")
Back to Archived Journals » Research and Reports in Biochemistry » Volume 5
The TREM2-DAP12 signaling pathway in Nasu–Hakola disease: a molecular genetics perspective
Authors Xing J, Titus A, Humphrey MB
Received 16 December 2014
Accepted for publication 29 January 2015
Published 17 March 2015 Volume 2015:5 Pages 89—100
DOI https://doi.org/10.2147/RRBC.S58057
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Nikolay Dokholyan
Junjie Xing,1,2 Amanda R Titus,1 Mary Beth Humphrey1–3
1Department of Medicine, 2Department of Microbiology and Immunology, University of Oklahoma Health Sciences Center, 3Department of Veteran's Affairs, Oklahoma City, OK, USA
Abstract: Nasu–Hakola disease or polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL) is a rare recessively inherited disease that is associated with early dementia and bone cysts with fractures. Here, we review the genetic causes of PLOSL with loss-of-function mutations or deletions in one of two genes, TYROBP and TREM2, encoding for two proteins DNAX-activating protein 12 (DAP12) and triggering receptor expressed on myeloid cells-2 (TREM2). TREM2 and DAP12 form an immunoreceptor signaling complex that mediates myeloid cell, including microglia and osteoclasts, development, activation, and function. Functionally, TREM2-DAP12 mediates osteoclast multi-nucleation, migration, and resorption. In microglia, TREM2-DAP12 participates in recognition and apoptosis of neuronal debris and amyloid deposits. Review of the complex immunoregulatory roles of TREM2-DAP12 in the innate immune system, where it can both promote and inhibit pro-inflammatory responses, is given. Little is known about the function of TREM2-DAP12 in normal brain homeostasis or in pathological central nervous system diseases. Based on the state of the field, genetic testing now aids in diagnosis of PLOSL, but therapeutics and interventions are still under development.
Keywords: polycystic, leukoencephalopathy, Alzheimer's, lipomembranous, dementia, microglia
Introduction – clinical characteristics and pathology of Nasu–Hakola disease
Nasu–Hakola disease, also known as polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL), is a rare autosomal recessive leukodystrophy characterized by progressive early-onset dementia and recurrent bone fractures due to polycystic osseous lesions. In the early 1970s, Nasu and Hakola both independently described the disease.1,2 Thus far, about 200 cases have been diagnosed worldwide. Most cases have been from Finland and Japan, but there is a global distribution.3
The disease course is generally divided into four stages: latent, osseous, early neurologic, and late neurologic.4 After normal childhood development (latent stage), the disease begins manifesting during adolescence or young adulthood with polyarthralgias. The osseous stage is typically between ages 15 years and 30 years when patients develop pain in the hands, wrists, ankles, and feet followed by recurrent low-trauma bone fractures. Fractures occur in location of polycystic osseous and osteoporotic lesions in the limb bones. During the early neurologic stage, most often in the third or fourth decades of life, patients develop personality changes characteristic of a frontotemporal dementia (FTD). Initially, they experience mild memory disturbances and personality changes followed by progressive memory deficits. Generalized seizures are frequently observed. In the late neurologic stage, patients progress to a profound dementia and become bedridden. PLOSL leads to a progressive dementia that usually is fatal during the fifth decade of life. Occasionally, the disease presents with the neurologic symptoms preceding the osseous ones; therefore, early unexplained leukoencephalopathy warrants limb X-rays to aid in the diagnosis. Unfortunately, there is no curative treatment for the disease, and management is supportive including antiepileptic drugs to prevent seizures. Although pathological fractures occur and osseous lesion may be painful, conservative management is usually recommended.
The combination of early-onset FTD starting in the third or fourth decade and radiographic demonstration of cystic bone lesions is unique and facilitates the differentiation of PLOSL from other forms of familial and nonfamilial FTD. A biopsy of bone lesions is not necessary to confirm the diagnosis of PLOSL in the setting of this unique presentation. Radiographic imaging of PLOSL is characterized by lytic lesions in the distal bones of the extremities, most often in the metaphyseal and epiphyseal regions.5 During the course of the disease, trabecular bone loss develops in the distal portions of the long bones as well as symmetrical cystic lesions in the carpal and tarsal bones, heads of the metatarsal and metacarpal bones, and phalanges of the hand and feet. The lesions are often painful and frequently lead to pathologic fractures. The polyostotic lytic lesions may be surrounded by sclerotic rims or can be ill defined. Computed tomography (CT) imaging of the lytic lesions often shows smooth, marginated cystic lesions. Magnetic resonance imaging (MRI) of the osteolytic lesions reveals fat-equivalent content. If the bony lesions are biopsied, there is generally replacement of the marrow by fatty tissue separated by folded membranes which stain positive for periodic acid Schiff.6 Interestingly, these membranocystic lesions are seen not only in the bone marrow but also in systemic adipose tissues of the subepicardium, mediastinum, adrenal glands, testes, hepatic sinusoids, and pulmonary vascular lumens.4,7 Such lesions are characteristic of PLOSL but not specific for this disease and are seen in lipomembranous panniculitis and ischemic necrosis of fat.4,8 The occurrence of lipomembranous cystic lesion adipose tissues suggests a systemic inflammatory state, although this has not been clinically apparent in PLOSL patients.
As discussed above, PLOSL also results in a profound and rapidly progressive FTD with associated seizures. Brain CT and MRI reveal atrophy of the cerebral white matter, and frontal and polar regions of the temporal lobes are most affected.7 On MRI, there is thinning of the corpus callosum and ventricular enlargement not attributable to atrophy.9 Bilateral calcifications of the basal ganglia are often found on CT imaging. Positron emission tomography scanning reveals decreased glucose metabolism in the frontal white matter and basal ganglia suggesting a state of hypometabolism.10 On histologic examination, there is loss of axons and myelin, as well as fibrillary gliosis.7 Areas of demyelination with accumulation of lipid granules termed sudanophilic leukodystrophy may be present around vessels and inside tissue. The axons are swollen and deformed into spheroid shapes termed axonal spheroids. In the spinal cord, neuronal loss and destruction of neuronal cell bodies or chromatolysis are seen in the anterior horn.4 Microglial activation in the frontal and temporal white matter areas is also prominent.3 Additionally, there is an accumulation of lipid-laden macrophages and free fatty acids in the brain.7 In the majority of patients, vascular abnormalities are seen in the deep frontal and temporal white matter. There is concentric thickening of the vascular walls of affected small arterioles and capillaries.3,11 The constellation of these clinical and histological characteristics has suggested a lipodystrophy, although none has been associated with PLOSL.
