")
Back to Journals » Clinical Interventions in Aging » Volume 15
The Role of Selected Pro-Inflammatory Cytokines in Pathogenesis of Ischemic Stroke
Authors Pawluk H , Woźniak A, Grześk G , Kołodziejska R , Kozakiewicz M , Kopkowska E, Grzechowiak E, Kozera G
Received 8 October 2019
Accepted for publication 13 February 2020
Published 23 March 2020 Volume 2020:15 Pages 469—484
DOI https://doi.org/10.2147/CIA.S233909
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Walker
Hanna Pawluk,1 Alina Woźniak,1 Grzegorz Grześk,2 Renata Kołodziejska,1 Mariusz Kozakiewicz,3 Ewa Kopkowska,1 Elżbieta Grzechowiak,4 Grzegorz Kozera4,5
1Department of Medical Biology and Biochemistry, Faculty of Medicine, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University in Toruń, Bydgoszcz, Poland; 2 2nd Department of Cardiology, Faculty of Health Sciences, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University in Toruń, Bydgoszcz, Poland; 3Department of Geriatrics, Division of Biochemistry and Biogerontology, Faculty of Health Sciences, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University in Toruń, Bydgoszcz, Poland; 4Department of Neurology, Faculty of Medicine, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University in Toruń, Bydgoszcz, Poland; 5Medical Stimulation Center, Medical University of Gdańsk, Gdańsk, Poland
Correspondence: Hanna Pawluk
Department of Medical Biology and Biochemistry, Faculty of Medicine, Collegium Medicum in Bydgoszcz, Nicolaus Copernicus University in Toruń, Karłowicza 24, Bydgoszcz 85– 092, Poland
Tel +48 52 585 37 55
Email [email protected]
Abstract: Stroke is currently one of the most common causes of death and disability in the world, and its pathophysiology is a complex process, involving the oxidative stress and inflammatory reaction. Unfortunately, no biochemical factors useful in the diagnostics and treatment of stroke have been clearly established to date. Therefore, researchers are increasingly interested in the inflammatory response triggered by cerebral ischemia and its role in the development of cerebral infarction. This article gives an overview of the available literature data concerning the role of pro-inflammatory cytokines in acute stroke. Detailed analysis of their role in cerebral circulation disturbances can also suggest certain immune response regulatory mechanisms aimed to reduce damage to the nervous tissue in the course of stroke.
Keywords: cytokines, ischemic stroke, old age disease
Introduction
Ischemic stroke is the most common cause of disability in elderly people (over 65 years of age) and the third most common cause of death in the world. The World Health Organization estimates that one in six people globally will suffer from stroke in their lifetime.1 The annual incidence of stroke in the general population is approximately 0.2%, while the mean value for Poland is estimated at an average of 150 per 100,000 residents.2,3 Ischemic stroke is the most frequent kind of stroke affecting approximately 85–90% of patients, most commonly caused by cardiogenic embolism, cerebral microcirculatory impairment (cerebral microangiopathy), atherosclerosis of extra- and intracranial arteries, as well as blood clotting disorders.4–6
The pathophysiology of stroke is a complex process and it is suggested that oxidative stress and inflammatory reaction constitute the key mechanisms involved in neuronal damage. During ischemia and asphyxia, brain and immune cells produce reactive oxygen species (ROS), which stimulate endothelial cells and cause oxidative stress. The activity of ROS during ischemic strokes not only causes primary vascular damage, but also triggers the development of the inflammatory response connected with the acute immune response. Glial cells (microglia, astrocytes) activated in ischemia, along with blood cells (leukocytes) and endothelial cells, synthesize a number of biochemical mediators and markers of inflammation, ie, cytokines, chemokines and pro-inflammatory enzymes.7–9 The development of inflammation in stroke is also affected by many factors, among which a significant role is played by genetic factors responsible for inflammatory reactions.10–12 It has also been demonstrated that many genes can contribute to both the etiology of stroke and the size of the ischemic area, and consequently the patient’s late outcome.13,14 In turn, chronic activation of inflammation is a factor which can significantly influence the advancement and impact of stroke risk factors, thereby also modifying the course of the acute phase of stroke.
Despite the broad range of molecular diagnostic methods, biochemical factors with a proven benefit for the prevention, diagnostics and treatment of stroke have not yet been clearly identified. A particular object of controversy is the impact of inflammatory factors on prognosis in re-perfusion therapies of ischemic stroke (ICA) involving intravenous thrombolytic therapy using recombinant tissue plasminogen activator (rtPA) and endovascular therapy (mechanical thrombectomy).15,16 Therefore, numerous studies of the diagnostic and prognostic value of pro-inflammatory factors of neuronal, glial and neuroendocrine origin, as well as inflammatory markers in ischemic stroke, are currently ongoing. Cytokines, as regulatory proteins produced by different types of cells, are an object of particular interest to researchers. They are recognized and bound by specific receptors located on cell membranes17,18 and by binding to the receptor, they transduce signal to the cell nucleus in which expression of genes encoding acute phase proteins (APPs). They can be triggered by the transcription factor (NF-κB) or regardless of the factor. Several groups of pro-inflammatory cytokines are known: chemokines, interleukins, interferons, as well as cell differentiation and growth factors.
Moreover, the inflammatory response in the brain changes over time. This is why the knowledge concerning if and when the level of cytokine increases or decreases in the course of the acute phase of cerebral stroke may help to determine a possible role of pro-inflammatory cytokines as early biomarkers and the object of a prospective therapeutic intervention in the acute phase of cerebral stroke.
This is why this paper consists of current theories concerning the role of pro-inflammatory cytokines as early inflammatory factors, which can be detected and determined both in serum and cerebrospinal fluid during the acute phase of cerebral stroke. Cytokines may also induce further phases of inflammatory markers and as a result, influence the final prognosis. Authors have methodically undertaken and checked the literature from latest years regarding pro-inflammatory cytokines in acute phase of stroke and assessed its potential clinical relevance.
Pathomechanism of Ischemic Brain Damage
The biochemical ground of pathophysiological processes in ischemic stroke of the brain is a complex cycle of interconnected molecular and cellular mechanisms (Figure 1).
 |
Figure 1 Pathomechanism of ischemic brain damage. |
At the time of getting sick, there is a severe focal hyperperfusion (a decrease in regional blood flow in the zone of irreversible ischemia <22 mL/100 g/min) and consequently - a decrease in pO2, increase in pCO2, development of tissue acidosis, bioenergetic insufficiency, excitotoxicity, occurrence of oxidative stress causing further microvascular damages and activation of neural and glial cells and secondary migration of leukocytes through a damaged blood-brain barrier (BBB).8,19 This cascading reaction ultimately leads to global death of neurons, glial cells and endothelium in necrosis or apoptosis mechanism.19
An inflammatory condition plays a vital role at all stages of the ischemic cascade, starting from the cessation of flow to the late regenerative processes associated with the repair of ischemic tissues. The energy deficit caused by the loss of neurons ability to synthesize ATP (adenosine triphosphate) is the main mechanism of cell death in the area of the cerebral infarction. The activity of Na+/K+ - ATPase decreases and there is an increased extracellular concentration of K+ ions and the uncontrolled influx of Na+, Ca2+, Cl− ions. Gradual depolarization of the cell membrane and loss of membrane potential are observed, which in turn increases the flow of sodium and allows for an osmotic transport of water to cells leading to the development of cytotoxic edema. As a consequence, the accumulation of Na+ and Ca2+ ions leads to organelle degeneration, loss of membrane integrity and cell death.20,21 The impairment of ATP synthesis also causes a reduction in the reuptake of glutamate, and its excessive extracellular accumulation leads to the ischemic death of neurons in the penumbra area. Overactivation of glutamate receptors causes excitotoxicity and accumulation of Ca2+ ions, leading to mitochondrial failure and apoptosis.22,23 The influx of Ca2+ ions activates catabolic enzymes by producing arachidonic acid and increasing the production of ROS mainly in neurons.20,24 An increased production of ROS enables the passage through the membrane not only of ions, but also of low-molecular-weight substances causing necrosis and, depending on the degree of neurons damage, a programmed death of the nerve cell. The excitotoxicity and growth of ROS activates microglia and astrocytes that secrete cytokines, chemokine and matrix metalloproteinases (MMP).16,25 These inflammatory mediators induce the expression of cell adhesion molecules on the endothelial surface (P-selectin, E-selectin, endothelial-leukocyte adhesion molecule (ELAM-1) and intercellular cell adhesion molecules-1 (ICAM-1)), which enable the neutrophils to infiltrate ischemic areas of the brain.8,25 In addition, endothelial cells increase the expression of chemokines in order to bring the leukocytes to the site of damage.9 Infiltrating immune cells, regardless of their beneficial role, can also damage the ischemic brain by producing various destructive immune cytotoxic mediators (including NO, ROS and prostanoids16,26), which prolong the inflammatory response, increasing the brain damage. They can also lead to secondary complications such as swelling and hemorrhagic transformation.8,27 The constant influx of leukocytes leads to lymphocytopenia, contributing to significant immunodepression which increases the risk of infection after stroke. In addition, the disproportionate concentration of pro-inflammatory mediators activates the autonomic nervous system, which leads to the inhibition of pro-inflammatory pathways and stimulation of anti–inflammatory mechanisms by the release of interleukins and growth factors.25,27 Furthermore, necrotic neurons release “hazard signals” that activate the immune system and secrete molecular patterns (DAMP).19,27 DAMP induces the Toll-like receptors (TLRs) on microglia, which in turn stimulates the NF-κB to synthesize most of the pro-inflammatory cytokines. NLRP3 inflammasome activates caspase-1 which eventually leads to the maturation and secretion of IL-1β and IL-18 cytokine .28 M1 microglia produce pro-inflammatory cytokines (IL-1β, interleukin 6 (IL-6), IL-18 and tumor necrosis factor (TNF-α)) and chemokines, while M2 microglia produce anti-inflammatory cytokines (interleukin 10 (IL-10), interleukin 4 (IL-4) and transforming growth factor-beta (TGF-β)),25 which are released a few days after acute brain damage leading to inhibition of inflammation.9,25
MMP, secreted by microglia, astrocytes and neurons, mediates in the destruction of the base plate, increasing BBB permeability and facilitating the entrance of additional peripheral immune cells into the brain area affected by stroke. This is because acute ischemia of the brain not only causes a local inflammatory reaction, but also triggers a systemic immune response.25,27
During the stroke, both congenital and adaptive immunity are involved, which do not affect the acute phase of damage. Nevertheless, modulation of adaptive immunity has an unusual protective effect on the ischemic brain and creates prospects for new stroke therapies.8,28 Immunomodulation is not devoid of harmful side effects, and a better understanding of the interaction between the immune system and the ischemic brain is necessary to exploit the full therapeutic potential of stroke immunology.
