")
Back to Journals » OncoTargets and Therapy » Volume 12
The Roles Of Angiogenesis And Cancer Stem Cells In Sorafenib Drug Resistance In Hepatocellular Carcinoma
Authors Cheng CC, Chao WT, Liao CC, Shih JH, Lai YS, Hsu YH, Liu YH
Received 28 May 2019
Accepted for publication 1 September 2019
Published 4 October 2019 Volume 2019:12 Pages 8217—8227
DOI https://doi.org/10.2147/OTT.S217468
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Chiung-Chi Cheng,1,2 Wei-Ting Chao,3 Chen-Chun Liao,3 Jing-Hao Shih,3 Yih-Shyong Lai,1 Yung-Hsiang Hsu,4 Yi-Hsiang Liu1,4
1Department of Pathology, Chang Bing Show-Chwan Memorial Hospital, Changhua 505, Taiwan; 2Center for General Education, Providence University, Taichung City 433, Taiwan; 3Department of Life Science, Tunghai University, Taichung City 407, Taiwan; 4Department of Pathology, Tzu Chi University, Hualien 97004, Taiwan
Correspondence: Yi-Hsiang Liu
Department of Pathology, Chang Bing Show-Chwan Memorial Hospital, 6 Lugong Road, Lukang Zhen, Changhua County 505, Taiwan
Tel +886 4 7813888 ext. 71181
Fax +886 4 7073235
Email [email protected]
Background: An increasing number of studies support cancer stem cells as the reason for chemoresistance to sorafenib therapy in hepatocellular carcinoma (HCC), but the mechanism is still unclear. In this study, the mechanism of sorafenib resistance in cancer stem cells was examined by in vitro experiments and xenograft mouse model.
Methods: The expression of cancer stem cell markers in the Chang liver cell line and PLC/PRF/5 and HepG2 hepatoma cell lines were compared by immunoblot assay before and after sorafenib treatment in vitro. As a xenograft mouse model, subcutaneous injection of hepatoma cells followed by sorafenib therapy was performed in NU/NU mice. The effects of sorafenib therapy on tumor growth and cancer stem cell markers were studied. Angiogenesis associated with cancer stem cells was studied by immunoblot and immunohistochemistry assay.
Results: The expression of cancer stem cell markers was higher in PLC/PRF/5 and HepG2 cells than Chang liver cells, indicating that these hepatoma cells had more stemness-related characteristics. The cancer stem cell markers were upregulated in the hepatoma cell lines following sorafenib treatment in vitro. In the xenograft model, tumors from PLC/PRF/5 and HepG2 cells with high E-cadherin expression were more resistance to sorafenib therapy. However, the expression of cancer stem cell markers was not significantly different after sorafenib therapy in these tumors. Furthermore, we found that sorafenib therapy induced angiogenesis within tumors from high E-cadherin expressing hepatoma cells.
Conclusion: The mechanism of chemoresistance in sorafenib therapy in HCC may be the tumor angiogenesis associated with high E-cadherin expression in cancer stem cells.
Keywords: angiogenesis, cancer stem cells, cytokeratin, E-cadherin, hepatocellular carcinoma, sorafenib
Introduction
Hepatocellular carcinoma (HCC) is a cancer with high incidence and high mortality in Taiwan as well as worldwide.1 Despite advances in therapeutic treatments, such as radiotherapy, chemotherapy, surgical resections, and liver transplantation, the prognosis of patients with HCC remains poor due to the high incidence of tumor recurrence and metastasis.2 For those patients with unresectable or metastatic disease, systemic therapy targeting cancer cells is an alternative option for the management of HCC in addition to palliative treatment.
Sorafenib is a multi-targeted tyrosine kinase inhibitor that blocks tumor cell proliferation by targeting the activity of Raf family serine/threonine kinases, and inhibits tumor angiogenesis by targeting the vascular endothelial growth factor receptor (VEGFR).3 Currently, sorafenib therapy is the standard treatment for late-stage HCC patients.4 However, not all patients with advanced HCC respond well to sorafenib therapy. It has been reported that many advanced HCC patients are not responsive to sorafenib therapy, and the effect of sorafenib is often hampered by the occurrence of drug resistance.5,6 As a consequence of the lack of more effective compounds or treatment strategies, a major aim of biological research is to improve the efficacy of sorafenib against HCC. Therefore, there is an urgent need to explore the mechanisms of resistance to sorafenib therapy.
Cytokeratin (CK) is a cytoskeletal intermediate filament (IF) component. Different epithelial cells express characteristic combinations of CK polypeptides. In human hepatocytes, CK8 and CK18 form a CK pair that makes up IF are required for maintaining the integrity of hepatocytes.7 Plectin, a cytolinker protein, exhibits binding sites accessible to IF and cytoskeletal components to maintain the integrity of the cytoskeletal network.8 E-cadherin renders strong mechanical attachment between adherent epithelial cells and plays an essential role in orienting collective migration of large epithelial sheets.9 Recently we found that hepatoma cells expressing high-level E-cadherin and low-level plectin are more sensitive to sorafenib treatment and have a higher rate of collective cell migration.10 In accordance with these findings, we suggest that individual levels of E-cadherin and CK in patients with HCC may be correlated with their sensitivity to sorafenib therapy.