Genetic causes of Nasu–Hakola disease
Over the last decade, tremendous achievements have been made in the understanding of the molecular aspect of Nasu–Hakola. Paloneva et al were the first to report the association of TYROBP mutations with PLOSL in 2000.12 They found that presenile dementia with bone cysts in Finnish patients was associated with loss-of-function mutations in TYROBP at chromosome 19q13.1. TYROBP contains five exons encoding DNAX-activating protein 12 (DAP12), a transmembrane signaling adapter protein found in a variety of immune cells. They subsequently reported the association of mutations in TREM2, encoding an immunoreceptor, triggering receptor expressed on myeloid cells-2 (TREM2), in several additional families with PLOSL.13 This was the first report of loss-of-function mutations in two proteins of the same signaling complex leading to the same disease.13 DAP12 is a 12 kDa, 113-amino acid (aa), transmembrane adaptor protein which is composed of a 27 aa leader, 14 aa extracellular domain, 24 aa transmembrane domain, and a 48 aa cytoplasmic region.14 TREM2 is a 40 kDa, 230 aa membrane glycoprotein with a single extracellular immunoglobulin-like ligand-binding domain, a transmembrane domain, and a short cytoplasmic tail without a signaling motif.15 A transmembrane domain positively charged lysine (aa186) of TREM2 couples to negatively charged aspartic acid residue in the transmembrane domain of DAP12 through electrostatic interactions (Figure 1).14,16 In addition to TREM2, DAP12 associates with a variety of immunoglobulin-like receptors, such as TREM-1 (TREM1) and signal-regulatory protein beta 1 (SIRPβ1), as well as lectin receptors including myeloid DAP12-associating lectin 1 (MDL1).17
In the initial report of genetic association of PLOSL with TYROBP, 26 patients were found to carry a homozygous genomic deletion of 5,265 bp encompassing the 5′ UTR and exons 1–4 of TYROBP (Table 1).12 This deletion results in no detectable DAP12. Additional point mutations in exons 1, 3, or 4 have been identified that produce truncated or nonfunctional DAP12 (Table 1).13,18–20 Klunemann et al also reported a 14 aa insertion in DAP12.21 Interestingly, one Japanese patient has been identified with compound heterozygous mutations in TYROBP, each with a point mutation predicted to produce nonfunctional truncated DAP12 polypeptides.19 Thus, deletion or production of nonfunctional DAP12 is associated with PLOSL.
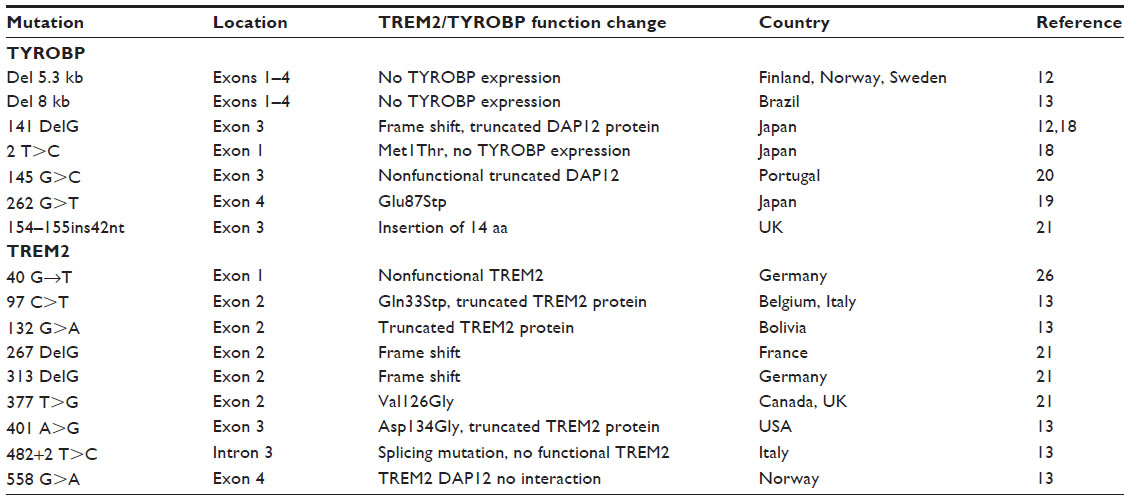 | Table 1 TREM2 and TYROBP mutations identified in PLOSL |
Additional analysis of ~20% of Nasu–Hakola patients having normal TYROBP revealed a surprising finding that they had deletions or mutations in TREM2 (Table 1).13 The TREM2 gene, found at human chromosome 6p21.1, consists of five exons encoding TREM2, a transmembrane cell surface receptor found on many myeloid cells including macrophages, dendritic cells, osteoclasts (OCs), and microglia. Each of these patients had one of several point mutants that are predicted to lead to a truncated TREM2 protein or to a specific aa change in the transmembrane domain leading to inability of TREM2 to couple to DAP12 (Table 1).13,21,22 PLOSL families with mutations in TREM2 have been described in the USA, Sweden, Italy, Norway, and Bolivia (Table 1). Thus, multiple mutations within TREM2 that lead to nonfunctional TREM2 proteins are associated with PLOSL.
Paloneva et al have further investigated other DAP12-associated receptors for genetic association with PLOSL and excluded TREM1, lymphocyte antigen-95 homolog (Ly95), SIRPβ1, MDL1, CD94, killer immunoglobulin-like receptor 2DS2 (KIR2DS2), and NKG2C (CD159c).13 Downstream intracellular kinases, spleen tyrosine kinase (Syk) and Zeta-chain-associated protein kinase 70 (ZAP70), have also been excluded.13 Thus, mutations in either the ligand-binding receptor or the signaling adapter protein of a myeloid cell immunoreceptor signaling complex, TREM2 or DAP12, are associated with Nasu–Hakola disease.
TREM2 and DAP12 expression
DAP12 is expressed in most innate immune cells including macrophages, monocytes, dendritic cells, granulocytes, and natural killer cells.17 DAP12 is also found to be expressed in some cells of the adaptive immune system, including subsets of T cells include γδ T cells and CD8+ αβ T cells17,23 and some activated B cells where DAP12 negatively mediates B cell immune responses.17,24 Interestingly, γδ T cells function as innate-type T cells with an invariant T cell receptor and the ability to respond to pathogen-associated molecular patterns similar to innate immune cells. OCs, also derived from myeloid precursors, express DAP12.25 Like DAP12, TREM2 is expressed in cells of the myeloid lineage including macrophages, OCs, and dendritic cells.
In the brain, DAP12 and TREM2 are mainly expressed in microglial cells derived from myeloid precursors.26 Microglia are the most abundant cells in the brain and responsible for immune surveillance of the brain. TREM2 is highly transcribed in resting unstimulated microglia and is downregulated by lipopolysaccharides and interferon-γ.27 In humans, TREM2 expression in brain tissue is highest in white matter and lowest in cerebellum and increases with age in the substantia nigra, thalamus, and medulla.28 In mice, TREM2 expression in microglia is more heterogeneous with the highest expression in the cingulate cortex and lateral entorhinal cortex and relatively little expression in regions with an incomplete blood–brain barrier including the hypothalamus, circumventricular organs, and the median eminence.27 In humans, DAP12 expression closely parallels TREM2 gene expression with the highest expression in the putamen, caudate nucleus, substantial medulla oblongata, and corpus callosum.13 TREM2 is detected in microglia associated with neurons and in microglial clusters.29 In the brains of Alzheimer’s patients, microglial-surrounding amyloid plaques have intense staining of TREM2.30 However, not all microglia express TREM2, supporting the notion that there are a variety of microglial phenotypes similar to the diversity of macrophages and dendritic cells in the periphery.