The Role of NLRP3 Inflammasome
Intracellular multiprotein complexes called inflammasomes play an important role in the innate immune response.
The best-known inflammasome is NLRP3 made of NLRP3 protein, ASC adaptor protein (apoptosis-associated speck-like protein containing a carboxy-terminal CARD) and procaspase-1.29–31
Activation of the NLRP3 inflammasome takes place in two stages: stage one (starting state) – preparation (signal 1) and stage two – activation stage (signal 2).
In the first stage, there is a release of molecular patterns (DAMP) which activate transmembrane TLRs, on immune cells, as a result of the activity of the signal from the damaged cells.27 TLR activation induces the expression of inactive protein NLRP3 and pro-IL-1β via NF-κB (signal 1).28,29 The NLRP3 resting form is placed at the endoplasmic reticulum (ER), and after activation, it is transferred to the cell nucleus region. There, together with other components of inflammasome, it contacts with ER and mitochondria.
In the second stage, the activation stage (signal 2), the NLRP3 central domain induces the oligomerization of an effector domain (PYD) that is a girder for the ASC adaptor protein, which is combined with procaspase-1 to form a protein complex – inflammasome NLRP3. Active inflammasome NLRP3 transforms (using the CARD domain) procaspase-1 into its active form, which triggers a pro-inflammatory cellular response by splitting the precursors IL-1β and IL-18 into their mature forms.29,31,32 This leads to a specific type of cell death called pyroptosis. In this way, inflammasomes contribute to the activation and promotion of congenital immunity, but also to the deterioration of tissue damage.19
The Role of Innate and Adaptive Immune Cells in the Production of Pro-Inflammatory Cytokines
There are ongoing clinical and experimental studies which highlight the complex role of the immune system in pathophysiological changes that occur after acute ischemic stroke of the brain. Molecular signals, which are generated by cerebral ischemia, activate components of the congenital immune system, leading to amplification of inflammatory cascades and tissue damage. At the same time, these processes stimulate a potentially harmful and cytoprotective immune adaptive response.20,28,33
Cells involved in the production of cytokines are representatives of the congenital and adaptive immune response:
- Microglia are a type of resident marrow cells of central nervous system (CNS). Their original role comes down to removing debris, repairing damaged cells and acting as CNS homeostasis regulator. They produce pro-inflammatory mediators such as TNF‐α, IL‐1β, IL-6 and ROS, NO after a stroke. It is also believed that they contribute to the inhibition of inflammation and tissue repair.28,34,35
- Perivascular macrophages occur between the vascular primary membrane and the brain surface. M1 macrophages produce pro-inflammatory cytokines (IL-1β, interleukin 12 (IL-12), interleukin 23 (IL-23) and TNF-α), chemokine, ROS and NO, thereby promoting the immune response of helper T cells type 1 (Th1).28
- Mast cells, mastocytes (MC) are found in meninges and cerebral vessels. MC granules store vasoactive substances (histamine), anticoagulants (heparin), protease (MMP2, MMP9) and cytokines (TNF‐α). They are capable of phagocytosis, antigen presentation and can modulate the adaptive immune response.28,36
- Blood monocytes are direct precursors of tissue macrophages. We distinguish two different populations: classical and pro-inflammatory monocytes that participate in the production of TNF‐α.28 After ischemia of the brain, inflammatory monocytes are quickly recruited to the site of damage where they activate macrophages.37
- Dendritic cells (DC) are cells that are a combination of congenital and adaptive immunity. Antigen-presenting cells (APC), mainly DC, are cells presenting antigen, containing a MHC class II antigen complex. They play a fundamental role in stimulating lymphocytes, especially virgin ones. After the presentation of the T-cells antigen CD4+, they undergo a clonal expansion in lymphatic organs, which is a process promoted by autocrine interleukin 2 (IL-2) production.28,34
- T cells cause ischemic damage after entering the brain parenchyma by releasing pro-inflammatory agents and cytokines such as reactive oxygen species, interferon γ (IFN-γ), TNF-α, IL‐1β, interleukin 17 (IL‐17) and interleukin 21 (IL‐21), which can cause cell destruction of neurovascular unit (NVU) or directly damage neurons.35
- Auxiliary T cells (Th) are responsible for coordinating and modulating immune responses. These include effector and regulator cells. Depending on molecular signals, there may be a development of different subpopulations of Th-cell effectors producing cytokines. IFN-γ and TNF are secreted by type 1 (Th1) cells, and their development is supported by IL-12. These cells lead to cytotoxicity.28,38
- Cells T γδ, activated by IL-23 are released from microglia/macrophages. They produce cytotoxic cytokine IL-17 and contribute to acute ischemic brain damage.39
- Natural killer T (NKT) cells cause cytotoxic effects by releasing IL-2, TNF.28
Interleukins
The main pro-inflammatory cytokines responsible for inducing the synthesis of APPs are: IL-6, IL-1 and tumor necrosis factor (TNF-alpha and TNF-beta).
IL-6 is released by different cells, including microglial cells, astrocytes, leukocytes and endothelial cells as part of a response to brain damage. It stimulates hepatocytes to synthesize APPs, mainly CRP (C-reactive protein) and fibrinogen.25,40,41 IL-6 activates APPs and affects the phosphorylation of the NF-IL-6 transcription factor responsible for enhancing the transcription of many genes. The synthesis of IL-6 requires IL-1 and TNF which stimulate fibroblasts, endothelial cells and keratinocytes, and increase the expression of IL-6.42 It is believed that IL-6 is a pleiotropic pro-inflammatory cytokine which increases the migration of leukocytes and regulates the production of chemokines and expression of adhesion molecules. It has also been shown that IL-6 is an important mediator of neurovascular dysfunction, neurodegeneration and neuritis.41,43 A number of reports also indicate that IL-6 inhibits the reciprocal release of TNF-α7,40,44 and IL-1, and stimulates the synthesis of their antagonists.
In recent years, a number of studies have been conducted to clarify the role of interleukins in the etiology and development of stroke. Although IL-6 is a pro-inflammatory cytokine, it has been suggested that it plays an important role in cerebral ischemia not only as a mediator of inflammatory process in the acute phase of stroke, but also as a neurotrophic factor in the late phase of the development of cerebral ischemia.45 It has been proven that IL-6 is actually a key inflammatory marker in stroke; numerous studies proved a significant increase in the concentration of IL-6 in serum, which occurred within a few hours after the onset of ischemia, and lasted for up to 90 days after the stroke.43,46-53 It has also been observed that the mean concentration of IL-6 in serum in patients with a symptomatic ICA stenosis is significantly higher than in healthy individuals in the period between 15th and 154th day after the onset of cerebral ischemia symptoms.54 Another study showed a similar phenomenon for up to 3 months after the onset of symptoms.55
There have been reports indicating that IL-6 is characterized by a high heterogeneity in humans. There is also little conclusive data on the role of this cytokine in the early (<4 to 6 hrs) post-stroke phase.
The presence of the IL-6-174 GC genotype and a higher concentration of IL-6 receptors increase the likelihood of a stroke12,56 as is the case in an acute myocardial infarction and other vascular diseases.56–58
The relationship between the level of IL-6 in serum, the severity of neurological deficit as well as prognosis concerning patients with an acute ischemic stroke and the volume of an ischemic stroke is an object of interest of many researchers. Many authors indicate that the increase in the concentration of IL-6 in serum within the first 24 hrs following a stroke is associated with a deterioration of patients’ functional state43,55,59,60 and greater spectrum covered by the lesion volume61,62 and they consider the level of IL-6 to be a reliable prognostic factor in the acute phase of ischemic stroke. It has been shown that the increase in the concentration of IL-6 in serum on the 1st and 6th day after the ischemic stroke is associated with long-term post-stroke prognosis post-stroke disability degree and mortality at 3 months and/or 1 year following a stroke.43 Based on a group of patients suffering from an ischemic stroke, Shenhar-Tsarfaty et al52,55 demonstrated that an increase IL-6 in serum within the first 24 hrs after the hospital admission is a highly sensitive stroke biomarker and also is an important prognostic factor for the survival of stroke patients within first year.55 Moreover, the authors demonstrated that the IL-6 level of 6.47 pg/mL is a threshold above which the chance of survival concerning patients with an acute stroke is reduced.52 The baseline level of IL-6 can impact the severity of stroke and should be considered as a prognostic biomarker for the development of clinical complications arising from acute ischemia.