Cancer stem cells (CSCs), a small subpopulation of cancer cells, can self-renew, differentiate, and proliferate. They play a central role in tumor development, chemotherapy resistance, metastasis, and recurrence, and are considered to be a therapeutic target in cancer therapy.11 Recently, it was reported that the majority of HCC tumor cells undergo apoptosis following treatment with sorafenib, whereas CSCs remain, demonstrating their resistance to sorafenib and leading to tumor metastasis.12 Many biological markers are used to standardize the CSCs of HCC, including CD133, CD90, CD44, CD13, E-cadherin, EpCAM, ICAM-1 and Sal-like protein 4 (SALL4).13,14 SALL4 is an oncofetal protein that is expressed in the human fetal liver and silenced in the adult liver, but it is re-expressed in HCC with a poor prognosis.15 Elevated expression of SALL4 in tumors is associated with poor survival and resistance to chemotherapy in HCC patients.16 It has been reported that CD90+ CSCs are highly resistant to sorafenib treatment of HCC.17 Dissemination of CSCs happens not through single-cell migration by epithelial–mesenchymal transition, but rather through the collective migration of tumor clusters. Using E-cadherin expression to maintain the connection between cells, the leader cells of the CSC clusters direct their collective migration.18 Altered expression of CK genes is associated with liver diseases, and aberrant CK19 expression has been reported as a CSC marker correlated with poor prognosis in HCC.19
HCC is a highly vascularized tumor, and angiogenesis is critical for tumor growth and progression; this process predominantly involves vascular endothelial growth factor (VEGF) and endothelial cells.20 Cross-talk between endothelial cells and CSCs supports angiogenesis via secretion of pro-angiogenic factors, and promotes the expansion, maintenance, and survival of phenotypic CSC.21 CSCs preferentially upregulate pro-angiogenic factors such as VEGF to drive the angiogenesis process.22 CSCs can also be involved in angiogenesis by transdifferentiation into functional endothelial cells, shown in breast cancer23 and glioma.24 Thus, our approach to the development of anti-angiogenic therapy may need to be re-strategized as we begin to unravel the lineage plasticity of CSCs capable of creating their own vascular system to maintain stemness and tumorigenicity.
An increasing number of studies support CSCs as the reason for chemoresistance to sorafenib therapy in HCC, but the mechanism is still unclear. The aim of this study is to explore the mechanism of sorafenib resistance attributed to CSCs. The expression of CSC markers in HCC cell lines was investigated and compared before and after sorafenib treatment in vitro. In a xenograft mouse model, HCC cells were injected subcutaneously into both sides of the same nude mouse. Half the mice were then treated with sorafenib. The remaining mice did not receive any treatment as a control group. Animals were sacrificed at the end of the experiment, and the tumor tissues were collected for the study.
Materials and Methods
Antibodies
The commercially available primary antibodies used for immunohistochemical and immunoblot assays included anti-CK7 and -CK19 (Abcam, Cambridge, MA, USA), anti-E-cadherin (BD Transduction Laboratories, Franklin Lakes, NJ, USA), anti-SALL4 (Proteintech North America, Rosemont, IL, USA), anti-CD90 (Cell Signaling Technology, Boston, MA, USA), anti-vascular endothelial growth factor receptor 1 (VEGFR-1, Cell Signaling Technology, Boston, MA, USA), and anti-CD31 (Santa Cruz Biotechnology, Dallas, TX, USA) monoclonal antibodies. Secondary antibodies including horseradish peroxidase-conjugated rabbit antibodies used for immunoblotting analysis as well as biotin-conjugated rabbit antibodies used for immunohistochemistry were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Sorafenib (Nexavar) was purchased from Bayer Pharmaceuticals (Bayer AG, Leverkusen, Germany).
Cell Culture
Chang liver cells, PLC/PRF/5 cells, and HepG2 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Chang liver cells are originally derived from human normal liver tissue with subsequently HeLa cell contamination. PLC/PRF/5 cells are human hepatoma cell line containing hepatitis B virus and secrete hepatitis B virus surface antigen. HepG2 cells are derived from liver HCC without evidence of hepatitis B virus genome in this cell line. Dulbecco’s minimum essential medium supplemented with 10% fetal bovine serum, 50 units/mL penicillin, non-essential amino acids, and HEPES were used for culture at 37°C in the presence of 5% CO2. Medium was replaced and cells were split regularly upon confluent growth.
Measurement Of Effect For Sorafenib Treatment On CSC Marker Expression
Sorafenib was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM and stored at −20°C. The working solution contains DMSO at a concentration of 0.1% to prevent background effects on cell growth. The Chang liver, PLC/PRF/5, and HepG2 cells were seeded in 6 cm culture dishes at a density of 1×106 cells per dish and treated with 8 μM of sorafenib for 48-hr incubation. The cultures were incubated with MTT reagent according to the manufacturer’s protocol, then OD values were read at 570 nm using an ELISA reader. Expression of CSC markers was then determined by Western blotting analysis. Each experiment was repeated in triplicate.
Xenograft Mouse Model
NU/NU mice, aged 4 to 5 weeks, were purchased from BioLASCO Taiwan (Taipei, Taiwan). All animal experiments were approved by the Institutional Animal Care and Use Committee of Tunghai University following the “Guideline for the Care and Use of Laboratory Animals” publicized by Council of Agriculture, Executive Yuan, Republic of China. For each Chang liver, PLC/PRF/5 and HepG2, 5×106 cells suspended in 100 μL phosphate-buffered saline were mixed with 100 μL Martrigel and injected subcutaneously into each side of the same nude mouse. The body weight of mice and the tumor sizes were measured every 2 days until the tumor had grown larger than 150 mm3. After that, half of the mice were treated with sorafenib by intraperitoneal injection at a dose of 30 mg/kg for 21 days. The remaining mice did not receive any treatment as a control group. Three animals were used in each group and the total number of animal is 18 (n=18). The mice were sacrificed at the end of the experiment, and the tumor tissues were collected for analysis.