TREM2 and DAP12 expression detected by immunohistochemistry has also been reported on a fraction of neurons and oligodendrocytes.31–33 However, there is controversy as to whether TREM2 and DAP12 are expressed in oligodendrocytes with some studies reporting expression in mouse and the human oligodendrocytes and others reporting the absence of expression.31,33 The differences in these studies could be due to several possibilities including alternative approaches with in vivo tissue staining versus cell culture of primary cells that may have microglial contamination. Additionally, a soluble form of TREM2 is generated, and it may bind to ligands, such as heat shock protein 60 (HSP60), on neurons and oligodendrocytes and would be detected as TREM2 surface staining by immunohistochemistry.34 Neither TREM2 nor DAP12 expression has been detected in astrocytes.33 Thus, TREM2 and DAP12 are expressed in microglia and OCs, the two cell types believed to play substantial roles in PLOSL pathogenesis.
TREM2 and DAP12 intracellular signaling
TREM2 and DAP12, associated via charged residues within the transmembrane region, are expressed on the plasma membrane of myeloid cells and microglia. DAP12 forms a homodimer linked by two cysteines in the extracellular domain and is believed to associate with one TREM2 receptor based on previous studies of other DAP12-associated receptors (Figure 1).16 The cytoplasmic domain of DAP12 contains an immunoreceptor tyrosine-based activation motif (ITAM), with the consensus sequence YxxL/Ix6-8YxxL/I (where x represents any aa), that facilitates recruitment and activation of downstream effector molecules.35,36 As diagramed in Figure 2, following receptor activation, Src protein tyrosine kinases phosphorylate DAP12 at the two conserved tyrosine residues within the ITAM providing a docking site for the Src homology 2 (SH2) domains of Syk and ZAP70 kinases.37 Stimulation of TREM2 also induces the co-localization of DAP10 with DAP12 and is required for TREM2-dependent activation of phosphatidylinositol-3 kinase (PI3K).38 PI3K then converts phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-trisphosphate (PIP3) leading to the recruitment of the scaffolding protein, linker for the activation of B cells (LAB). LAB subsequently recruits a protein complex consisting of phospholipase C gamma (PLC-γ), SH2 domain-containing leukocyte protein of 76 kDa (SLP-76), guanine nucleotide exchange factor VAV, Tec kinases, growth factor-receptor-bound protein 2 (Grb2), and son of sevenless homolog1 (SOS1). PLC-γ hydrolyzes the membrane lipid PIP2 to secondary messengers diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3). The production of DAG activates two major signaling molecules, protein kinase C (PKC) and Ras guanyl nucleotide-releasing proteins (RasGRPs), which initiate the NF-κB and mitogen-activated protein kinase (MAPK) pathways. IP3 increases calcium mobilization leading to activation of calcium-dependent kinases such as calmodulin-dependent kinase (CaMK), Pyk2, and calcineurin which dephosphorylates members of the nuclear factor-activated T-cell (NFAT) family and induces their nuclear translocation. Thus, DAP12 signaling induces activation of downstream signals including MAPK, PI3K, PKC, and intracellular calcium flux.25 Other studies have shown that TREM2 and DAP12 are required for reactive oxygen species formation in response to bacteria.39 These signaling events in myeloid cells lead to cellular differentiation, activation, migration, cytokine production, and phagocytosis.
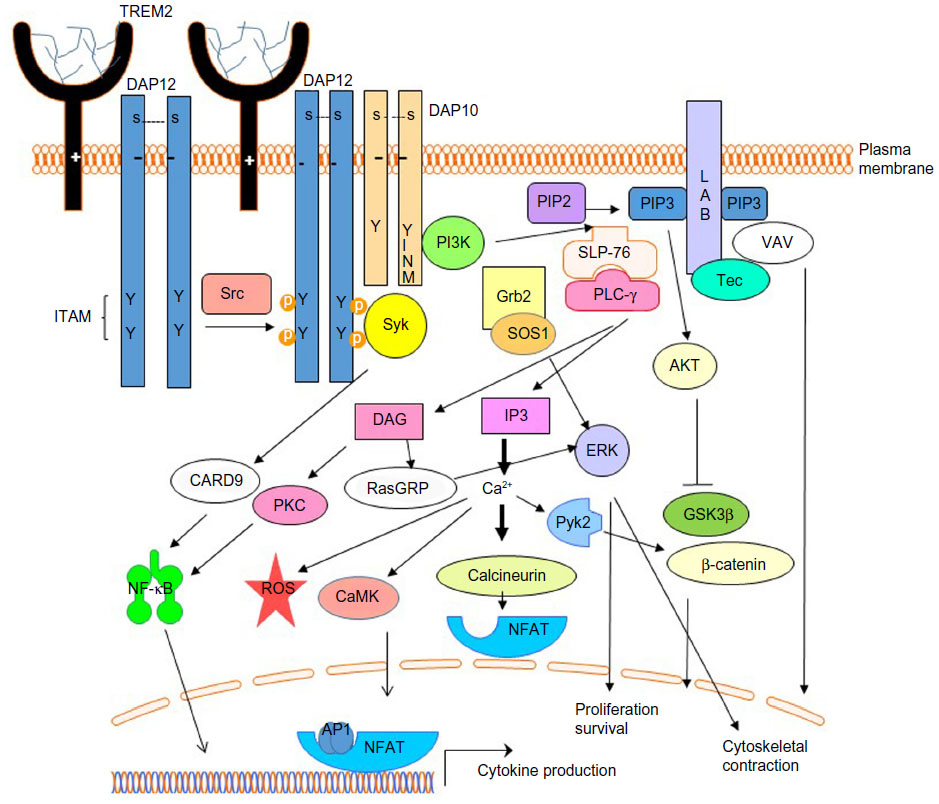 | Figure 1 Multimeric or high-affinity ligand-induced TREM2 signaling promotes myeloid cell activation. |
Interestingly, in macrophages and OCs, TREM2 and DAP12 can also recruit SH2 domain-containing inositol phosphatase-1 (SHIP1) to DAP12’s mono- and bi-phosphorylated ITAM.38 During tonic ITAM signaling induced by weak or monovalent ligands, SHIP1 prevents PI3K recruitment to limit excessive downstream activation (Figure 2). During strong receptor activation by TREM2 receptor cross-linking, SHIP1 and Syk are recruited to DAP12 and compete for ITAM binding.38 In the absence of SHIP1, Syk and PI3K activation is increased leading to enhanced ERK, VAV, and AKT activation as well as calcium flux.38 In macrophages, TREM2 and DAP12 signaling also recruits downstream of kinases 3 (DOK3).40 DOK3 subsequently binds Grb2 and SOS1, sequestering them, and limiting ERK activation. CBL-b, activated by Syk, then ubiquitinates DOK3 leading to DOK3, Grb2, and SOS1 degradation in the proteasome.40 Cbl also ubiquitinates Syk leading to degradation of Syk in the proteasome.41 In B cells, SH2 domain-containing protein tyrosine phosphatase 1 (SHP-1) can also be recruited to DAP12 and also serves an inhibitory function.24 Whether these downstream regulatory mechanisms contribute to microglial and OC dysfunction in PLOSL has not been explored.