On the other hand, Nakase et al63 proved that the assessment of IL-6 concentration on admission and in 28 days acute cerebral ischemia to establish a prognosis within 1 year from the stroke onset. What is more, Fahmi and Elsaid64 demonstrated a synergistic effect of the level of IL-6 in serum and the size of an ischemic focus as assessed in CT and MRI on a prognosis in stroke patients. Rodríguez-Yáñez and Castillo65 also observed that an increase in the levels of IL-6 and TNF-α in serum is associated with the degree of neurological deficit and the increase in the ischemic volume. A similar relationship has been established for IL-6 in the cerebrospinal fluid of stroke patients. Beridze et al66 demonstrated that the increased concentration of IL-6 in cerebrospinal fluid in hour 6 after the onset of stroke correlates with the degree of neurological deficit, as assessed on days 1 and 7 after the stroke using the National Institute of Health Stroke Scale (NIHSS), and with the functional deficit measured using the Barthel Index (BI) scale. Recent reports also indicate a relationship between a stroke etiology and the levels of pro-inflammatory cytokines in serum. The highest concentration of IL-6 in plasma and other immune-inflammatory and thromboembolic/fibrinolytic agents (PAI-1, TPA) was found in patients with cardiogenic stroke caused by a cardiac embolism.48,67
This group of patients was observed with not only an increased immune-inflammatory activity but also with a greater degree of neurological deficit as assessed according to the Scandinavian Stroke Scale (SSS).48 In a lacunar stroke, which is caused by cerebral microangiopathy, lower levels of pro-inflammatory cytokines (IL-6 and TNF-α) in plasma and lower P-selectin and ICAM-1 (intercellular adhesion molecule-1) were observed.49
Not all available literature reports confirm the relationship between the increase in the concentration of IL-6 and prognosis in stroke patients. For example, Worthmann et al51 based on their assessment of the level of inflammatory markers in 69 patients with a transient ischemic attack (TIA) or an ischemic stroke conducted 6, 12 and 24 hrs, and 3 and 7 days after the onset of symptoms, proved that a gradual increase in the levels of IL-6, MPC-1 and S100 protein (marker of damage to the BBB) is not associated with the degree of neurological and functional deficit assessed 90 days after the stroke. Similarly, Oto et al68 and Ormstad et al44 observed that IL-1, IL-6, IL-10 and TNF-α are not significant prognostic factors in neurological assessments conducted more than 1 month after a stroke, despite the observed high concentrations of IL-6 in serum in the early phase of hemorrhagic strokes. Whiteley et al also reported that the levels of IL-6 and N-terminal pro-brain natriuretic peptide (NT-pro-BNP) are not prognostic factors in the clinical assessment of stroke patients.69,70
Other conclusions were presented by Sotgiu et al47 who noticed a negative correlation between the level of IL-6 and the size of cerebral infarction. The authors concluded that in the context of the complex activity of pro-inflammatory network in an ischemic stroke, IL-6 is a neuroprotective and not a neurotoxic factor. In the literature, there have also been data from experimental models of a stroke that indicate a neuroprotective function of IL-6. Studies conducted using the pMCAo and tMCAo stroke models in mice have shown, eg, that although administration of IL-6 immediately after the reperfusion does not decrease the ischemic volume, it improves the functional status,71 which also confirms the neuroprotective effect of IL-6. Grønhøj et al72 demonstrated that intravenous administration of IL-6 reduces the stroke volume in mice and improves functional status. It has also been shown that IL-6 produced by damaged brain cells furthers angiogenesis after a stroke.73 IL-6 can not only be an interesting and important prognostic biomarker for the outcome after cerebral ischemia, but also can be used as a neuroprotective biomolecule as part of a therapy.
Several regulatory molecules are involved in inflammation, but the most known inflammatory mediator for acute brain injury is cytokine - interleukin-1. After stroke, IL-1β stimulates the expression of other inflammatory mediators such as cytokines and adhesion molecules.
IL-1 is a pro-inflammatory cytokine that acts as a messenger molecule. It occurs as two forms: IL-1α (intracellular) and IL-1β (secreted). In reference to the plausible role of the IL-1 family as stroke biomarkers, there is some controversy. In addition to the neurotoxic effect, it can also activate astrocytes for the production of survival-promoting factors.27 Distinct studies have revealed that ischemic stroke patients have higher circulating levels of IL-1β within 24 hrs after the ischemic event when compared with controls.44 However, two other studies have reported that IL-1β levels in serum or plasma are not higher in stroke patients when compared with healthy controls, at 12, 24 and 72 hrs intervals after the onset of symptoms.74
The clinical potential of the recombinant human IL-1 receptor antagonist (IL-1Ra - anakinra) in ischemic stroke was tested in clinical studies.
Patients receiving anakinra (100 mg loading dose over 60 s, followed by a 2 mg/kg/hr infusion over 72 hrs) had a reduced inflammatory response, with lower levels of leukocytes, CRP and IL-6 in the blood. The obtained clinical results were better than in the placebo group, this fact may be caused by the reversal of peripheral immunosuppression, because in patients treated with anakinra the concentration of pro-inflammatory mediators: TNFα, IL-6 and IL-10 are reduced.75
Similarly, subcutaneous administration of IL-1Ra significantly reduced the inflammatory response, but no improvement in clinical outcomes was seen compared to the placebo group.76 Further experimental studies are needed to assess the effectiveness of anakinra in reducing infarction volume to better understand the role of IL-1 in stroke pathophysiology.25,26
TNF-α in Strokes
TNF-α, previously called cachectin or differentiation factor, is one of the main protein involved in an inflammatory and immune response. This pro-inflammatory cytokine is produced by monocytes, T cells, mast cells, macrophages, neutrophils, keratinocytes and fibroblasts.77–79 It has been demonstrated that TNF-α stimulates brain pericytes (stem cells covering the surface of capillary blood vessels) to increase the synthesis of IL-6 by activating NF-κB.25,41 TNF-α has two forms: a transmembrane form (tmTNF-α), which regulates inflammation locally via cell–cell interactions, and a soluble, biologically active form (sTNF-α)80 generated by the tumor necrosis factor-alpha converting enzyme (TACE). The soluble form acts systemically by increasing the phagocytic and cytotoxic activity of macrophages and upregulating the expression of other cytokines, such as IL-6 and IL-1. TNF does not only act systemically, it also acts locally in the brain. TNF-α acts via receptors (TNFR-1 and TNFR-2) that differ in their affinity for TNF-α and the degree of glycosylation. sTNF-α is mediated by TNFR1, while tmTNF-α is mediated by TNFR-2 and TNFR-1.
It is believed that TNF-α plays an important role in the pathophysiology of stroke. With regard to the experimental models of strokes, TNF-α is probably one of the most widely studied cytokines.25 The results of these studies indicate both a neurotoxic and a neuroprotective effect of TNF-α in strokes.81,82
The process of binding and inhibiting TNF-α and its receptors in mice, using intravenous or intraperitoneal TNFbp (TNFR-1 linked with polyethylene glycol) has resulted in a more extensive ischemic area in the experimental MCAo model cerebral ischemia, which formed a basis to assume a neuroprotective role of TNF-α. It has also been demonstrated that the pathway involving TNF-α, its receptor (TNFR-1) and NF-κB is linked with intracerebral neuroprotection.81 While Khaksar et al83 show that cannabidiol administration to rats is neuroprotective in part by inhibiting TNFR-1 signaling.
On the other hand, Arango-Dávila et al84 observed a reduction in brain ischemia volume in rats after injection of protein binding TNF-α. The suggested neurotoxic effect of TNF-α depends on TNFR-1 which triggers processes leading to the release of cytochrome c from mitochondria into cytosol and, consequently, to the loss of nerve cells.85
The results of studies looking at concentration of TNF-α in serum are also ambiguous. A number of studies have shown that the level of TNF-α does not change both within the first 24 and 72 hrs, and during the first week following the onset of ischemia.44,86 Moreover, Puz et al54 did not observe any significant differences in the TNF-α concentration in case of a presence or absence of a carotid artery stenosis, or by comparing patient groups with a symptomatic and asymptomatic stenosis. A peripheral inflammatory response to acute cerebral ischemia should not be expected to occur only within the first few hours following a sudden failure in cerebral circulation; therefore, no relationship between TNF-α and the majority of the analyzed inflammatory parameters (S100, fibrinogen, leukocytes) has been observed in stroke patients.86
In the literature, there have also been reports indicating that TNF-α is one of the first cytokine to appear as part of an inflammatory response to ischemic brain damage47–49,87 and contributes to triggering the cascade of other inflammatory components in both blood serum and the cerebrospinal fluid.88 The authors claim that TNF-α is a sensitive parameter determining the onset of an inflammatory response and a useful indicator for the assessment of prognosis.65,89 An increase in the concentration of TNF-α is observed in stroke patients as soon as 6–12 hrs after the onset of symptoms.47 An increase in TNF-α concentration within 24 and 48 hrs after a stroke has also been shown, and the observed slow decrease occurring within 72 and 144 hrs after a stroke correlates with a clinical improvement in patients in the acute phase of ischemic stroke.53 Similar correlations have been observed for long-term prognosis in a group of 286 patients who were followed up on admission to the hospital, as well as after 2 weeks and up to 1 year after the stroke. With regards to these patients, higher TNF-α concentrations and the presence of the −850 and −308 AT alleles were associated with a poor prognosis concerning the patient’s status after 1 year from the stroke.90 TNF-alpha is an immunomodulating cytokine involved in neuroinflammation and neuronal damage in response to ischemia. Therefore, the use of its inhibitors in the treatment of ischemic stroke should be considered.