Western Blotting Analysis
Total cellular lysates from each sample were separately loaded and run in 10% sodium dodecyl sulfate polyacrylamide gels. Following electrophoresis, the gels were blotted onto polyvinylidene difluoride (PVDF) membranes by semi-dry transfer (Bio-Rad Laboratories, Hercules, CA, USA). The blotted PVDF membranes were blocked with 5% non-fat milk in phosphate-buffered saline with Tween-20 (PBST) buffer for 1 hr, and then washed three times for 5 mins each with PBST prior to adding the diluted primary antibody (CK7, CK19, E-cadherin, SALL4, CD90, VEGFR-1 or CD31; all at 1:1000 in PBST). The membranes were incubated for 1 hr, and the hybridized membranes were washed three times for 5 mins each with PBST prior to adding the diluted secondary antibody (1:10,000 in PBST). Finally, the enhanced chemiluminescent reagent (Merck Millipore, Burlington, MA, USA) was added to the washed membrane, and the image was developed using the Chemiluminescence Imaging System (Multigauge version 2.2, Fujifilm, Tokyo, Japan) as per the manufacturer’s instructions.
Immunohistochemistry Assay
The deparaffinized sample sections were rehydrated and treated with 3% H2O2 for 10 mins to eliminate the endogenous peroxidase activity. Non-specific binding was blocked with bovine serum albumin for 10 mins. The sample sections were incubated with anti-VEGFR-1 and CD31 primary monoclonal antibodies (at 1:500 dilutions) for 1 hr at room temperature. The biotinylated secondary antibody was then added (at a 1:500 dilution) and incubated at room temperature for 1 hr. Specific antibody binding was detected by adding avidin-conjugated peroxidase and observing the sections under a light microscope (BX51; Olympus, Tokyo, Japan) in the presence of the substrate reagent. For each specimen, the assay was repeated on separate triplicate sample sections.
Statistical Analysis
Quantitative results were presented as mean±standard deviation. Statistical analyses were performed by using a two-tailed unpaired t-test (between two groups) or one-way ANOVA (between three groups) using SAS 9.2 software (SAS Institute, Cary, NC, USA). P<0.05 was considered to indicate a statistically significant difference between values.
Results
Stemness Characterization Of Hepatoma Cell Lines
Western blotting analysis demonstrated different levels of E-cadherin, SALL4 and CD90 expression among Chang liver, PLC/PRF/5, and HepG2 cells. The expression of E-cadherin and SALL4 was higher in PLC/PRF/5 and HepG2 cells than in Chang liver cells. However, there was no visible difference in CD90 expression among these cells (Figure 1A). Quantification of these data (Figure 1B) confirmed that PLC/PRF/5 cells exhibited the highest amount of E-cadherin, while the lowest level was found in Chang liver cells (P<0.05). PLC/PRF/5 and HepG2 cells exhibited similar SALL4 expression, which was significantly higher than in Chang liver cells (P<0.05). CD90 expression was similar among these cells. These data indicate that PLC/PRF/5 and HepG2 cells possess more significant expression of E-cadherin, which is characteristic of stemness, than Chang liver cells.
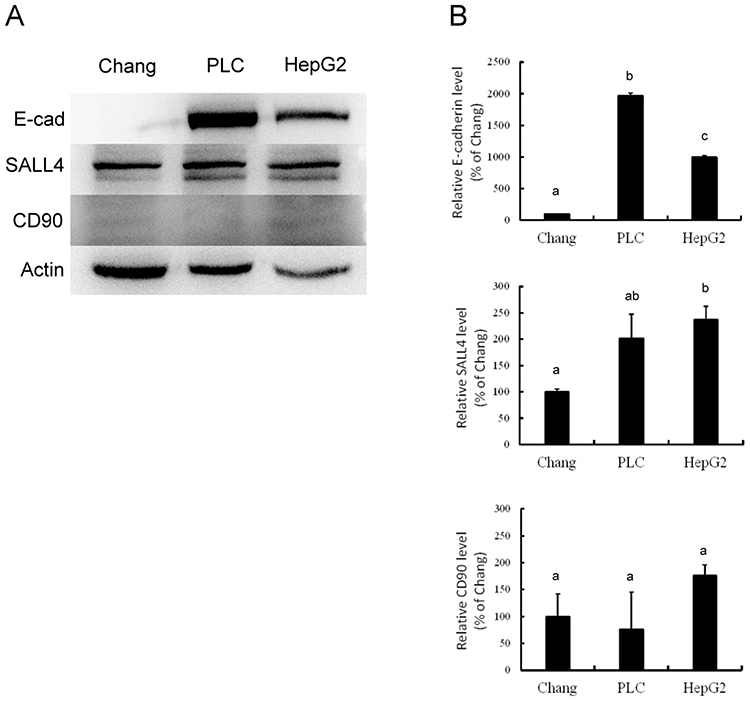 |
Figure 1 Western blotting (A) and associated statistical analysis (B) for the expression of E-cadherin (E-cad), SALL4 and CD90 in Chang liver cells (Chang), PLC/PRF/5 cells (PLC) and HepG2 cells (HepG2). The characters in this figure (a, b, c) indicate which percentages differ. Percentages with a same character are not statistically significant while percentages with different characters are statistically significant. Abbreviations: CK, cytokeratin; CSCs, cancer stem cells; DMSO, dimethyl sulfoxide; HCC, hepatocellular carcinoma; IF, intermediate filament; PBST, phosphate-buffered saline with Tween-20; PVDF, polyvinylidene difluoride; SALL4, Sal-like protein 4; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor; VEGFR-1, vascular endothelial growth factor receptor 1. |
Expression Of CSC Markers Affected By Sorafenib Treatment In Vitro
The hepatoma cells were treated with 8 μM sorafenib for 48-hr incubation. The expression of CSC markers was then investigated (Figure 2). The results revealed that the expression of CSC markers was elevated following sorafenib treatment in hepatoma cells. In Chang liver cells, sorafenib treatment significantly increased the expression of CD90 (P<0.05), but E-cadherin and SALL4 expression remained unchanged. The expression of all three CSC markers was increased in PLC/PRF/5 cells, especially E-cadherin and CD90 (P<0.05). In HepG2 cells, E-cadherin expression was significantly increased (P<0.05), but the levels of SALL4 and CD90 were not altered by sorafenib treatment. These results determined that sorafenib treatment may promote the expression of stemness-related proteins by hepatoma cells in vitro.