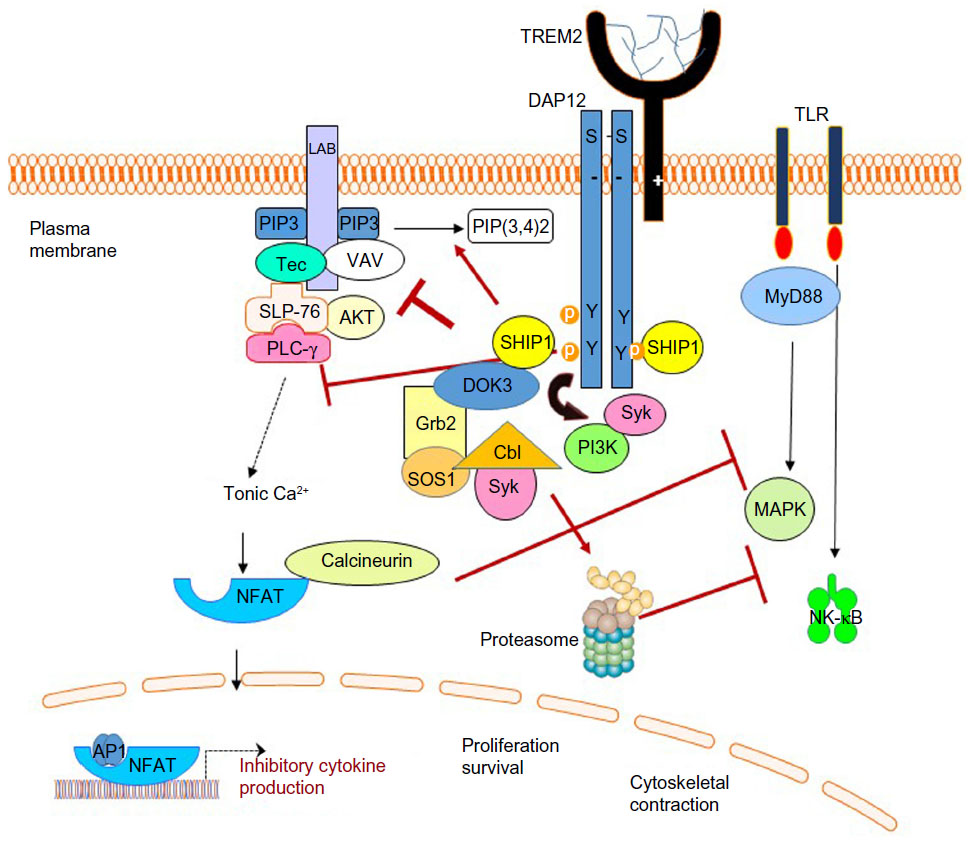 | Figure 2 Monovalent or weak ligand-induced TREM2 signaling is anti-inflammatory. |
TREM2 and DAP12 molecular complex also has significant cross talk with other signaling pathways including growth factor receptors, integrins, and Toll-like receptors (TLRs).25 Macrophage colony-stimulating factor (MCSF) binding to its receptor, c-Fms, induces Src activation, DAP12 phosphorylation, and Syk activation leading to myeloid proliferation.42,43 Integrin signaling also induces phosphorylation of DAP12 leading to activation of ERK, PLC-γ, and VAV required for cytoskeletal rearrangement.44,45 Additionally, TREM2 and DAP12 inhibit low-level TLR ligand-induced cytokine responses in myeloid cells leading to reduced pro-inflammatory cytokines and inflammation. Thus, TREM2 and DAP12 play several key roles in many vital signaling pathways controlling cellular differentiation, survival, or function.
TREM2 receptor cycling
In microglia, TREM2 is localized on the plasma membrane and in the Golgi complex. Phagocytic receptor recycling or ectodomain shedding regulates cell surface expression of TREM2, a process that leads to soluble TREM2 (sTREM2) release from the cell.46 TREM2 is recycled to and from the cell surface in endocytic and exocytic vesicles upon stimulation by ionomycin or interferon-γ.47 TREM2 is also actively regulated by ectodomain shedding of the extracellular ligand-binding domain by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) followed by intramembranous cleavage of the remaining C-terminal transmembrane domain and intracellular domain by γ-secretase.46,48 Inability to fully process the TREM2 C-terminal domain by blocking γ-secretase activity leads to accumulation of membrane-bound nonfunctional TREM2 fragments lacking the extracellular domain inappropriately coupled to DAP12, thereby decreasing the availability of DAP12 to couple with functional TREM2 molecules.48 This is predicted to result in a dominant-negative TREM2-DAP12 complex. Indeed, accumulation of these dysfunctional complexes leads to reduced DAP12 phosphorylation and decreased PI3K activation.48
Recent studies by Park et al have revealed that TREM2 mutations seen in PLOSL have altered trafficking from the endoplasmic reticulum (ER) and Golgi to the plasma membrane.49 The defective trafficking is associated with impaired TREM2 glycosylation with complex oligosaccharides. As a result, mutant TREM2 proteins accumulate in the ER or Golgi and have reduced plasma membrane expression. In comparison, TREM2 mutant R74H, associated with Alzheimer’s disease (AD), has near normal expression and trafficking but altered glycosylation.49 The severely reduced TREM2 receptor expression seen in PLOSL mutants likely contributes to the early onset of central nervous system (CNS) disease compared to late onset with the more normal Alzheimer’s-associated TREM2 mutant R47H.
The role of the cleaved sTREM2 in the cerebrospinal fluid (CSF) is not known, but levels appear to be altered in correlation with CNS diseases. CSF sTREM2 levels are markedly elevated in patients with multiple sclerosis and inflammatory CNS disease compared to those with non-inflammatory CNS disease.50 Additionally, patients with AD and FTD have significantly decreased sTREM2 in CSF, suggesting that reduced TREM2 function may contribute to increased risk for these neurodegenerative disorders as well.46 Indeed, heterozygous missense mutations in TREM2 are related to AD, Parkinson’s disease, amyotrophic lateral sclerosis, and FTD.46 Whether TREM2 receptor is processed correctly in PLOSL patients lacking DAP12 is unknown.
TREM2 and DAP12 mediate OC development and function
TREM2-DAP12 signaling plays an important role in both the innate and the adaptive immune systems.51 In microglia, TREM2-DAP12 is involved in cell survival, phagocytosis, and actin reorganization required for cell activation and phagocytic activity.42,52–55 TREM2-DAP12 signaling promotes OC and microglia differentiation by modulating precursor responsiveness to certain cytokines and growth factors along with reinforcing cytokine-induced and integrin-triggered signaling pathways.53
DAP12 and TREM2 play an important role in osteoclastogenesis and bone-remodeling homeostasis. Loss of function in human DAP12 or TREM2 peripheral monocytes results in inefficient and delayed OC differentiation in response to receptor activator of NF-κB ligand (RANKL).26 These defective OCs also have reduced bone resorption capacity in vitro; however, PLOSL bone lesions are of an osteoporotic nature with loss of trabeculae.26 Similar to OC derived from PLOSL patients, mice deficient in DAP12 have defective OC development with decreased OC multi-nucleation and decreased function in vitro.31,56,57 However, these mice fail to develop cystic osseous lesions in the bones and have mild osteopetrosis in vivo.31,56,57 Thus, the role of DAP12 in OC is similar in vitro but differs in vivo between mouse and man.
