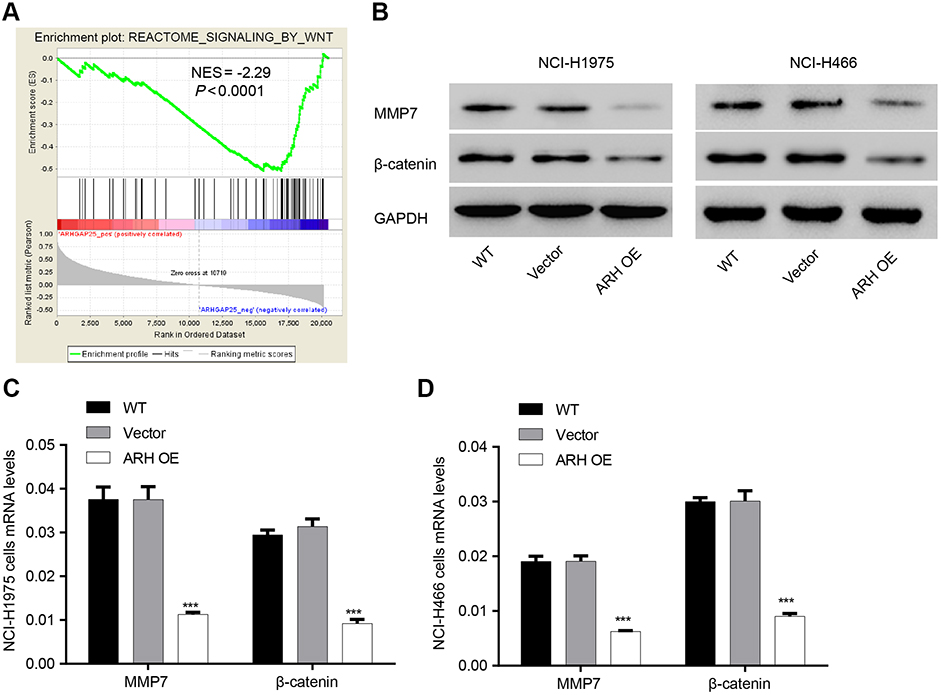 |
Figure 4 Negative correlation between ARHGAP25 and the Wnt/β-catenin signaling pathway. (A) Gene set enrichment analysis (GSEA) analysis was conducted to display a series of genes negatively associated with Wnt signaling pathway (NES=−2.29, P<0.0001). (B) Western blot was conducted in NCI-H1975 and NCI-H446 cells in which ARHGAP25 was overexpressed. (C and D) qRT-PCR was conducted in NCI-H1975 (C) and NCI-H446 (D) in which ARHGAP25 was overexpressed. Wild type group was used as control group. ***P<0.001. |
Role of HOXA4 in the regulation of ARHGAP25 expression
Transcription factors encoded by HOX family are involved with cell fate and embryo formation.20 HOXA4 has been proved to be involved with reduced cell growth and motility in lung cancer which means HOXA4 may function as an important transcription factor in lung cancer progression.25 Therefore, we examined whether HOXA4 increased expression of ARHGAP25 by regulating the transcriptional activity of ARHGAP25. By performing qRT-PCR and Western blotting assays, we found that overexpression of HOXA4 could significantly up-regulate the expression levels of ARHGAP25 at protein and mRNA levels, respectively (Figure 5A and B). Further, knockdown of HOXA4 dramatically decreased the transcriptional activity of the ARHGAP25 promoter in a luciferase assay, while HOXA4 overexpression significantly up-regulated the transcriptional activity of the ARHGAP25 promoter (Figure 5C). These results implied that HOXA4 might up-regulate the expression of ARHGAP25 through modulation of the transcriptional activity of ARHGAP25. In addition, the CHiP assay also demonstrated that HOXA4 might directly bind to the promoter of ARHGAP25 and further regulate the transcriptional activity of ARHGAP25 (Figure 5D).