Chemokines
Chemokines, ie, chemotactic cytokines, are one of the major groups of biologically active pro-inflammatory molecules. Their main function is stimulation of leukocyte chemotaxis and control of leukocyte migration from blood into tissues, which is of particular importance in the development of inflammation and suggests participation of these proteins in the etiology of many diseases.91–93 Chemokines are a family of over 50 proteins with a molecular weight of 8–12 kDa. Their structure, consists of at least 3 subunits: N-terminal domain, β-sheet subunit and C-terminal α-helix. Despite differences in the amino-acid sequence, these proteins present a similar tertiary structure that is stabilized by disulfide bonds.94
Chemokines contain 2–4 cysteine residues. Depending on the number and location of these residues, they are divided into the following groups:95
- CC – two cysteine residues, immediately adjacent,
- CXC – two cysteine residues separated by one amino acid,
- CX3C – two cysteine residues separated by three amino acids.
Chemokines bind their target cells via specific chemokine receptors and activate cellular response. Approximately 20 chemokine receptors have been identified.96 Their structure consists of 7 transmembrane domains coupled with G protein (G protein-coupled receptors, GPCRs).91,97,98
Based on their ability to bind ligand, chemokine receptors can be divided into the following classes: CRCCR, CXC-R, CX3C-R. It has been demonstrated that one receptor can bind many chemokines, and every ligand can show affinity for more than one receptor.99
Selected Chemokines in Strokes
BBB plays a key role in the central nervous system in which it maintains homeostasis and constitutes a system of protection by providing a selective connection between the circulatory system, nervous tissues and cerebrospinal fluid (blood–cerebrospinal fluid barrier). Ischemic strokes are caused by blood clot for example and result in the disruption of the BBB, which leads to a massive migration of not only morphotic blood elements, but also mediators of inflammatory response, including chemotactic cytokines, between the nervous tissue and blood vessels.100–102
In recent years, more attention has been paid to these markers, confirming their significant contribution to the occurrence of cerebral ischemic lesions.103–105
Chemokines and their receptors act as both trophic and protective modules in the nervous system, thus increasing the survival of neurons. What is important, they not only regulate neuronal metabolism, but also affect their synaptic transmission. Chemokines can be released by neurons, astrocytes, microglial cells, oligodendrocytes and the endothelial cells of brain blood vessels.106,107 An increased expression of mRNA for the CCL5 (RANTES) protein was recently noted in the neurons producing dopamine.108
Chemokines show specificity for different types of leukocytes, which is particularly important in pathological conditions. Their contribution has been found in, eg, cardiovascular diseases, strokes and many inflammatory and autoimmune diseases, as well as in brain damage.103,109-114
Increased expressions of chemokine genes in the brain can be caused not only by ischemia, but also by, eg, axonal damage or neurotoxins.115
Although the contribution of chemokines to the pathogenesis of stroke has been demonstrated in both animal and human models, this issue still remains controversial. Mirabelli-Badenier et al104 suggest that CC- and CXC-type chemokines direct the inflammatory response during a stroke and modulate the migration of stem cells into tissues, which furthers tissue repair and could be used in the future treatment of stroke patients. In experimental in vivo studies in rats in which an ischemic stroke was induced by occluding the middle cerebral artery (MCAo), Jiang et al observed that after 24 hrs from the MCAo, an increase in expression of CCL2 and CCL3 occurred in the ischemic region of the brain, as well as did an increase in infiltration of the damaged part by HUCBCs (human umbilical cord blood cells).116
Knocking out the CCR2 receptor of chemokine MCP-1 (CCL2) in animal models results in the abolishment of cell migration and the reduction of size of the ischemic area. An increased expression of mRNA for the chemokines CCL2 and CCL3 was observed after 2 hrs of ischemia, there was an increase in the concentration of these chemokines in the brain during the following 48 hrs. Similar conclusions were drawn for CCR2 (-/-) knockout mice, in which transient focal ischemia was induced and reduced inflammatory response resulting in a decreased size of an ischemic lesion, decreased brain edema and significantly lower permeability of the blood-brain barrier was observed.117
A decreased stroke volume and attenuated cellular response have been observed in rats with mutated form of MCP-1 via adenovirus in the lateral ventricle after induction of ischemic stroke, which clearly indicates that the anti-MCP-1 gene therapy can be a promising approach to the treatment of ischemic stroke.118
Another chemokine with a potentially significant role in strokes is CX3CL1 (fractalkine), along with its receptor CX3CR1. In an earlier study, Tarozzo et al119 demonstrated a transient increase in the CX3CL concentration in the infarct (necrotic) focus 12 hrs after the ischemia, and an increase in the CX3CL1 concentration in the penumbra region within 48 hrs of the hypoperfusion in rats in which strokes were induced with the use of the MCAo model. In the further follow-up, ie within 7 days of the MCAo, a decrease in the CX3CL concentration to the baseline value was observed. An increase in the CX3CL concentration was also detected in vascular endothelial cells in the period between the 2nd and 7th day of ischemia, and the presence of CX3CR1 was found in microglial cells 24 hrs, 48 hrs and 7 days after the stroke.
The role of fractalkine in the pathogenesis of ischemic stroke is not fully understood, but experimental studies suggest that it can be both a pro-inflammatory mediator and a neuroprotective agent.120 Based on their study, Dénes et al demonstrated that animals with a CX3CR1 deficiency (±) present a significantly lower risk of ischemic stroke (56%) and risk of damage to the blood-brain barrier comparing to wild-type mice and CX3CR1 (±) heterozygous mice.121
These results indicate a possible role of fractalkine in the increase of ischemic injuries and mortality after transient focal cerebral ischemia. Cipriani et al122 conducted a similar study in CX3CL1 (-/-) and CX3CR1 (-/-) mice. They observed a reduction in the degree of stroke lesion volume after MCAo in both CX3CL1 (-/-) and CX3CR1 (-/-) mice. The same authors also showed a neuroprotective effect of exogenous fractalkine, which was administered into wild-type mice in a narrow range of concentrations, shortly before MCAo. This resulted in a reduction of the next infarct volume. In the case of CX3CR1 (-/-) mice, fractalkine did not affect the size of infarction, while in CX3CL1 (-/-) mice, an increased size of infarction was observed. The neuroprotective activity of CX3CL1 depends on its effect on microglia which, under in vitro conditions, requires activation of adenosine A1 receptor (A1R). The neuroprotective effect of CX3CL1 in MCAo was abolished based on the presence of the A1R antagonist 1,3-dipropyl-8-cyclopentyl xanthine and in A1R (-/-) mice.122 Based on the presented results, it can be stated that a disrupted communication system between neurons and microglia can have an impact on the function of fractalkine in ischemic damage, which suggests that fractalkine CX3CR1 signaling in microglia is important for the proper function of these cells.
On the other hand, Terao et al123 revealed a decreased volume of the ischemic focus and a significantly limited BBB permeability, along with a reduced leukocyte and platelet adhesion, 60 mins after the MCAo in CCL5-knockout mice in comparison with wild-type mice. It has also been found that the protein encoded by the CCL5 gene increases brain damage by reactivating other strong pro-inflammatory cytokines123 and induces proliferation of oligodendrocyte precursor-like cells (Oli-neu).124 The expression of CCL5 mRNA and protein along with its receptors, in selected populations of brain cells also suggests the participation of this chemokine in the communication between neurons and glial cells.56 A neuroprotective effect of CCL5 has also been hypothesized.125–127
Molecular studies in animals (mouse) have shown that CCL5 affects neurons via the CCR3 and CCR5 receptors and can have a direct or indirect protective effect on neurons by inducing the release of neurotrophic factors in the pericerebral area.128 It has been demonstrated that removal of the CCR5 receptor gene has a protective effect against cerebral ischemia and reperfusion injury.129 Administration of a synthetic cyclic peptide based on CCL5 (MKEY), which is an inhibitor of the CXCL4–CCL5 heterodimer formation and enhances of the activity of CCL5 in the signal transduction, adhesion, and migration of monocytes during an inflammatory process in mice, reduces the volume of ischemic tissue by decreasing the intensity and strength of the inflammatory process.130 Reports from recent years revealed that CCR5 is important when it comes to regulatory T cell (Treg)-dependent protection of the BBB, which is an important factor in optimizing the therapy for the acute-phase of cerebral stroke.130 Tregs up-regulate PDL1 at the cell surface, which allow them to interact with PD1, expressed at the surface of neutrophils, thereby inhibiting MMP9 expression, and protecting the BBB.131
In a study in stroke patients, Montecucco et al132 observed an increase in the serum concentration of CCL5 in the first month after an ischemic stroke. However, they did not find a correlation between the concentration of CCL5 and the degree of neurological deficit. Zaremba et al133 in a study in 27 patients did not observe an increase in the concentration of CCL5 and CCL2, but observed an increase in CCL3 within the first 3 days following the occurrence of a stroke. The levels of CCL2 and CCL3 determined in blood serum on the 1st, 2nd, and 3rd day of the stroke correlated negatively with the degree of functional deficit, which was measured on the 28th day with the use of the BI. The levels of CCL2 assessed on the 2nd and 3rd day of the stroke also correlated with the BI score on 14th day. Interestingly, a negative correlation between the serum CCL5 level determined on the 2nd day of the stroke and the BI score on the 28th day was also found. No similar correlation was observed for this and other chemokines in relation to the degree of neurological deficit evaluated using the SSS. Other researchers Canoui-Poitrine et al134 also demonstrated that the increase in the concentration of CCL5 in serum constitutes an independent prognostic factor with regards to an ischemic stroke. Similarly, a higher concentration of CCL5 was observed in the acute-phase in a group of 171 patients suffering from ischemic strokes compared to a control group of healthy individuals.128
The characteristics of inflammatory mediators and their results are presented in Table 1.