TREM2 and DAP12 also mediate OC fusion and migration.26,56,58 When TREM2 is highly expressed in RAW264.7 pre-osteoclastic cells, TREM2 cross-linking leads to increased multi-nucleation, migration, and survival of mouse OCs.58 Similarly, when TREM2 is reduced by RNA interference, RAW264.7 have significantly decreased OC potential and reduced multi-nucleation.58 However, TREM2 deficiency in mice leads to accelerated osteoclastogenesis in vitro and osteopenia in vivo.55 TREM2-deficient mice, however, do not develop osseous bone lesions. Of note, although TREM2-deficient mice do not produce full-length TREM2, sTREM2 may be present and may have additional effects on OC in vitro and in vivo. The differences in bone and OC phenotypes in DAP12-deficient and TREM2-deficient mice compared to PLOSL patients are not fully understood at this time. Alternative immunoreceptor and adapter proteins expressed between human and mouse cells or the effects of sTREM2 could contribute to the different pathologies seen.
TREM2 and DAP12 mediate innate immune responses
The function of TREM2 and DAP12 in innate immune cells is complex with both immune activation and inhibitory functions (Figures 1 and 2). While DAP12 is expressed constitutively in most myeloid cells, TREM2 expression is upregulated in M2-polarized macrophages that promote tissue repair.59 TREM2 expression is also upregulated in tissue macrophages infiltrating across an endothelial barrier and by MCSF and interleukin-4 important for regulation of chronic inflammation. Additionally, TREM2-DAP12 plays a role in maturation and survival of human dendritic cells, as well as in dendritic cell migration.60
Since DAP12 promotes pro-inflammatory responses when coupled to other DAP12-associated receptors such as TREM1 and MDL1, TREM2-DAP12 was expected to enhance pro-inflammatory responses.61 Surprisingly, TREM2-DAP12 inhibits TLR-induced pro-inflammatory cytokine responses in macrophages and dendritic cells (Figure 2).61–63 Whereas DAP12 mediates the majority of TLR ligand responses, TREM2 is required to dampen more selective TLR ligands including lipopolysaccharides (TLR4), zymosan (TLR2/6), and CpG (TLR9).59,62 The inhibition of TLR responses by TREM2 is dependent on the expression and coupling to functional DAP12.62 Thus, TREM2 and DAP12 function together to limit TLR-induced pro-inflammatory responses in macrophages and dendritic cells.
TREM2-DAP12 mediates microglia function
Perhaps in an analogous manner, TREM2 expression on microglia promotes phagocytosis of apoptotic neurons and bacteria without eliciting an immune response. TREM2 expression promotes phagocytosis of apoptotic neurons presumably by recognition of a ligand expressed during apoptosis.52,54 When TREM2 is expressed on microglia, it has enhanced phagocytosis of apoptotic neurons and does so with minimal pro-inflammatory cytokine production. In the absence of TREM2, apoptotic neurons promote excessive microglial activation and pro-inflammatory responses leading to CNS inflammation. TREM2-DAP12 is also important for efficient microglial phagocytosis of amyloid deposits.46 Thus, abnormalities in TREM2 or DAP12 in human PLOSL microglia are expected to inhibit clearance of apoptotic neurons and amyloid deposits.
Importantly to its role in promoting and inhibiting immune responses, TREM2 functions as a pattern-recognition receptor and can bind to a variety of bacterial cell wall products, lipids, and anionic ligands.64 Engagement of these ligands with TREM2 promotes phagocytosis.65 Despite recognition of TREM2 ligands on macrophages, neuroblastomas, and apoptotic neurons by TREM2 fusion proteins, endogenous TREM2 ligands remain largely unknown.52,65 One exception is HSP60, a mitochondrial protein chaperone, which becomes expressed on the plasma membranes of apoptotic neurons.34 TREM2-dependent microglial clearance of apoptotic neurons is significantly increased by HSP60 and may serve as a trigger for TREM2-mediated phagocytosis.34 Interestingly, HSP60 is also an endogenous ligand for TLR4 and induces tumor necrosis factor-α and nitric oxide in macrophages.66 Therefore, HSP60 could induce an inflammatory response bound to TLR4 or could lead to suppression of TLR4-induced cytokines when bound to TREM2. Additional studies are required to test this hypothesis, but recently studies have found mutations in Hspd1, the gene encoding HSP60, associated with two human nervous system diseases including spastic paraplegia67,68 and an autosomal recessively inherited hypomyelinating leukodystrophy termed MitCHAP60.69 Supporting the role of HSP60 in normal brain function, mice haploinsufficient for HSP60 were recently shown to develop late-onset neurodegeneration.70 More studies are needed to determine the role of HSP60 in microglial responses.
TREM2 has also been shown to pair with plexin-A1, a semaphorin 6D (Sema6D)-binding receptor important for axon guidance, cardiac morphogenesis, and dendritic cell function.71–73 TREM2 and plexin-A1 can associate independently of DAP12; thus, TREM2 appears to function as a bridge to bring plexin-A1 together with DAP12.71 Sema6D enhances osteoclastogenesis in vitro, but Sema6D binding to OCs does not require TREM2 indicating that Sema6D is not a TREM2 ligand per se.71 However, plexin-A1-deficient mice develop high bone mass due to decreased OCs similar to DAP12-deficient mice.71 Importantly, no nervous system changes were found in plexin-A1-deficient mice indicating that TREM2-DAP12 coupled with plexin-A1 is not required for brain homeostasis. Further studies are required to define a role, if any, of this tri-molecular complex in microglia.
TREM2-DAP12 dysfunction in PLOSL
Although the exact molecular mechanisms of the development of leukoencephalopathy in brains of PLOSL patients are unknown, abnormalities in TREM2-DAP12 regulation of microglial inflammatory responses and clearance of neuronal and amyloid debris appear to be central to the cause. In PLOSL, TREM2 or DAP12 deficiency is postulated to lead to excessive microglial pro-inflammatory activation with decreased clearance of neuronal debris leading to neurodegeneration. DAP12- or TREM2-deficient microglia fail to remove apoptotic neurons, and thus induce inflammation of the brain, amyloid plaque deposition, and early FTD seen in PLOSL.