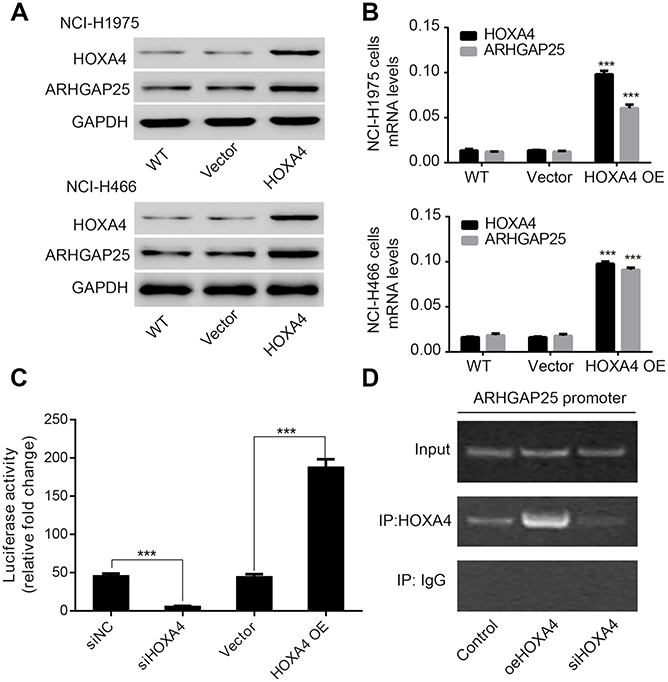 |
Figure 5 Role of HOXA4 in the regulation of ARHGAP25 in lung cancer. Western blot (A) and qRT-PCR (B) were conducted in NCI-H1975 and NCI-H446 cells in which HOXA4 was overexpressed. (C) The transcriptional activity of ARHGAP25 promoter was detected by luciferase assay after transfection of HOXA4 siRNA or overexpressed plasmid in NCI-H1299 cells. (D) ARHGAP25 DNA was detected in the chromatin sample immunoprecipitated from NCI-H1299 cells using an antibody against HOXA4.Note: ***P<0.001. |
Role of ARHGAP25 was mediated by the Wnt/β-catenin signaling pathway
More and more investigations indicated that the Wnt/β-catenin signaling pathway was involved with lung cancer cell growth and mobility.11,12 To further investigate the relationship between the Wnt/β-catenin signaling pathway and roles of ARHGAP25, ARHGAP25 knockdown cells were additionally transfected with Wnt inhibitor, XAV939. Three specific shRNAs were designed to knockdown ARHGAP25 expression (Figure S3), and we found that sh#1 showed the highest knockdown efficiency which was then used for further experiments.
We found that cell growth rate (Figure 6A) and mobility (Figure 6B) of NCI-H1299 cells remarkably increased when ARGHAP25 was knocked down which could be compromised when XAV939 was added. Also, ARHGAP25 knockdown significantly up-regulated β-catenin and MMP-7 which could be abrogated by adding XAV939 (Figure 6C). In addition, knockdown of HOXA4 also significantly strengthened the proliferation which could be abrogated by ARHGAP25 (Figure 6A) as well as migration and invasion (Figure 6B). HOXA4 knockdown could up-regulate β-catenin and MMP-7 which was compromised by ARHGAP25. These results suggested that Wnt/β-catenin signaling pathway could mediate the inhibitory effects of ARHGAP25 on cell growth, migration, and invasion, and these effects of ARHGAP25 could be regulated by HOXA4.