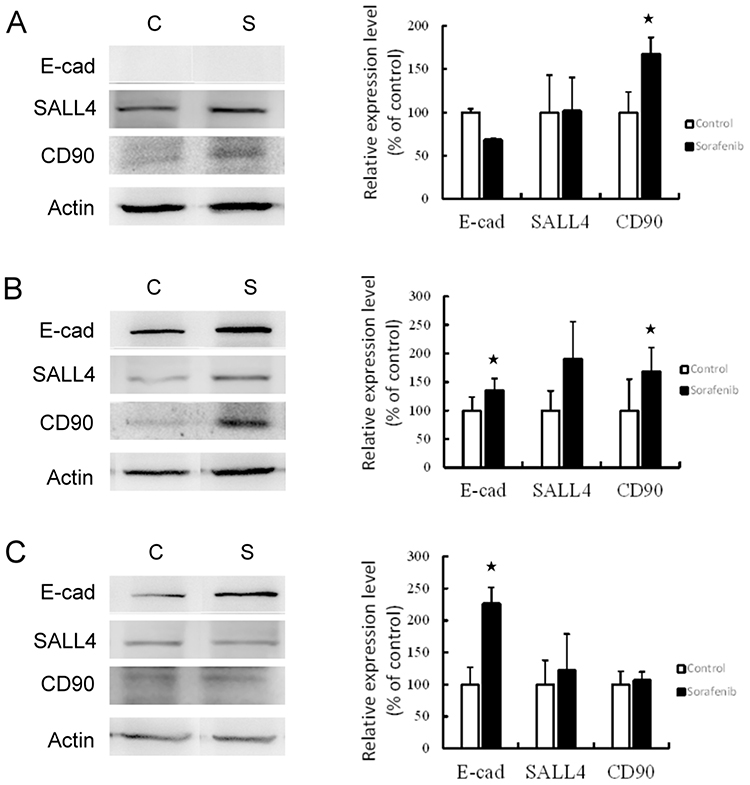 |
Figure 2 Western blotting (left panel) and associated statistical analysis (right panel) for the expression of CSC markers (E-cad, SALL4 and CD90) affected by sorafenib treatment in Chang liver cells (A), PLC/PRF/5 cells (B), and HepG2 cells (C). *P<0.05, compared to control group. Abbreviations: C, control group; S, sorafenib treated group. |
Tumor Growth Inhibited By Sorafenib Therapy In Xenograft Mouse Model
Chang liver, PLC/PRF/5, or HepG2 cells (5×106 cells each) were injected subcutaneously into nude mice. Half of the mice were treated with sorafenib at a dose of 30 mg/kg for 21 days. The remaining mice did not receive any treatment as a control group. A higher rate of hepatoma cell xenograft tumor growth was observed in the control group relative to the sorafenib treated group. Mice that underwent sorafenib therapy had smaller tumors at endpoint than the control group (Figure 3A and B). While tumor volumes of the sorafenib treated group were smaller than those of the control group, no significant difference was found between the PLC/PRF/5 and HepG2 xenograft tumor volumes. Only Chang liver xenograft tumors were significantly smaller in volume throughout the experimental period (P<0.05) (Figure 3C).