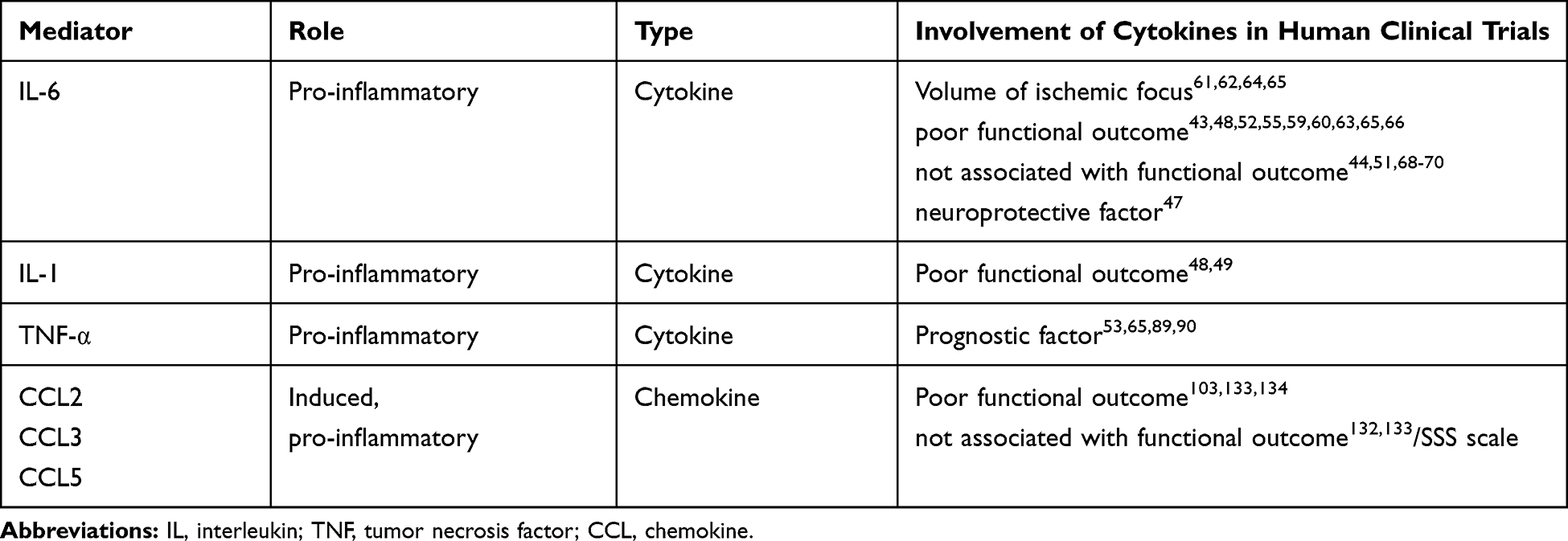 |
Table 1 Inflammatory Mediators and Outcomes in Human Stroke |
It should be mentioned that cytokines are not parameters for which clinical norm ranges are determined.90 Table 2 presents some concentration values for IL-6, IL-1β and TNF-α and chemokines. These markers of inflammation are determined at different time intervals, with different methods and for different types of stroke.
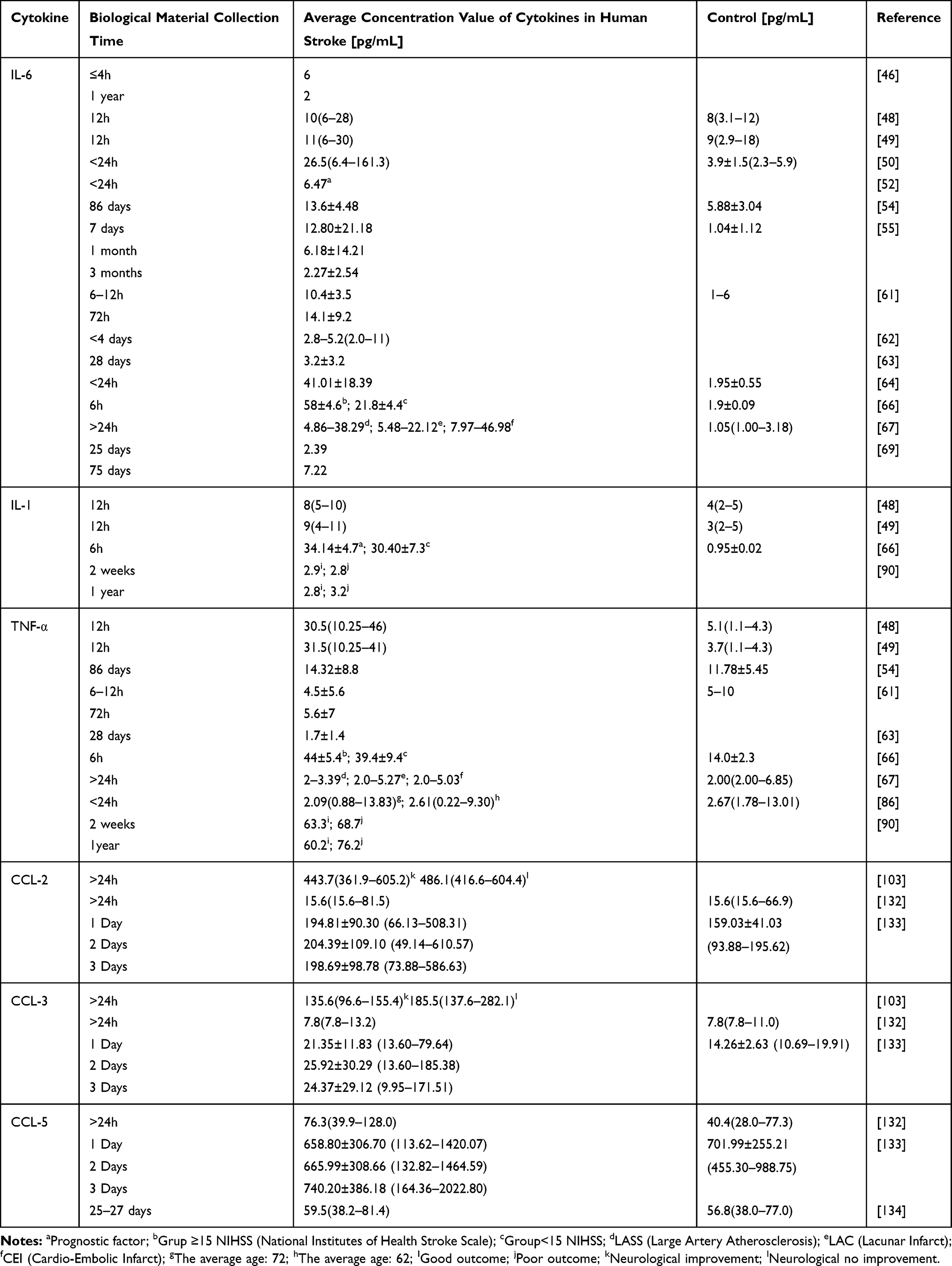 |
Table 2 Some Values of Cytokine Concentrations at Different Times for Different Types of Strokes |
Conclusions
The inflammatory response to stroke is extremely complex, with multiphasic pro-inflammatory responses. Determination of cytokine levels in the acute phase of stroke can contribute to a thorough understanding of their role in the immune response observed during brain ischemia. It should also allow the identification of interactions between the immune system and brain cells during post-stroke recovery and regeneration. This knowledge might permit the modification of immune response in order to limit the damage to the nervous tissue, which may affect outcome after stroke. Therefore, they would serve as prognostic factor in acute cerebral ischemia. Particularly, IL-6 potentially appears as a good prognostic factor in stroke and potential biomarker of acute cerebral ischemia. However, further research is necessary to assess the therapeutic implications of IL-6. It has been shown in experimental models that inhibition of TNF-α, CCL-2, CCL-3 and CCL-5 could be a promising tool in the treatment of acute stroke. Moreover, administration of the recombinant human IL-1 receptor antagonist a significantly may have important in reducing the inflammatory response. In order to evaluate the therapeutic the cytokines potential inhibition, clinical trials in patients with ischemic stroke are necessary.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Feigin VL, Norrving B, Mensah GA. Global burden of stroke. Circ Res. 2017;20:439–448.