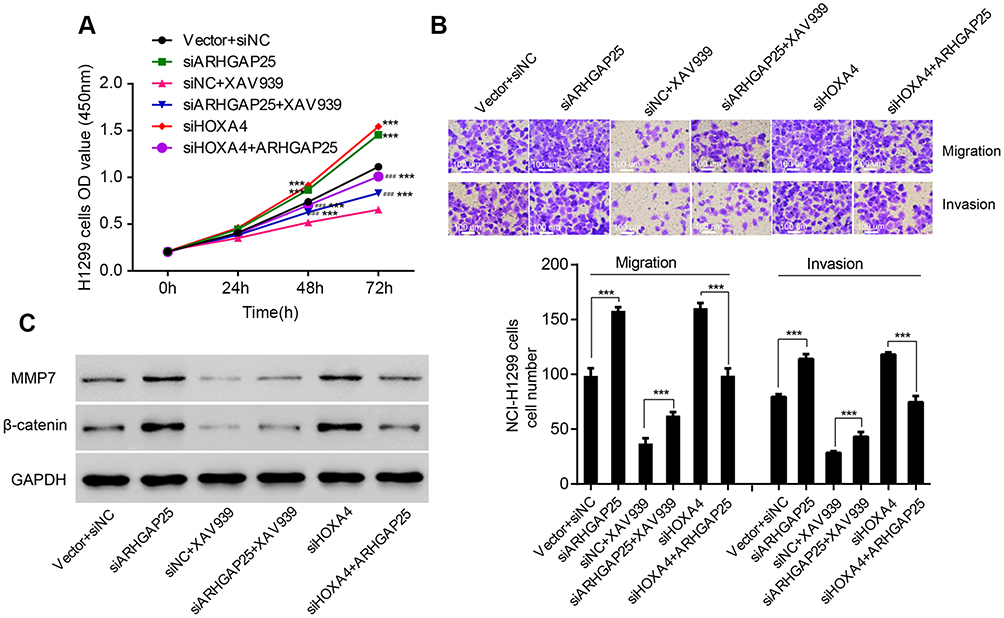 |
Figure 6 Regulation of HOXA4 on the biological function of ARHGAP25 through the Wnt/β-catenin signaling pathway. Cell proliferation assay (A), Transwell assay (B) and Western blot (C) were conducted in designed groups including vector plus siNC, ARHGAP25 siRNA, siNC plus β-catenin inhibitor XAV939, ARHGAP25 siRNA plus β-catenin inhibitor XAV939, HOXA4 siRNA and HOXA4 siRNA plus ARH OE. ***Indicated significance of siNC (P<0.001); ###indicated significance of siNC plus XAV939 (P<0.001). |
Discussion
ARHGAP25 was first reported to be correlated with cancer by Thuault et al.7 Recently, ARHGAP25 has also been found to be involved with hematopoietic cells and played an important role in regulating phagocytosis and corresponding superoxide production.8,26 In this research, lower expression of ARHGAP25 was identified in lung cancer (Figure 1). To explore the biological function of ARHGAP25, we transfected lentivirus into lung cancer cells to overexpress ARHGAP25. Our findings indicated aberrant expression of ARHGAP25 could reduce cell growth rate (Figure 2C and D) and mobility (Figure 3) of lung cancer cells, while the contrary effects could be observed when ARHGAP25 was knocked down (Figure 6A and B). These results suggest that ARHGAP25 may suppress tumor progression by acting as a tumor suppressor.
Further, we tried to clarify the underlying mechanism of how ARHGAP25 correlated with lung cancer development and progression. Initially, we identified negative relationship between the expression of ARHGAP25 and the activity of the Wnt/β-catenin signaling pathway (Figure 4A). The Wnt/β-catenin signaling pathway has been reported to mediate the proliferation, survival and metastasis of lung cancer by regulating the transcription of downstream effectors.11,12 As an important molecule in Wnt/β-catenin signaling pathway, β-catenin has been found highly expressed in lung cancer.11 Down-regulation of β-catenin would contribute to worse survival of lung cancer.13,14 Our results indicated that β-catenin expression was significantly reduced when ARHGAP25 was overexpressed in NCI-H1975 and NCI-H446 cells (Figure 4B). Additionally, MMP-7, a critical target gene of the Wnt/β-catenin signaling pathway, was simultaneously reduced when ARHGAP25 was overexpressed. MMP-7 leads to the degradation of extracellular matrix and is correlated with the mobility of cancer cells.18,19 The reduced expression of MMP-7 may help to interpret the reduced cell growth rate and mobility of lung cancer cells when ARHGAP25 is overexpressed.
We also observed that effect of ARHGAP25 knockdown such as increased cell growth, migration and invasion can be abrogated by β-catenin inhibitor, XAV939 (Figure 6A and B). Our results indicated that the Wnt/β-catenin signaling pathway might participate in the biological process of ARHGAP25. As for the mechanism, we found that HOXA4 might serve as a transcription factor by regulating the expression of ARHGAP25. The overexpression of HOXA4 could significantly up-regulate the expression levels of ARHGAP25 on mRNA and protein levels. The luciferase assay indicated that HOXA4 could activate the transcription of ARHGAP25. Further ChIP assay suggested that HOXA4 could directly connect with the promoter of ARHGAP25. Besides, previous study has suggested HOXA4 could inhibit cell proliferation and metastasis by regulating the Wnt/β-catenin signaling in lung cancer.18 Altogether, these findings imply HOXA4 controls the transcriptional activation of ARHGAP25, and further ARHGAP25 functions as a suppressor gene through Wnt/β-catenin signaling pathway.