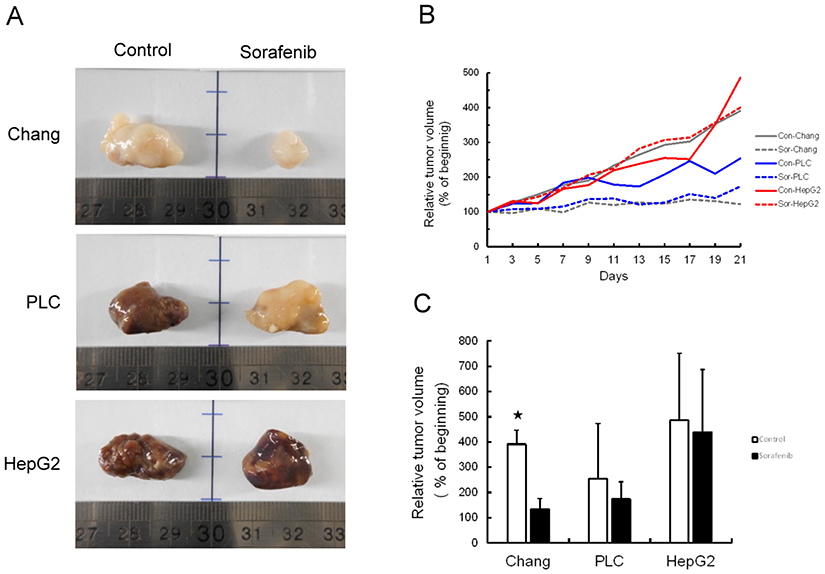 |
Figure 3 Tumor growth was inhibited by sorafenib treatment in xenograft mouse model. (A) Representative images of tumors from each group with (sorafenib) or without (control) treatment in hepatoma cell xenograft tumor growth. (B) Relative tumor volume was measured every day for 21 days following treatment. (C) Statistical analysis of the efficiency of sorafenib therapy based on differences in tumor growth. *P<0.05, compared to control group. |
CSC Marker Expression Affected By Sorafenib Therapy In Vivo
An effect of sorafenib therapy on the expression of CSC markers was observed in vivo. In Chang liver xenograft tumors, the expression of E-cadherin and CD90 were slightly elevated but this difference was not statistically significant (Figure 4A). In PLC/PRF/5 (Figure 4B) and HepG2 (Figure 4C) xenograft tumors, there was no significant change in CSC marker expression after sorafenib therapy. Unexpectedly, we found that the in vivo data were not correlated with our in vitro results. As CK19 has been reported as a CSC marker of HCC, we checked the expression of CK7 and CK19 in this study to identify potential changes in expression following sorafenib therapy. In Chang liver xenograft tumors, the expression of CK7/19 was not affected by sorafenib therapy (Figure 4D). In PLC/PRF/5 xenograft tumors, CK7/19 expression was slightly elevated, but this was not significant (Figure 4E). In HepG2 xenograft tumors, CK19 expression was significantly increased by sorafenib therapy (P<0.05), but CK7 was not changed (Figure 4F). According to these results, we speculate that CSCs alone cannot explain the mechanism of chemoresistance to sorafenib therapy in HCC.
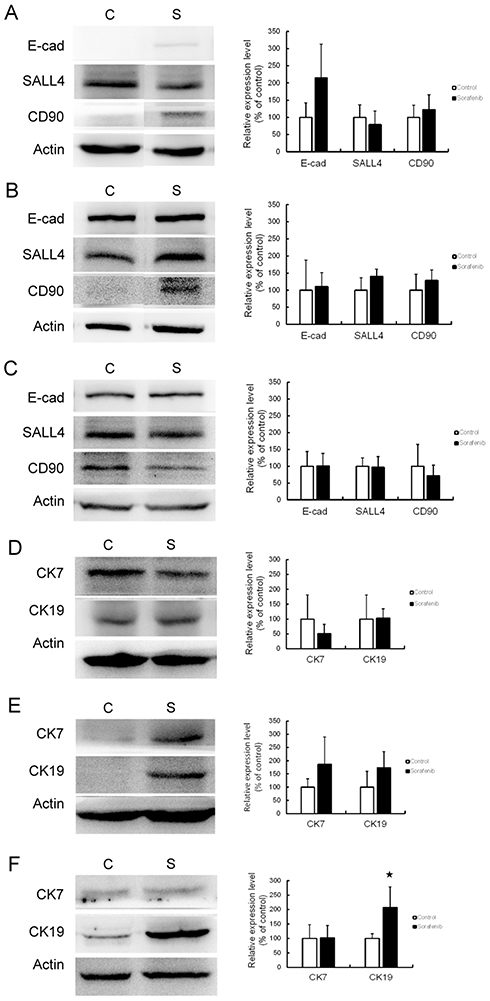 |
Figure 4 CSC marker expression was affected by sorafenib therapy in the xenograft mouse model. Western blotting (left panel) and associated statistical analysis (right panel) for the expression of E-cadherin (E-cad), SALL4, and CD90 with or without sorafenib therapy in Chang liver (A), PLC/PRF/5 (B), and HepG2 (C) xenograft tumors revealed no significant change in CSC marker expression. Western blotting (left panel) and statistical analysis (right panel) for the expression of cytokeratin 7 (CK7) and cytokeratin 19 (CK19) with or without sorafenib therapy in Chang liver (D), PLC/PRF/5 (E) and HepG2 (F) xenograft tumors also revealed no significant change, except for significantly increased CK19 expression in HepG2 xenograft tumors. Abbreviations: C, control group; S, sorafenib treated group. |
Angiogenesis In HCC Promoted By Sorafenib Therapy
We used Western blotting analysis and immunohistochemistry assay to investigate the expression of VEGFR-1 and CD31 before and after sorafenib therapy, with the aim of clarifying whether angiogenesis is involved in the chemoresistance to sorafenib therapy in HCC. The results of Western blotting analysis revealed that VEGFR-1 expression was not changed, but CD31 expression was significantly increased by sorafenib therapy in Chang liver (P<0.05) (Figure 5A), PLC/PRF/5 (P<0.05) (Figure 5B), and HepG2 (P < 0.01) (Figure 5C) xenograft tumors. By immunohistochemistry assay (Figure 6), the expression of VEGFR-1 and CD31 were significantly increased by sorafenib therapy in PLC/PRF/5 and HepG2 xenograft tumors, while Chang liver xenograft tumors showed a mild increased VEGFR-1 and CD31 expression. Therefore, we determined that sorafenib therapy promoted angiogenesis in mouse xenograft tumors from HCC cells.