2. Grupa Ekspertów Sekcji Chorób Naczyniowych Polskiego Towarzystwa Neurologicznego. Postępowanie w udarze mózgu Wytyczne Grupy Ekspertów Sekcji Chorób Naczyniowych Polskiego Towarzystwa Neurologicznego. Neurol Neurochir Pol. 2012;46.
3. Kozera G, Sobolewski P, Serafin Z. Doświadczenie dwóch dekad leczenia trombolitycznego udaru niedokrwiennego mózgu: aktualne pytania i odpowiedzi. Gdansk: Wydawnictwo AsteriaMed; 2017.
4. Wein T, Lindsay MP, Côté R, et al. Canadian Stroke Best Practice Canadian stroke best practice recommendations: secondary prevention of stroke, sixth edition practice guidelines, update 2017. Int J Stroke. 2018;13:420–443. doi:10.1177/1747493017743062
5. Lackland DT, Roccella EJ, Deutsch AF, et al. Factors influencing the decline in stroke mortality: a statement from the American heart association/American stroke association. Stroke. 2014;45:315–353. doi:10.1161/01.str.0000437068.30550.cf
6. Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics —2018 update: a report from the American Heart Association. Circ Res. 2018;137:67–492.
7. Zaremba J, Losy WJ, Selmaj K. Immunologiczne aspekty udaru mózgu, Neuroimmunologia Kliniczna Czelej (Lublin). 2007;14:261–279.
8. Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009;7:97–107.
9. Mizum A, Yenari MA. Anti-inflammatory targets for the treatment of reperfusion injury in stroke. Front Neurol. 2017;8:467–487. doi:10.3389/fneur.2017.00467
10. Kim NS, Ko MM, Cha MH, et al. Age and sex dependent genetic effects of neuropeptide Y promoter polymorphism on susceptibility to ischemic stroke in Koreans. Clin Chim Acta. 2010;411:1243–1247. doi:10.1016/j.cca.2010.04.026
11. Pera J. Polski Przegląd badań genetycznych i neuroobrazowych w diagnostyce udarów mózgu o rzadkiej etiologii. Pol Prz Neurol. 2017;13:1–9.
12. Bazina A, Sertić J, Mišmaš A, et al. PPARγ and IL-6 −174G>C gene variants in Croatian patients with ischemic stroke. Gene. 2015;560:200–204. doi:10.1016/j.gene.2015.02.003
13. Gromadzka G. Genetyczne uwarunkowania udaru mózgu. Pol Prz Neurol. 2011;7:53–72.
14. Kim DH, Yoo SD, Chon J, et al. Interleukin-6 receptor polymorphisms contribute to the neurological status of korean patients with ischemic stroke. J Korean Med Sci. 2016;31:430–434. doi:10.3346/jkms.2016.31.3.430
15. Powers WJ, Rabinstein AA, Ackerson TI, et al. American Heart Association Stroke Council: 2018 Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2018;49:46–110. doi:10.1161/STR.0000000000000158
16. Simats A, García-berrocoso T, Montaner J. Neuroinflammatory biomarkers: from stroke diagnosis and prognosis to therapy. BBA Mol Basis Dis. 2016;1862:411–424. doi:10.1016/j.bbadis.2015.10.025
17. Rose JS, Scheller J, Elson G, et al. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukocyte Biol. 2006;80:227–236. doi:10.1189/jlb.1105674
18. Abayev M, Rodrigues JP, Srivastava G, et al. The solution structure of monomeric CCL5 in complex with a doubly sulfated N-terminal segment of CCR5. FEBS J. 2018;285:1988–2003. doi:10.1111/febs.14460
19. Vidale S, Consolib A, Arnaboldia M, et al. Postischemic Inflammation in Acute Stroke. J Clin Neurol. 2017;13(1):1–9. doi:10.3988/jcn.2017.13.1.1
20. Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568.
21. Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67:181–198. doi:10.1016/j.neuron.2010.07.002
22. Zhang QG, Xu YL, Li HC, et al. NMDA receptor/LVGCC-dependent expression and AMPA/KA receptor-dependent activation of c-Jun induced by cerebral ischemia in rat hippocampus. Neurosci Lett. 2006;398:268–273. doi:10.1016/j.neulet.2006.01.005
23. Li XM, Yang JM, Hu DH, et al. Contribution of downregulation of L-type calcium currents to delayed neuronal death in rat hippocampus after global cerebral ischemia and reperfusion. J Neurosci. 2007;27:5249–5259. doi:10.1523/JNEUROSCI.0802-07.2007
24. Kahlert S, Zündorf G, Reiser G. Glutamate-mediated influx of extracellular Ca2+ is coupled with reactive oxygen species generation in cultured hippocampal neurons but not in astrocytes. J Neurosci Res. 2005;79:262–271. doi:10.1002/jnr.v79:1/2
25. Ramiro L, Simats A, Berrocoso TG, et al. Inflammatory molecules might become both biomarkers and therapeutic targets for stroke management. Ther Adv Neurol Diso. 2018;11:1–24.
26. Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147:232–240. doi:10.1038/sj.bjp.0706400
27. Bustamante A, Simats A, Vilar-bergua A, et al. Blood/brain biomarkers of inflammation after stroke and their association with outcome: from C-reactive protein to damage-associated molecular patterns. Neurotherapeutics. 2016;13:671–684. doi:10.1007/s13311-016-0470-2
28. Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi:10.1038/nm.2399
29. Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res. 2015;8:15–27. doi:10.2147/JIR.S51250
30. Hong P, Gu RN, Li FX, et al. NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J Neuroinflamm. 2019;16:121. doi:10.1186/s12974-019-1498-0
31. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10:128–139. doi:10.1038/s41419-019-1413-8
32. ChS Y, Kim JJ, Kim TS, et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat Commun. 2015;6:6115–6126. doi:10.1038/ncomms7115
33. Chamorro Á, Meisel A, Planas AM, et al. The immunology of acute stroke. Nat Rev Neurol. 2012;8:401–410. doi:10.1038/nrneurol.2012.98
34. Amantea D, Micieli G, Tassorelli C, et al. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front Neurosci. 2015;9:1–19. doi:10.3389/fnins.2015.00147
35. Rayasam A, Hsu M, Kijak JA, et al. Immune responses in stroke: how the immune system contributes to damage and healing after stroke and how this knowledge could be translated to better cures? Immunology. 2018;154(3):363–376. doi:10.1111/imm.2018.154.issue-3
36. Rao KN, Brown MA. Mast cells: multifaceted immune cells with diverse roles in health and disease. Ann N Y Acad Sci. 2008;1143:83–104. doi:10.1196/nyas.2008.1143.issue-1
37. Felger JC, Abe T, Kaunzner UW, et al. Brain dendritic cells in ischemic stroke: time course, activation state, and origin. Brain Behav Immun. 2010;24(5):724–737. doi:10.1016/j.bbi.2009.11.002
38. Wan YY. Multi-tasking of helper T cells. Immunology. 2010;130:166–171. doi:10.1111/imm.2010.130.issue-2
39. Shichita T, Sugiyama Y, Ooboshi H, et al. Pivotal role of cerebral interleukin-17-producing gamma delta T cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15(8):946–950. doi:10.1038/nm.1999
40. Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. 2012;8:1254–1266. doi:10.7150/ijbs.4679
41. Matsumoto J, Dohgu S, Takata F, et al. TNF-α-sensitive brain pericytes activate microglia by releasing IL-6 through cooperation between IjB-NFjB and JAK-STAT3 pathways. Brain Res. 2018;1692:34–44. doi:10.1016/j.brainres.2018.04.023
42. Gołąb J, Jakóbisiak M, Lasek W, et al. Cytokiny. Immunologia PWN Warszawa; 2017:108–151.
43. Shaafi S, Sharifipour E, Rahmanifar R, et al. Interleukin-6, a reliable prognostic factor for ischemic stroke. Iran J Neurol. 2014;13:70–76.
44. Ormstad H, Aass HCD, Lund-sørensen N, et al. Serum levels of cytokines and C-reactive protein in acute ischemic stroke patients, and their relationship to stroke lateralization, type, and infarct volume. J Neurol. 2012;258:677–685. doi:10.1007/s00415-011-6006-0
45. Suzuki S, Tanaka K, Suzuki N. Ambivalent aspects of interleukin-6 in cerebral ischemia: inflammatory versus neurotrophic aspects. J Cereb Blood Flow Metab. 2009;29:464–479. doi:10.1038/jcbfm.2008.141
46. Waje-andreassen U, Kråkenes J, Ulvestad E, et al. IL-6: an early marker for outcome in acute ischemic stroke. Acta Neurol Scand. 2005;111:360–365. doi:10.1111/ane.2005.111.issue-6
47. Sotgiu S, Zanda B, Marchetti B, et al. Inflammatory biomarkers in blood of patients with acute brain ischemia. Eur J Neurol. 2006;13:505–513. doi:10.1111/ene.2006.13.issue-5
48. Tuttolomondo A, Sciacca RD, Raimondo DD, et al. Plasma levels of inflammatory and thrombotic/fibrinolytic markers in acute ischemic strokes. Relationship with TOAST subtype, outcome and infarct site. J Neuroimmunol. 2009;215:84–89. doi:10.1016/j.jneuroim.2009.06.019
49. Licata G, Tuttolomondo A, Raimondo DD, et al. Immuno-inflammatory activation in acute cardio-embolic strokes in comparison with other subtypes of ischaemic stroke. J Thromb Haemost. 2009;101:929–937. doi:10.1160/TH08-06-0375
50. Cojocaru IM, Cojocaru M, Tănăsescu R, et al. Expression of IL-6 activity in patients with acute ischemic stroke. Rom J Intern Med. 2009;47:393–396.