In conclusion, ARHGAP25 was down-regulated in lung cancer. ARHGAP25 may inhibit cell proliferation, migration and invasion mediated by the Wnt/β-catenin signaling pathway. HOXA4 could increase ARHGAP25 expression by regulating the transcriptional activity of ARHGAP25. ARHGAP25 also might serve as a therapeutic target of lung cancer and more investigations should be focused on it.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. Cancer J Clin. 2015;65(2):87–108. doi:10.3322/caac.21262
2. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
3. Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015;16(4):e165–e172. e165–e172. doi:10.1016/S1470-2045(14)71180-5
4. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi:10.3322/caac.20107
5. Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. (1081-0706 (Print)). doi:10.1146/annurev.cellbio.21.020604.150721
6. Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690. doi:10.1038/nrm2476
7. Thuault S, Comunale F, Hasna J, et al. The RhoE/ROCK/ARHGAP25 signaling pathway controls cell invasion by inhibition of Rac activity. Mol Biol Cell. 2016;27:2653–2661. (1939-4586 (Electronic)). doi:10.1091/mbc.e16-01-0041
8. Csepanyi-Komi R, Sirokmany G, Geiszt M, Ligeti E. ARHGAP25, a novel Rac GTPase-activating protein, regulates phagocytosis in human neutrophilic granulocytes. Blood. 2012;119:573–582. (1528-0020 (Electronic)). doi:10.1182/blood-2010-12-324053
9. Schlam D, Bagshaw RD, Freeman SA, et al. Phosphoinositide 3-kinase enables phagocytosis of large particles by terminating actin assembly through Rac/Cdc42 GTPase-activating proteins. Nat Commun. 2015;6. (2041-1723 (Electronic)). doi:10.1038/ncomms9623
10. Pongracz JE, Stockley RA. Wnt signalling in lung development and diseases. Respir Res. 2006;7(1):15. doi:10.1186/1465-9921-7-15
11. Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. JNCI. 2014;106(1). doi:10.1093/jnci/djt356
12. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11. doi:10.1038/nrc3419
13. Xu X, Sun P-L, Li J-Z, Jheon S, Lee C-T, Chung J-H. Aberrant Wnt1/β-catenin expression is an independent poor prognostic marker of non-small cell lung cancer after surgery. J Thorac Oncol. 2011;6(4):716–724. doi:10.1097/JTO.0b013e31820c5189
14. Hommura F, Furuuchi K, Yamazaki K, et al. Increased expression of β‐catenin predicts better prognosis in nonsmall cell lung carcinomas. Cancer. 2002;94(3):752–758. doi:10.1002/cncr.10213
15. Crawford HC, Fingleton BF, Rudolph-Owen LA, et al. The metalloproteinase matrilysin is a target of beta-catenin transactivation in intestinal tumors. Oncogene. 1999;18(18):2883. (0950-9232 (Print)).
16. Marchenko GN, Marchenko NF, Leng J, Leng JF, Strongin AY. Promoter characterization of the novel human matrix metalloproteinase-26 gene: regulation by the T-cell factor-4 implies specific expression of the gene in cancer cells of epithelial origin. Biochemical J. 2002;363(12):253–262. (0264-6021 (Print)).
17. Wu B, Crampton SF, Hughes CCW, Hughes CC. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity. 2007;26:227–239. (1074-7613 (Print)). doi:10.1016/j.immuni.2006.12.007
18. Basu S, Thorat R, Dalal SN. MMP7 is required to mediate cell invasion and tumor formation upon Plakophilin3 loss. PLoS One. 2015;10:e0123979. (1932-6203 (Electronic)). doi:10.1371/journal.pone.0123979
19. Shiomi T, Okada Y. MT1-MMP and MMP-7 in invasion and metastasis of human cancers. Cancer Metastasis Rev. 2003;22(2–3):145–152. (0167-7659 (Print)).
20. Krumlauf R. Hox genes in vertebrate development. Cell. 1994;78:191–201. (0092-8674 (Print)). doi:10.1016/0092-8674(94)90290-9
21. Packer AI, Mailutha KF, Ambrozewicz LA, Ambrozewicz LF, Wolgemuth DJ. Regulation of the Hoxa4 and Hoxa5 genes in the embryonic mouse lung by retinoic acid and TGFbeta1: implications for lung development and patterning. Dev Dyn. 2000;217(1):62–74. (1058-8388 (Print)).