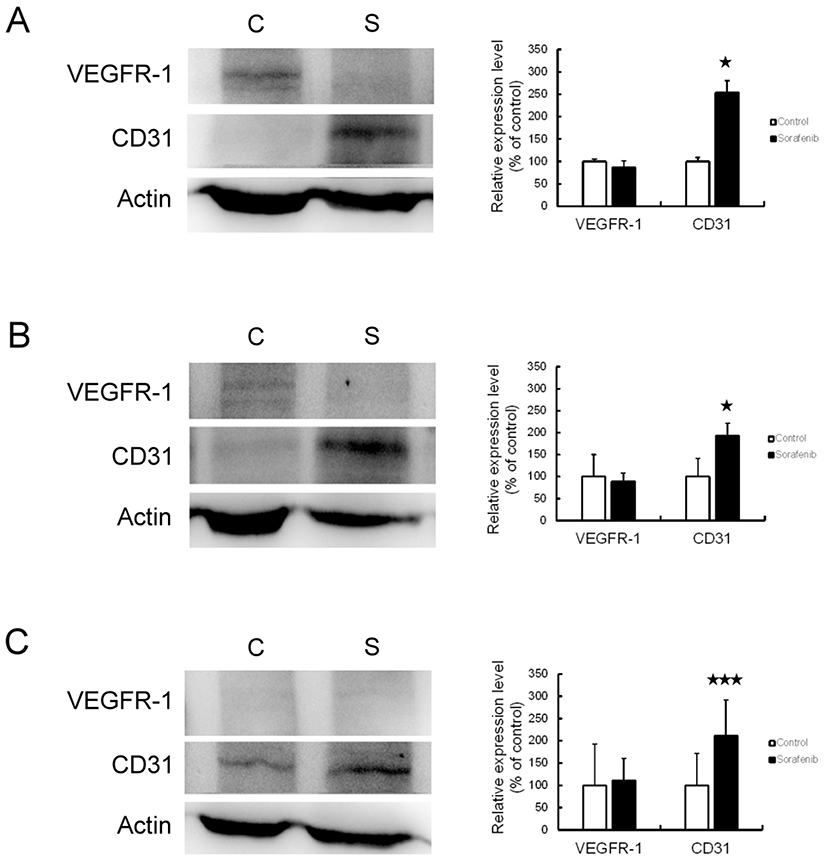 |
Figure 5 Angiogenesis was promoted by sorafenib therapy in the xenograft mouse model, based on Western blot. The expression of VEGFR-1 and CD31 was observed by Western blot analysis (left panel) and associated statistical analysis (right panel) in Chang liver (A), PLC/PRF/5 (B), and HepG2 (C) xenograft tumors. *P<0.05, ***P<0.01 compared to control group. Abbreviations: C, control group; S, sorafenib treated group. |
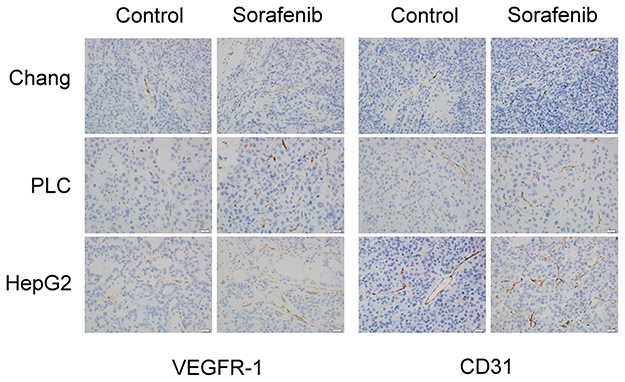 |
Figure 6 Angiogenesis was promoted by sorafenib therapy in the xenograft mouse model, based on immunohistochemistry. The expression of VEGFR-1 (left panel) and CD31 (right panel) was observed by immunohistochemistry assay. |
Discussion
In this study, we found that PLC/PRF/5 and HepG2 cells possessed more significant CSC marker expression than Chang liver cells. Sorafenib treatment promoted expression of proteins characteristic of CSCs in the three hepatoma cells in vitro. In animal experiments, sorafenib therapy was effective in inhibiting tumor growth against Chang liver xenograft tumor with lower CSC-associated protein expression. In PLC/PRF/5 and HepG2 xenograft tumors with higher CSC-associated protein expression, sorafenib was unable to effectively inhibit tumor growth. However, the expression of CSC markers was not significantly altered by sorafenib therapy in any of the xenograft tumor types. Moreover, we found that angiogenesis was promoted by sorafenib therapy in xenograft HCCs, and xenograft tumors with higher CSC-associated protein expression had higher levels of angiogenesis after sorafenib therapy.
It is well known that stemness or CSC is a promising mechanism of sorafenib resistance in HCC.25,26 Our in vitro experiments showed that sorafenib treatment promoted CSC-associated protein expression of the hepatoma cells, while the in vivo experiments showed that PLC/PRF/5 and HepG2 xenograft tumors with higher CSC-associated protein expression were more resistant to sorafenib therapy. Together these findings support our speculation that CSCs play a role in the mechanism of chemoresistance to sorafenib therapy in HCC. However, protein analysis showed that sorafenib therapy did not have a significant effect on altering the expression of CSC markers in xenografted HCCs (Figure 4). We also found resistance to sorafenib therapy in PLC/PRF/5 and HepG2 xenograft tumors, which was not consistent with our previous study which showed that PLC/PRF/5 and HepG2 cells were more sensitive to sorafenib treatment than Chang liver cells in vitro.10 The possible reason for this phenomenon may be that the extracellular conditions in vivo are more complicated than in vitro experiments, and that additional factors may be involved in the resistance to sorafenib therapy. Therefore, we speculate that CSCs may not play a very direct role in chemoresistance to sorafenib therapy in HCC.