51. Worthmann H, Tryc AB, Goldbecker A. The temporal profile of inflammatory markers and mediators in blood after acute ischemic stroke differs depending on stroke outcome. Cerebrovasc Dis. 2010;30:85–92. doi:10.1159/000314624
52. Shenhar‐tsarfaty S, Ben E, Assayag I, et al. Interleukin‐6 as an early predictor for one‐year survival following an ischaemic stroke/transient ischaemic attack. Int J Stroke. 2010;5:16–20. doi:10.1111/j.1747-4949.2009.00396.x
53. Nayak AR, Kashyap SR, Kabra D, et al. Time course of inflammatory cytokines in acute ischemic stroke patients and their relation to inter-alfa trypsin inhibitor heavy chain 4 and outcome. Ann Indian Acad Neurol. 2012;15:181–185. doi:10.4103/0972-2327.99707
54. Puz P, Lasek-bal A, Kazibutowska Z. Evaluation of the serum concentration of selected inflammatory cytokines in patients with carotid artery stenosis. Chir Pol. 2014;16:57–65.
55. Choudhary S, Chowdhur D, Mishra TK, et al. Temporal profile of serum levels of IL-6 in acute ischemic stroke and its relationship with stroke severity and outcome in indian population. Int J Intg Med Sci. 2018;5:555–560.
56. Boehme AK, McClure LA, Zhang Y, et al. Inflammatory markers and outcomes after lacunar stroke: levels of inflammatory markers in treatment of stroke study. Stroke. 2016;47:659–667. doi:10.1161/STROKEAHA.115.012166
57. Libby P, Rocha VZ. All roads lead to IL-6: a central hub of cardiometabolic signaling. Int J Cardiol. 2018;15:213–215. doi:10.1016/j.ijcard.2018.02.062
58. Groot HE, Lawien AA, Horst ICC, et al. Plasma interleukin 6 levels are associated with cardiac function after ST-elevation myocardial infarction. Clin Res Cardiol. 2019;108:612–621. doi:10.1007/s00392-018-1387-z
59. Bustamante A, Sobrino T, Giralt D, et al. Prognostic value of blood interleukin-6 in the prediction of functional outcome after stroke: a systematic review and meta-analysis. J Neuroimmunol. 2014;15:215–224.
60. Dobrucka-głowacka A, Sarecka-hujar B, Raczkiewicz D, et al. Interleukin-6 in cardiovascular disorders. Eur J Med Technol. 2018;3:23–30.
61. Martínez-sánchez P, Gutiérrez-fernández M, Fuentes B, et al. Biochemical and inflammatory biomarkers in ischemic stroke: translational study between humans and two experimental rat models. J Transl Med. 2014;3:220–229. doi:10.1186/s12967-014-0220-3
62. Hotter B, Hoffmann S, Ulm L, et al. IL-6 Plasma levels correlate with cerebral perfusion deficits and infarct sizes in stroke patients without associated infections. Front Neurol. 2019;10:1–8. doi:10.3389/fneur.2019.00083
63. Nakase T, Yamazaki T, Ogura N, et al. The impact of inflammation on the pathogenesis and prognosis of ischemic stroke. J Neurol Sci. 2008;271:104–109. doi:10.1016/j.jns.2008.03.020
64. Fahmi RM, Elsaid AF. Infarction size, interleukin-6, and their interaction are predictors of short-term stroke outcome in young egyptian adults. J Stroke Cerebrovasc Dis. 2016;25:2475–2481. doi:10.1016/j.jstrokecerebrovasdis.2016.06.021
65. Rodríguez-yáñez M, Castillo J. Role of inflammatory markers in brain ischemia. Curr Opin Neurol. 2008;21:353–357. doi:10.1097/WCO.0b013e3282ffafbf
66. Beridze M, Sanikidze T, Shakarishvilil R, et al. Selected acute phase CSF factors in ischemic stroke: findings and prognostic value. BMC Neurol. 2011;11:41–48. doi:10.1186/1471-2377-11-41
67. Lehmann MF, Kallaur AP, Oliveira SR, et al. Inflammatory and metabolic markers and short-time outcome in patients with acute ischemic stroke in relation to TOAST subtypes. Metab Brain Dis. 2015;30:1417–1428. doi:10.1007/s11011-015-9731-8
68. Oto J, Suzue A, Inui D, et al. Plasma proinflammatory and anti-inflammatory cytokine and catecholamine concentrations as predictors of neurological outcome in acute stroke patients. J Anesth. 2008;22:207–212. doi:10.1007/s00540-008-0639-x
69. Whiteley W, Jackson C, Lewis S, et al. Association of circulating inflammatory markers with recurrent vascular events afterstroke: a prospective cohort study. Stroke. 2011;42:10–16. doi:10.1161/STROKEAHA.110.588954
70. Whiteley W, Wardlaw J, Dennis M, et al. The use of blood biomarkers to predict poor outcome after acute transient ischemic attack or ischemic stroke. Stroke. 2012;43:86–91. doi:10.1161/STROKEAHA.111.634089
71. Meng C, Zhang JC, Shi RL, et al. Inhibition of interleukin-6 abolishes the promoting effects of pair housing on post-stroke neurogenesis. Neuroscience. 2015;307:160–170. doi:10.1016/j.neuroscience.2015.08.055
72. Grønhøj MH, Clausen BH, Fenger CD, et al. Beneficial potential of intravenously administered IL-6 in improving outcome after murine experimental stroke. Brain Behav Immun. 2017;65:296–311. doi:10.1016/j.bbi.2017.05.019
73. Gertz K, Kronenberg G, Kälin RE, et al. Essential role of interleukin-6 in post-stroke angiogenesis. Brain. 2012;135:1964–1980. doi:10.1093/brain/aws075
74. Emsley HCA, Smith CJ, Gavin CM, et al. Clinical outcome following acute ischaemic stroke relates to both activation and autoregulatory inhibition of cytokine production. BMC Neurol. 2007;7:1–12. doi:10.1186/1471-2377-7-5
75. Smith CJ, Emsley HC, Udeh CD, et al. Interleukin-1 receptor antagonist reverses stroke-associated peripheral immune suppression. Cytokine. 2012;58(3):384–389. doi:10.1016/j.cyto.2012.02.016
76. Smith CJ, Hulme S, Vail A, et al. SCIL-STROKE (Subcutaneous Interleukin-1 receptor antagonist in ischemic stroke): a randomized controlled Phase 2 trial. Stroke. 2018;49:1210–1216. doi:10.1161/STROKEAHA.118.020750
77. Grabarek B, Bednarczyk M, Mazurek U. The characterization of tumor necrosis factor alpha (TNF-α), its role in cancerogenesis and cardiovascular system diseases and possibilities of using this cytokine as a molecular marker. Folia Biol Oecol. 2017;13:1–8. doi:10.1515/fobio-2017-0001
78. Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood F Met. 2012;32:1677–1698. doi:10.1038/jcbfm.2012.88
79. Prakash S, Smith A. TNF-α. (Tumor Necrosis Factor-α) a paradox in thrombosis. Arterioscler Thromb Vasc Biol. 2018;38:2542–2543. doi:10.1161/ATVBAHA.118.311660
80. Bartsch JW, Wildeboer D, Koller G, et al. Tumor necrosis factor-α (TNF-α) regulates shedding of TNF-α receptor 1 by the metalloprotease-disintegrin ADAM8: evidence for a protease-regulated feedback loop in neuroprotection. J Neurosci. 2010;30:12210–12218. doi:10.1523/JNEUROSCI.1520-10.2010
81. Lambertsen KL, Clausen BH, Babcock AA, et al. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci. 2009;29:1319–1330. doi:10.1523/JNEUROSCI.5505-08.2009
82. Hasan N, McColgan P, Bentley P, et al. Towards the identification of blood biomarkers for acute stroke in humans: a comprehensive systematic review. Br J Clin Pharmacol. 2012;74:230–240. doi:10.1111/j.1365-2125.2012.04212.x
83. Khaksar S, Bigdeli MR. Intra-cerebral cannabidiol infusion-induced neuroprotection is partly associated with the TNF-α/TNFR1/NF-кB pathway in transient focal cerebral ischemia. Brain Injury. 2017;31:1932–1943. doi:10.1080/02699052.2017.1358397
84. Arango-Dávila CA, Vera A, Londoño AC, et al. Soluble or soluble/membrane TNF-α inhibitors protect the brain from focal ischemic injury in rats. Int J Neurosci. 2015;125:936–940. doi:10.3109/00207454.2014.980906
85. Doll DN, Rellick SL, Barr TL, et al. Rapid mitochondrial dysfunction mediates TNF-alpha-induced neurotoxicity. J Neurochem. 2015;132:443–445. doi:10.1111/jnc.13008
86. Masztalewicz M, Nowacki P, Turowska-kowalska J, et al. Peripheral blood indicators of inflammatory response during. The first twenty-four hours if ischemic stroke. Ann Acad Med Stetin. 2010;56:36–40.