22. Wang J, Zhang J, Xu L, Zheng Y, Ling D, Yang Z. Expression of HNF4G and its potential functions in lung cancer. Oncotarget. 2018;9(26):18018.
23. Subramanian A, Tamayo PF, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545–15550. (0027-8424 (Print)). doi:10.1073/pnas.0506580102
24. Fu X, Beer DF, Behar J, et al. cAMP-response element-binding protein mediates acid-induced NADPH oxidase NOX5-S expression in Barrett esophageal adenocarcinoma cells. J Biol Chem. 2006;281:20368–20382. (0021-9258 (Print)). doi:10.1074/jbc.M603353200
25. Cheng S, Qian F, Huang Q, Wei L, Fu Y, Du Y. HOXA4, down-regulated in lung cancer, inhibits the growth, motility and invasion of lung cancer cells. Cell Death Dis. 2018;9. (2041-4889 (Electronic)). doi:10.1038/s41419-018-0497-x
26. Lorincz AM, Szarvas G, Smith SM, Ligeti E. Role of Rac GTPase activating proteins in regulation of NADPH oxidase in human neutrophils. Free Radical Biol Med. 2014;68:65–71. (1873-4596 (Electronic)). doi:10.1016/j.freeradbiomed.2013.12.001
Supplementary materials
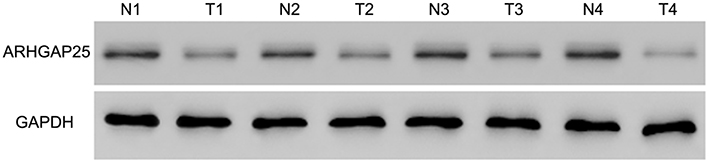 |
Figure S1 Western blot was conducted between paired cancerous and non-cancerous samples (n=8), referring to Figure 1C. |
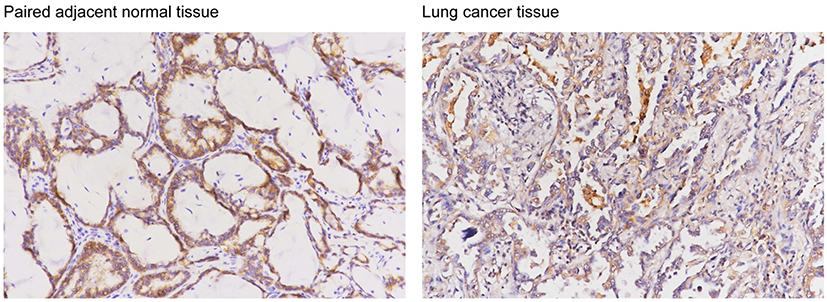 |
Figure S2 Representative images of ARHGAP25 expression in lung cancer and adjacent normal lung tissues using IHC assay in clinical specimen. |
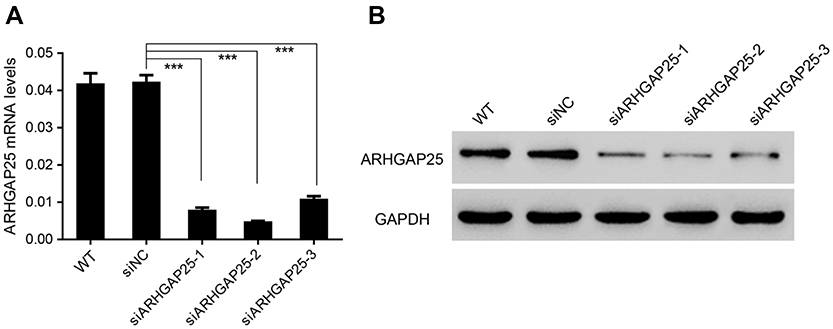 |
Figure S3 Down-regulated ARHGAP25 expression in NCI-H1299 cells. NCI-H1299 cells were transduced with ARHGAP25 shRNAs (sh#1, #2 and #3) or control shRNA (shNC). ARHGAP25 silencing was confirmed by qRT-PCR (A) and Western blotting (B). Experiments were repeated three times independently. ***P<0.001. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.