PLOSL brains also show loss of myelin, accumulation of axonal spheroids, and profound gliosis. During periods of cellular distress or tissue repair, oligodendrocyte autophagy is required to maintain myelin sheaths. Perhaps, the loss of myelin in PLOSL results from dysfunctional oligodendrocytes autophagy induced by abnormal TREM2- or DAP12-deficient microglia.74 Alternatively, loss of TREM2-DAP12 signaling in oligodendrocytes themselves could contribute to oligodendrocyte apoptosis or a defect in oligodendrocyte function leading to loss of myelin maintenance.74 In support of this hypothesis, DAP12-deficient mice and DAP12-loss-of-function (KΔ75) transgenic mice expressing mutation of tyrosine 75 (Y75) within the DAP12 ITAM have decreased myelin.57,75 DAP12-deficient mice also have decreased oligodendrocytes, whereas the KΔ75 mice have decreased microglia but normal oligodendrocytes.57,75 Interestingly, phosphorylated Syk, the downstream effector of DAP12 signaling, is significantly increased in neurons and microglia of PLOSL brains compared to control brains.76 This finding suggests uncontrolled Syk activation in the absence of TREM2 and DAP12 and is compatible with increased microglial activation.
The recent findings that rare TREM2 variants are associated with FTD without bone lesions, AD, and Parkinson’s disease provide additional insight into the role of TREM2 in brain homeostasis.77–80 Although it is not clear how TREM2 mutations contribute to AD, many of these rare variants lead to point mutations in the TREM2 ligand-binding domain or to the inability to effectively shed TREM2 from plasma membranes leading to accumulation of dysfunctional TREM2 fragment-DAP12 complexes.46 These TREM2 fragment-DAP12 complexes could function as a dominant negative form of the receptor complex. TREM2 is highly expressed in AD brain tissue, especially surrounding amyloid plaques. As discussed earlier, TREM2 can mediate amyloid plaque phagocytosis; therefore, it is tempting to speculate that dominant-negative TREM2 mutants seen in AD fail to phagocytose amyloid resulting in a similar amyloid-associated Alzheimer’s-type dementia seen in PLOSL when TREM2 or DAP12 is absent. Both conditions would lead to inappropriate pro-inflammatory microglial activation and accumulation of amyloid-β plaques.
To test this hypothesis, Ulrich et al recently examined the role of TREM2 haploinsufficiency in the APPPS1-21 mouse model of AD.81 They found that TREM2 haploinsufficient APPPS1-21 mice had decreased number and size of plaque-associated microglia at 3 months, suggesting that microglial responses to amyloid plaque are regulated by TREM2. However, no significant differences in inflammatory cytokines or total amyloid plaque burden were seen in TREM2 heterozygotes compared to TREM2+/+ mice. A recent study has shown that two point mutations in TREM2 seen in PLOSL have abnormal TREM2 glycosylation leading to accumulation in the ER and Golgi with decreased surface expression.49 The AD mutant R74H had normal trafficking but altered glycosylation compared to wild type. This suggests that the glycosylation pattern of TREM2 is critical for normal function and that it is not just the level of TREM2 expressed. These results would then predict normal brain function in TREM2 haploinsufficiency.
Amyloid-β deposits are recognized in part by a molecular complex of TLR4 and TLR6 in conjunction with CD36.82,83 This molecular complex is required for recognition and uptake of amyloid and induces activation of Src family kinase-mediated intracellular signals resulting in pro-inflammatory cytokine production.82 Given the strong evidence supporting the role of TREM2 and DAP12 inhibiting TLR signals, we speculate that in the absence of TREM2 or DAP12 in PLOSL, amyloid-stimulated TLR activation is uninhibited, leading to excessive activation of microglia with poor ability to phagocytize apoptotic neurons or amyloid deposits. Similarly, in AD, the accumulation of dysfunctional TREM2 fragment-DAP12 complexes or impaired ligand binding by TREM2 mutants would result in the inability to effectively inhibit the pro-inflammatory cytokine responses induced by amyloid-β. As discussed above, TREM2 and DAP12 associate with inhibitory proteins SHIP1 and DOK3 to mediate inhibition of Syk, PI3K, and ERK activation as well as limit TLR responses in macrophages. The role of this inhibitory pathway in PLOSL or AD has not been explored. Thus, further studies looking at the activation state and subcellular location of these proteins in PLOSL or AD microglia in response to amyloid may be informative as to the inhibitory function of DAP12 in these settings.
Recent studies have attempted to find biomarkers that could serve to aid the diagnosis of PLOSL. Giuliano et al performed proteomic analysis comparing Epstein–Barr virus-transformed B cells from seven Nasu–Hakola patients and five healthy controls.84 They found 21 differentially expressed proteins in the B cells from affected patients. Many of these proteins are involved in glucose metabolism and stress responses and suggest that energy metabolism of the brain may be altered in PLOSL. However, no definitive biomarkers have yet been identified specific for PLOSL.
Despite our significant knowledge on the roles of TREM2 and DAP12 in human and mouse osteoclastogenesis and mouse bone remodeling, it remains a mystery why PLOSL patients develop cystic bone lesions filled with folded membranes and degenerated fatty tissue. The lesions progress from early loss of trabeculae to a poorly formed, non-sclerotic cyst. The early progressive loss of trabeculae could be the result of excessive OC activity as seen in the TREM2 knock-out mice, but this does not explain the accumulation of lipid bilayers and fatty tissue. Additional molecular analysis of the contents of the cysts has not yet been performed, so it is not known if there are substantial adipocyte-associated macrophages present in the cystic fat tissues that could be pathological. TREM2 has been reported to be increased in adipose tissue from obese dogs compared to lean ones and in obese diabetic db/db mice.85,86 Therefore, it is possible that TREM2 could be regulated by adipocytes or participate in adipocyte regulation. If so, then TREM2 deficiency could lead to adipocyte degeneration or dysfunction. In support of this hypothesis, TREM2-transgenic mice fed a high-fat diet became more obese with increased adipocyte hypertrophy than control mice on a high-fat diet.87 The role of TREM2 and DAP12 in adipocytes, adipose tissue macrophages, or metabolic programming of myeloid cells has not been explored at this time.