It has been reported that during inhibition of tumor growth by angiogenesis inhibitors, tumors gradually develop resistance to the angiogenesis inhibitors, which eventually leads to angiogenesis and tumor recurrence.12 In our animal experiments, we observed more prominent hemorrhage on the xenograft tumors of PLC/PRF/5 and HepG2 with high sorafenib resistance. Therefore, we speculated that tumor angiogenesis may play a role in resistance to sorafenib therapy, and we conducted a study to clarify whether angiogenesis in xenograft tumors was inhibited or promoted after sorafenib therapy. The results showed that vascularity and the expression of VEGFR-1 and CD31 had significantly increased in xenograft tumors treated with sorafenib (Figures 5 and 6); that is, angiogenesis was promoted by sorafenib therapy in xenograft tumors. These data indicate that tumor angiogenesis may play an important role in the resistance mechanism.
Several prior studies have pointed out that tumor angiogenesis is important for CSCs to maintain their drug resistance properties, and that CSCs also promote tumor angiogenesis through VEGF so as to maintain the self-renewal and survival of the CSC population.22,27,28 Drug resistance by evasion of antiangiogenic targeting of VEGF signaling has also been reported in late-stage pancreatic islet tumors.29 In our study, PLC/PRF/5 and HepG2 xenograft tumors with higher CSC-associated protein expression were more resistant to sorafenib therapy and exhibited prominent tumor angiogenesis after sorafenib therapy; in contrast, in Chang liver xenograft tumor with lower CSC-associated protein expression, sorafenib therapy effectively inhibited tumor growth and milder tumor angiogenesis was noted. Hence, we posit that tumor angiogenesis promoted by sorafenib may be highly correlated with CSCs in HCC. In addition, increased expression of VEGFR-1 was found after sorafenib therapy; we therefore believe that CSCs in HCC promote tumor angiogenesis via VEGF signaling. It has been reported that the vascular niche plays an instructive role in promoting tumor growth,30 and that brain CSCs are maintained within vascular niches that are important targets for therapeutic approaches.31 Therefore, we speculate that HCC CSCs might promote vascular niche conditions, and anti-angiogenic therapy should be considered in the development of new treatments for HCC.
We found that sorafenib treatment enhanced CSC properties of the hepatoma cells in vitro. However, whether the expression of CSC-associated proteins was enhanced by sorafenib treatment through the induction of apoptosis in non-CSC hepatoma cells, resulting in a relative increase in the overall proportion of CSCs, or through direct enhancement of the CSC properties of hepatoma cells, needs to be further clarified. Besides ordinary CSC markers, we observed that the expression level of E-cadherin was higher in PLC/PRF/5 and HepG2 cells with high CSC properties (Figure 1). After sorafenib treatment, E-cadherin expression was also significantly higher in PLC/PRF/5 and HepG2 cells than Chang liver cells (Figure 2). We therefore consider E-cadherin as a favorable marker for stemness characterization of HCC. We recently demonstrated that hepatoma cells expressing higher level of E-cadherin by transient plectin knockdown are more sensitive to sorafenib treatment in vitro.10 Thus, the role of E-cadherin in HCC tumor progression may be simply as a CSC marker, or it may induce tumor angiogenesis by regulating cell–cell interactions; this is currently under investigation.
CK19 has been reported as a CSC marker of HCC and is correlated with clinicopathologic features of tumor aggressiveness and poor prognosis in HCC patients.19 In this study, we found that sorafenib therapy significantly increased CK19 expression in HepG2 xenograft tumors; CK19 was elevated to a non-significant degree in PLC/PRF/5 xenograft tumors, and there was no change in Chang liver xenograft tumor (Figure 4D–F). These results indicate that CK19 expression was promoted by sorafenib therapy in xenograft tumors with higher CSC-associated protein expression. Recently, we also reported a significant correlation between CK19 expression and SALL4 in human HCC, as well as poor prognosis in CK19-positive Taiwanese HCC patients.32 The key role of the CK19 molecule in the progression of human HCC through invasion and angiogenesis has also been reported.33 Based on these data, we speculate that in addition to being a useful marker for HCC CSCs, CK19 may be involved in chemoresistance to sorafenib therapy and might be a potential predictive factor for evaluating the effects of sorafenib therapy clinically.
Conclusion
In conclusion, our data demonstrated that the possible mechanism of sorafenib therapy in chemoresistance might be based on the promotion of tumor angiogenesis via VEGF signaling associated with CSCs in HCC. Tumor angiogenesis and chemoresistance were more prominent in xenograft tumors with high CSC-associated protein expression, also identified by high-level expression of E-cadherin and CK19. E-cadherin and CK19 may serve as CSC markers in HCC and be involved in the process of tumor angiogenesis in response to sorafenib therapy.