Summary
PLOSL is a complex disease with significant disorder in the brain and bones. Genetic analysis has identified mutations in two genes, TYROBP and TREM2, resulting in loss of function of the TREM2-DAP12 immunoreceptor signaling complex. Genetic analysis of the genes for TREM2 and DAP12 remains the current gold standard for identification of Nasu–Hakola. Early studies suggest that TREM2-DAP12 is required for microglia phagocytosis of neuronal debris and amyloid deposits. In the absence of this complex, microglia become excessively activated, amyloid deposits accumulate, and there is myelin loss in the brain. In the bones, cystic bone lesions filled with lipid bilayers develop and result in pathological fractures and pain. While helpful, mouse models of PLOSL recapitulate some but not all features of PLOSL. Additional studies are needed to understand the role of TREM2-DAP12 in normal and pathological CNS diseases.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Nasu TTY, Terayama K. A lipid metabolic disease – “membranous lipodystrophy” – an autopsy case demonstrating numerous peculiar membrane-structures composed of compound lipid in bone and bone marrow and various adipose tissues. Pathol Int. 1973;23:539–558. | |
Hakola H. Neuropsychiatric and genetic aspects of a new hereditary disease characterized by progressive dementia and lipomembranous polycystic osteodysplasia. Acta Psychiatr Scand Suppl. 1972;232(1):1–173. | |
Paloneva J, Autti T, Raininko R, et al. CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology. 2001;56: 1552–1558. | |
Kaneko M, Sano K, Nakayama J, Amano N. Nasu-Hakola disease: the first case reported by Nasu and review. Neuropathology. 2010;30:463–470. | |
Kilic SA, Oner AY, Yuce C, Ozlu IC. Imaging findings of Nasu-Hakola disease: a case report. Clin Imaging. 2012;36:877–880. | |
Madry H, Prudlo J, Grgic A, Freyschmidt J. Nasu-Hakola disease (PLOSL) report of five cases and review of the literature. Clin Orthop Relat Res. 2006;454:262–269. | |
Bianchin MM, Capella HM, Chaves DL, et al. Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy – PLOSL): a dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol Neurobiol. 2004;24(1):1–24. | |
Al-Brahim N, Ceballos K, Sur M, Daya D, Alowami S. Lipomembranous panniculitis: report of a case. Ann Diagn Pathol. 2007;11(4):282–284. | |
Bilgic B, Gelisin O, Guerreiro R, et al. Neuroimaging findings of Nasu-Hakola disease. Alzheimer Dement. 2014;10(4 Suppl):2415. | |
Ueki Y, Kohara N, Oga T, et al. Membranous lipodystrophy presenting with palilalia: a PET study of cerebral glucose metabolism. Acta Neurol Scand. 2000;102(1):60–64. | |
Kalimo H, Sourander P, Jarvi O, Hakola P. Vascular changes and blood-brain barrier damage in the pathogenesis of polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (membranous lipodystrophy). Acta Neurol Scand. 1994;89(5):353–361. | |
Paloneva J, Kestilä M, Wu J, et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet. 2000;25(3):357–361. | |
Paloneva J, Manninen T, Christman G, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71(3):656–662. | |
Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998;391(6668):703–707. | |
Daws MR, Lanier LL, Seaman WE, Ryan JC. Cloning and characterization of a novel mouse myeloid DAP12-associated receptor family. Eur J Immunol. 2001;31(3):783–791. | |
Call ME, Wucherpfennig KW, Chou JJ. The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat Immunol. 2010;11(11):1023–1029. | |
Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev. 2009;227(1):150–160. | |
Kondo T, Takahashi K, Kohara N, et al. Heterogeneity of presenile dementia with bone cysts (Nasu-Hakola disease): three genetic forms. Neurology. 2002;59(7):1105–1107. | |
Kuroda R, Satoh J, Yamamura T, et al. A novel compound heterozygous mutation in the DAP12 gene in a patient with Nasu-Hakola disease. J Neurol Sci. 2007;252(1):88–91. | |
Baeta E, Guarda C, Mendes I, et al. Síndroma Nasu-Hakola: um caso português. [Nasu- Hakola syndrome: a Portuguese case]. Sinapse. 2002;2:15–18. Portugese. | |
Klunemann HH, Ridha BH, Magy L, et al. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology. 2005;64(9):1502–1507. | |
Soragna D, Papi L, Ratti MT, Sestini R, Tupler R, Montalbetti L. An Italian family affected by Nasu-Hakola disease with a novel genetic mutation in the TREM2 gene. J Neurol Neurosurg Psychiatry. 2003; 74(6):825–826. | |
Angelini DF, Zambello R, Galandrini R, et al. NKG2A inhibits NKG2C effector functions of gammadelta T cells: implications in health and disease. J Leukoc Biol. 2011;89(1):75–84. | |
Nakano-Yokomizo T, Tahara-Hanaoka S, Nakahashi-Oda C, et al. The immunoreceptor adapter protein DAP12 suppresses B lymphocyte-driven adaptive immune responses. J Exp Med. 2011;208(8):1661–1671. | |
Long CL, Humphrey MB. Osteoimmunology: the expanding role of immunoreceptors in osteoclasts and bone remodeling. Bonekey Rep. 2012;1:59. | |
Paloneva J, Mandelin J, Kiialainen A, et al. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J Exp Med. 2003;198(4):669–675. | |
Schmid CD, Sautkulis LN, Danielson PE, et al. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J Neurochem. 2002;83(6):1309–1320. | |
Forabosco P, Ramasamy A, Trabzuni D, et al. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol Aging. 2013;34(12):2699–2714. | |
Lue LF, Schmitz C, Walker DG. What happens to microglial TREM2 in Alzheimer’s disease: immunoregulatory turned into immunopathogenic? Neuroscience. Epub 2014 Sept 5. | |
Lue LF, Schmitz CT, Serrano G, Sue LI, Beach TG, Walker DG. TREM2 protein expression changes correlate with Alzheimer’s disease neurodegenerative pathologies in post-mortem temporal cortices. Brain Pathol. Epub 2014 Sep 3. | |
Kaifu T, Nakahara J, Inui M, et al. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J Clin Invest. 2003;111(3):323–332. | |
Kiialainen A, Hovanes K, Paloneva J, Kopra O, Peltonen L. Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co-expressed in the CNS. Neurobiol Dis. 2005;18(2):314–322. | |
Sessa G, Podini P, Mariani M, et al. Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur J Neurosci. 2004;20(10):2617–2628. | |
Stefano L, Racchetti G, Bianco F, et al. The surface-exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J Neurochem. 2009;110(1):284–294. | |
Lanier LL, Bakker AB. The ITAM-bearing transmembrane adaptor DAP12 in lymphoid and myeloid cell function. Immunol Today. 2000;21(12):611–614. | |
Humphrey MB, Lanier LL, Nakamura MC. Role of ITAM-containing adapter proteins and their receptors in the immune system and bone. Immunol Rev. 2005;208:50–65. | |
Furlong MT, Mahrenholz AM, Kim KH, Ashendel CL, Harrison ML, Geahlen RL. Identification of the major sites of autophosphorylation of the murine protein-tyrosine kinase Syk. Biochim Biophys Acta. 1997; 1355(2):177–190. | |
Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal. 2010; 3(122):ra38. | |
Charles JF, Humphrey MB, Zhao X, et al. The innate immune response to Salmonella enterica serovar typhimurium by macrophages is dependent on TREM2-DAP12. Infect Immun. 2008;76(6):2439–2447. | |