Acknowledgments
This work was supported by the Chang Bing Show-Chwan Memorial Hospital: grant numbers RD-107038 and RD-107039. We greatly appreciate Ms You-Yin Chen’s skillful assistance in the laboratory. We would like to thank Uni-edit for editing and proofreading this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi:10.3322/caac.21387
2. Han K, Kim JH, Ko GY, Gwon DI, Sung KB. Treatment of hepatocellular carcinoma with portal venous tumor thrombosis: a comprehensive review. World J Gastroenterol. 2016;22(1):407–416. doi:10.3748/wjg.v22.i1.407
3. Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. doi:10.1158/0008-5472.CAN-04-1443
4. Bruix J, Raoul JL, Sherman M, et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: subanalyses of a phase III trial. J Hepatol. 2012;57(4):821–829. doi:10.1016/j.jhep.2012.06.014
5. Villanueva A, Llovet JM. Second-line therapies in hepatocellular carcinoma: emergence of resistance to sorafenib. Clin Cancer Res. 2012;18(7):1824–1826. doi:10.1158/1078-0432.CCR-12-0151
6. van Malenstein H, Dekervel J, Verslype C, et al. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett. 2013;329(1):74–83. doi:10.1016/j.canlet.2012.10.021
7. Omary MB, Ku NO, Toivola DM. Keratins: guardians of the liver. Hepatology. 2002;35(2):251–257. doi:10.1053/jhep.2002.31165
8. Castanon MJ, Walko G, Winter L, Wiche G. Plectin-intermediate filament partnership in skin, skeletal muscle, and peripheral nerve. Histochem Cell Biol. 2013;140(1):33–53. doi:10.1007/s00418-013-1102-0
9. Li L, Hartley R, Reiss B, et al. E-cadherin plays an essential role in collective directional migration of large epithelial sheets. Cell Mol Life Sci. 2012;69(16):2779–2789. doi:10.1007/s00018-012-0951-3
10. Cheng CC, Chao WT, Liao CC, et al. Plectin deficiency in liver cancer cells promotes cell migration and sensitivity to sorafenib treatment. Cell Adh Migr. 2018;12(1):19–27. doi:10.1080/19336918.2017.1288789
11. Chiba T, Iwama A, Yokosuka O. Cancer stem cells in hepatocellular carcinoma: therapeutic implications based on stem cell biology. Hepatol Res. 2016;46(1):50–57. doi:10.1111/hepr.12548
12. Chen J, Jin R, Zhao J, et al. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015;367(1):1–11. doi:10.1016/j.canlet.2015.06.019
13. Kamiya A, Inagaki Y. Stem and progenitor cell systems in liver development and regeneration. Hepatol Res. 2015;45(1):29–37. doi:10.1111/hepr.12349
14. Sun JH, Luo Q, Liu LL, Song GB. Liver cancer stem cell markers: progression and therapeutic implications. World J Gastroenterol. 2016;22(13):3547–3557. doi:10.3748/wjg.v22.i13.3547
15. Yong KJ, Gao C, Lim JS, et al. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N Engl J Med. 2013;368(24):2266–2276. doi:10.1056/NEJMoa1300297
16. Oikawa T, Kamiya A, Zeniya M, et al. Sal-like protein 4 (SALL4), a stem cell biomarker in liver cancers. Hepatology. 2013;57(4):1469–1483. doi:10.1002/hep.26159
17. Luo J, Wang P, Wang R, et al. The Notch pathway promotes the cancer stem cell characteristics of CD90+ cells in hepatocellular carcinoma. Oncotarget. 2016;7(8):9525–9537. doi:10.18632/oncotarget.6672
18. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. 2017;168(4):670–691. doi:10.1016/j.cell.2016.11.037
19. Kim H, Choi GH, Na DC, et al. Human hepatocellular carcinomas with “Stemness”-related marker expression: keratin 19 expression and a poor prognosis. Hepatology. 2011;54(5):1707–1717. doi:10.1002/hep.24559
20. Zhu AX, Duda DG, Sahani DV, Jain RK. HCC and angiogenesis: possible targets and future directions. Nat Rev Clin Oncol. 2011;8(5):292–301. doi:10.1038/nrclinonc.2011.30
21. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J Biomed Sci. 2018;25(1):20. doi:10.1186/s12929-018-0408-6
22. Folkins C, Shaked Y, Man S, et al. Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res. 2009;69(18):7243–7251. doi:10.1158/0008-5472.CAN-09-0167
23. Bussolati B, Grange C, Sapino A, Camussi G. Endothelial cell differentiation of human breast tumour stem/progenitor cells. J Cell Mol Med. 2009;13(2):309–319. doi:10.1111/j.1582-4934.2008.00338.x
24. Ricci-Vitiani L, Pallini R, Biffoni M, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468(7325):824–828. doi:10.1038/nature09557
25. Shi DM, Bian XY, Qin CD, Wu WZ. miR-106b-5p promotes stem cell-like properties of hepatocellular carcinoma cells by targeting PTEN via PI3K/Akt pathway. Onco Targets Ther. 2018;26(11):571–585. doi:10.2147/OTT.S152611
26. Chen Z, Jiang Z, Zhang W, He B. Silencing the expression of copine-III enhances the sensitivity of hepatocellular carcinoma cells to the molecular targeted agent sorafenib. Cancer Manag Res. 2018;29(10):3057–3067. doi:10.2147/CMAR.S167781
27. Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66(16):7843–7848.
28. Zhao Y, Bao Q, Renner A, et al. Cancer stem cells and angiogenesis. Int J Dev Biol. 2011;55(4–5):477–482. doi:10.1387/ijdb.103225yz
29. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8(4):299–309. doi:10.1016/j.ccr.2005.09.005
30. Butler JM, Kobayashi H, Rafii S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer. 2010;10(2):138–146. doi:10.1038/nrc2791
31. Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11(1):69–82. doi:10.1016/j.ccr.2006.11.020
32. Lai YS, Cheng CC, Lee MT, et al. The prognostic value of cytokeratin and sal-like protein 4 expression in hepatocellular carcinoma and intra-hepatic cholangiocarcinoma in Taiwan. Int J Med Sci. 2018;15(14):1746–1756. doi:10.7150/ijms.28440
33. Takano M, Shimada K, Fujii T, et al. Keratin 19 as a key molecule in progression of human hepatocellular carcinomas through invasion and angiogenesis. BMC Cancer. 2016;16(1):903. doi:10.1186/s12885-016-2949-y
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.