Peng Q, Long CL, Malhotra S, Humphrey MB. A physical interaction between the adaptor proteins DOK3 and DAP12 is required to inhibit lipopolysaccharide signaling in macrophages. Sci Signal. 2013; 6(289):ra72. | |
Lupher ML Jr, Rao N, Lill NL, et al. Cbl-mediated negative regulation of the Syk tyrosine kinase. A critical role for Cbl phosphotyrosine-binding domain binding to Syk phosphotyrosine 323. J Biol Chem. 1998;273(52):35273–35281. | |
Otero K, Turnbull IR, Poliani PL, et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol. 2009;10(7):734–743. | |
Faccio R, Zou W, Colaianni G, Teitelbaum SL, Ross FP. High dose M-CSF partially rescues the Dap12-/- osteoclast phenotype. J Cell Biochem. 2003;90(5):871–883. | |
Mocsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006; 7(12):1326–1333. | |
Zou W, Kitaura H, Reeve J, et al. Syk, c-Src, the alphavbeta3 integrin, and ITAM immunoreceptors, in concert, regulate osteoclastic bone resorption. J Cell Biol. 2007;176(6):877–888. | |
Kleinberger G, Yamanishi Y, Suárez-Calvet M, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6(243):243ra286. | |
Prada I, Ongania GN, Buonsanti C, Panina-Bordignon P, Meldolesi J. Triggering receptor expressed in myeloid cells 2 (TREM2) trafficking in microglial cells: continuous shuttling to and from the plasma membrane regulated by cell stimulation. Neuroscience. 2006;140(4):1139–1148. | |
Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma-secretase-dependent intramembranous cleavage. J Biol Chem. 2013;288(46):33027–33036. | |
Park JS, Ji IJ, An HJ, et al. Disease-associated mutations of TREM2 alter the processing of N-linked oligosaccharides in the Golgi apparatus. Traffic. 2015. | |
Piccio L, Buonsanti C, Cella M, et al. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain. 2008;131(pt 11):3081–3091. | |
Tomasello E, Vivier E. KARAP/DAP12/TYROBP: three names and a multiplicity of biological functions. Eur J Immunol. 2005;35(6):1670–1677. | |
Hsieh CL, Koike M, Spusta SC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. 2009;109(4):1144–1156. | |
Colonna M. DAP12 signaling: from immune cells to bone modeling and brain myelination. J Clin Invest. 2003;111(3):313–314. | |
Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005;201(4):647–657. | |
Otero K, Shinohara M, Zhao H, et al. TREM2 and beta-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J Immunol. 2012;188(6):2612–2621. | |
Humphrey MB, Ogasawara K, Yao W, et al. The signaling adapter protein DAP12 regulates multinucleation during osteoclast development. J Bone Miner Res. 2004;19(2):224–234. | |
Nataf S, Anginot A, Vuaillat C, et al. Brain and bone damage in KARAP/DAP12 loss-of-function mice correlate with alterations in microglia and osteoclast lineages. Am J Pathol. 2005;166(1):275–286. | |
Humphrey MB, Daws MR, Spusta SC, et al. TREM2, a DAP12-associated receptor, regulates osteoclast differentiation and function. J Bone Miner Res. 2006;21(2):237–245. | |
Turnbull IR, Gilfillan S, Cella M, et al. Cutting edge: TREM-2 attenuates macrophage activation. J Immunol. 2006;177(6):3520–3524. | |
Bouchon A, Hernandez-Munain C, Cella M, Colonna MA. DAP12-mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med. 2001;194(8):1111–1122. | |
Hamerman JA, Tchao NK, Lowell CA, Lanier LL. Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol. 2005;6(6):579–586. | |
Hamerman JA, Jarjoura JR, Humphrey MB, Nakamura MC, Seaman WE, Lanier LL. Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol. 2006;177(4):2051–2055. | |
Ito H, Hamerman JA. TREM-2, triggering receptor expressed on myeloid cell-2, negatively regulates TLR responses in dendritic cells. Eur J Immunol. 2012;42(1):176–185. | |
Daws MR, Sullam PM, Niemi EC, Chen TT, Tchao NK, Seaman WE. Pattern recognition by TREM-2: binding of anionic ligands. J Immunol. 2003;171(2):594–599. | |
N’Diaye EN, Branda CS, Branda SS, et al. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol. 2009;184(2):215–223. | |
Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the Toll-like receptor-4 complex. J Immunol. 2000;164(2):558–561. | |
Hansen JJ, Dürr A, Cournu-Rebeix I, et al. Hereditary spastic paraplegia SPG13 is associated with a mutation in the gene encoding the mitochondrial chaperonin Hsp60. Am J Hum Genet. 2002;70(5):1328–1332. | |
Bross P, Li Z, Hansen J, et al. Single-nucleotide variations in the genes encoding the mitochondrial Hsp60/Hsp10 chaperone system and their disease-causing potential. J Hum Genet. 2007;52(1):56–65. | |
Magen D, Georgopoulos C, Bross P, et al. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am J Hum Genet. 2008;83(1):30–42. | |
Magnoni R, Palmfeldt J, Christensen JH, et al. Late onset motoneuron disorder caused by mitochondrial Hsp60 chaperone deficiency in mice. Neurobiol Dis. 2013;54:12–23. | |
Takegahara N, Takamatsu H, Toyofuku T, et al. Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat Cell Biol. 2006;8(6):615–622. | |
Toyofuku T, Zhang H, Kumanogoh A, et al. Guidance of myocardial patterning in cardiac development by Sema6D reverse signalling. Nat Cell Biol. 2004;6(12):1204–1211. | |
Takamatsu H, Takegahara N, Nakagawa Y, et al. Semaphorins guide the entry of dendritic cells into the lymphatics by activating myosin II. Nat Immunol. 2010;11(7):594–600. | |
Satoh J, Motohashi N, Kino Y, et al. LC3, an autophagosome marker, is expressed on oligodendrocytes in Nasu-Hakola disease brains. Orphanet J Rare Dis. 2014;9:68. | |
Roumier A, Bëchade C, Poncer JC, et al. Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J Neurosci. 2004; 24(50):11421–11428. | |
Satoh J, Tabunoki H, Ishida T, et al. Phosphorylated Syk expression is enhanced in Nasu-Hakola disease brains. Neuropathology. 2012;32(2):149–157. | |
Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368(2):107–116. | |
Rayaprolu S, Mullen B, Baker M, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener. 2013;8:19. | |
Abduljaleel Z, Al-Allaf FA, Khan W, et al. Evidence of trem2 variant associated with triple risk of Alzheimer’s disease. PLoS One. 2014;9(3):e92648. | |
Jin SC, Benitez BA, Karch CM, et al. Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum Mol Genet. 2014;23(21):5838–5846. | |
Ulrich JD, Finn MB, Wang Y, et al. Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener. 2014;9:20. | |
Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11(2):155–161. | |
Moore KJ, El Khoury J, Medeiros LA, et al. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. 2002;277(49):47373–47379. | |
Giuliano S, Agresta AM, De Palma A, et al. Proteomic analysis of lymphoblastoid cells from Nasu-Hakola patients: a step forward in our understanding of this neurodegenerative disorder. PLoS One. 2014;9(12):e110073. | |
Fujimoto S, Goda T, Mochizuki K. In vivo evidence of enhanced di-methylation of histone H3 K4 on upregulated genes in adipose tissue of diabetic db/db mice. Biochem Biophys Res Commun. 2011;404(1):223–227. | |
Grant RW, Vester Boler BM, Ridge TK, Graves TK, Swanson KS. Adipose tissue transcriptome changes during obesity development in female dogs. Physiol Genomics. 2011;43(6):295–307. | |
Park M, Yi JW, Kim EM, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) promotes adipogenesis and diet-induced obesity. Diabetes. Epub 2014 Aug 11. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.