87. Bokhari FA, Shakoori TA, Butt A, et al. TNF-alpha: a risk factor for ischemic stroke. J Ayub Med Coll Abbottabad. 2014;26:111–114.
88. Maas MB, Furie KL. Molecular biomarkers in stroke diagnosis and prognosis. Biomar Med. 2009;3:363–383. doi:10.2217/bmm.09.30
89. Huţanu A, Iancu M, Bălaşa R, et al. Predicting functional outcome of ischemic stroke patients in Romania based on plasma CRP, sTNFR-1, D-Dimers, NGAL and NSE measured using a biochip array. Acta Pharmacol Sin. 2018;39:1228–1236. doi:10.1038/aps.2018.26
90. Kim JW, Park MS, Kim JT, et al. The impact of tumor necrosis factor-α and interleukin-1β levels and polymorphisms on long-term stroke outcomes. Eur J Neurol. 2018;79:38–44. doi:10.1159/000484599
91. Waśniowska K. Chemokines–future therapeutic targets. Postepy Hig Med Dosw. 2004;58:37–46.
92. Bielecki B, Głąbiński A. Chemokines and their receptors in pathogenesis of multiple sclerosis. Aktual Neurol. 2007;7:223–231.
93. Bašić-Kes V, Simundic AM, Nikolac N, et al. Pro-inflammatory and anti-inflammatory cytokines in acute ischemic stroke and their relation to early neurological deficit and stroke outcome. Clin Biochem. 2008;41:1330–1334. doi:10.1016/j.clinbiochem.2008.08.080
94. Stone MJ, Hayward JA, Huang C. Mechanisms of regulation of the chemokine-receptor network. Int J Mol Sci. 2017;18:342–375. doi:10.3390/ijms18020342
95. Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–242. doi:10.1146/annurev.immunol.18.1.217
96. Murphy PM. International Union of Pharmacology. XXX. Update on chemokine receptor nomenclature. Pharmacol Rev. 2002;54:227–229. doi:10.1124/pr.54.2.227
97. Wang Y, Huang J, Li Y. Roles of chemokine CXCL12 and its receptors in ischemic stroke. Curr Drug Targets. 2012;13:166–172. doi:10.2174/138945012799201603
98. Moser B, Wolf M, Walz A, et al. Chemokines: multiple levels of leukocyte migration control. Trends Immunol. 2004;25:75–84. doi:10.1016/j.it.2003.12.005
99. Baggiolini M. Chemokines in pathology and medicine. J Intern Med. 2001;250:91–104. doi:10.1046/j.1365-2796.2001.00867.x
100. Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. 2012;33:579–589. doi:10.1016/j.it.2012.07.004
101. Morancho A, Rosell A, García-bonilla L, et al. Metalloproteinase and stroke infarct size: role for anti-inflammatory treatment? Ann NY Acad Sci. 2010;1207:123–133. doi:10.1111/j.1749-6632.2010.05734.x
102. Lee Y, Lee SR, Choi SS, et al. Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. Biomed Res Int. 2014;2014:1–9.
103. García-berrocoso T, Giralt D, Lombart V, et al. Chemokines after human ischemic stroke from neurovascular unit to blood using protein arrays. Transl Proteom. 2014;3:1–9. doi:10.1016/j.trprot.2014.03.001
104. Mirabelli-badenier M, Braunersreuther V, Viviani GL. CC and CXC chemokines are pivotal mediators of cerebral injury in ischaemic stroke. Thromb Haemost. 2011;105:409–420. doi:10.1160/TH10-10-0662
105. Kapurniotu A, Gokce O, Bernhagen J. The multitasking potential of alarmins and atypical chemokines. Front Med (Lausanne). 2019;6:3. doi:10.3389/fmed.2019.00003
106. Rostène W, Dansereau MA, Godefroy D, et al. Neurochemokines: a menage a trois providing new insights on the functions of chemokines in the central nervous system. J Neurochem. 2011;118:680–694. doi:10.1111/j.1471-4159.2011.07371.x
107. Rostène W, Guyon A, Kular L, et al. Chemokines and chemokine receptors: new actors in neuroendocrine regulations. Front Neuroendocrin. 2011;32:10–24. doi:10.1016/j.yfrne.2010.07.001
108. Lanfranco MF, Mocchetti I, Burns MP, et al. Glial- and neuronal-specific expression of CCL5 I. mRNA in the rat brain. Front Neuroanat. 2018;12:1–13. doi:10.3389/fnana.2018.00001
109. Pharoah DS, Varsani H, Tatham RW, et al. Expression of the inflammatory chemokines CCL5, CCL3 and CXCL10 in juvenile idiopathic arthritis, and demonstration of CCL5 production by an atypical subset of CD8+ T cells. Arthritis Res Ther. 2006;8:1–11. doi:10.1186/ar1913
110. White GE, Iqbal AJ, Greaves DR. CC chemokine receptors and chronic inflammation - therapeutic opportunities and pharmacological challenges. Pharmacol Rev. 2013;65:47–89. doi:10.1124/pr.111.005074
111. Siniscalchi A, Gallelli L, Malferrari G, et al. Cerebral stroke injury: the role of cytokines and brain inflammation. J Basic Clin Physiol Pharmacol. 2014;25:131–137. doi:10.1515/jbcpp-2013-0121
112. Venencia A, Subramanian A, Agrawal D, et al. RANTES levels in peripheral blood, CSF and contused brain tissue as a marker for outcome in traumatic brain injury (TBI) patients. BMC Res Notes. 2017;10:139–148. doi:10.1186/s13104-017-2459-2
113. Villapol S, Loane D, Burns MP. Sexual dimorphism in the inflammatory response to traumatic brain injury. Glia. 2017;65:1423–1438. doi:10.1002/glia.v65.9
114. Vajen T, Koenen RR, Werner I, et al. Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci Rep. 2018;8:1–11. doi:10.1038/s41598-018-29026-0
115. Minami M, Satoh M. Role of chemokines in ischemic neuronal stress. Neuromol Med. 2005;7:149–155. doi:10.1385/NMM:7:1-2:149
116. Jiang L, Newman M, Saporta S, et al. MIP-1α and MCP-1 induce migration of human umbilical cord blood cells in models of stroke. Curr Neurovasc Res. 2008;5:118–124. doi:10.2174/156720208784310259
117. Dimitrijevic OB, Stamatovic SM, Keep R, et al. Effects of the chemokine CCL2 on blood–brain barrier permeability during ischemia–reperfusion injury. J Cereb Blood Flow Metab. 2006;26:797–810. doi:10.1038/sj.jcbfm.9600229
118. Kumai Y, Ooboshi H, Takada J, et al. Anti–monocyte chemoattractant protein-1 gene therapy protects against focal brain ischemia in hypertensive rats. J Cereb Blood Flow Metab. 2004;24:1359–1368. doi:10.1097/01.WCB.0000143534.76388.3C
119. Tarozzo G, Campanella M, Ghiani M, et al. Expression of fractalkine and its receptor, CX3CR1, in response to ischaemia-reperfusion brain injury in the rat. Eur J Neurosci. 2002;15:1663–1668. doi:10.1046/j.1460-9568.2002.02007.x
120. Sheridan GK, Murphy KJ. Neuron–glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol. 2013;3:1–14. doi:10.1098/rsob.130181
121. Dénes Á, Ferenczi S, Halász J, et al. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi:10.1038/jcbfm.2008.64
122. Cipriani R, Villa P, Chece G, et al. CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J Neurosci. 2011;31:16327–16335. doi:10.1523/JNEUROSCI.3611-11.2011
123. Terao S, Yilmaz G, Stokes KY, et al. Blood cell-derived RANTES mediates cerebral microvascular dysfunction, inflammation, and tissue injury after focal ischemia-reperfusion. Stroke. 2008;39:2560–2570. doi:10.1161/STROKEAHA.107.513150
124. Kadi L, Selvaraju R, de Lys P, et al. Differential effects of chemokines on oligodendrocyte precursor proliferation and myelin formation in vitro. J Neuroimmunol. 2006;174:133–146. doi:10.1016/j.jneuroim.2006.01.011
125. Ignatov A, Robert J, Gregory-evans C, et al. RANTES stimulates Ca2+ mobilization and inositol trisphosphate (IP3) formation in cells transfected with G protein-coupled receptor 75. Br J Pharmacol. 2006;149:490–497. doi:10.1038/sj.bjp.0706909
126. Rozzi SJ, Borelli G, Ryan K, et al. PACAP27 is protective against tat-induced neurotoxicity. J Mol Neurosci. 2014;54:485–493. doi:10.1007/s12031-014-0273-z
127. Campbell LA, Avdoshina V, Day C, et al. Pharmacological induction of CCL5 in vivo prevents gp120-mediated neuronal injury. Neuropharmacology. 2015;92:98–107. doi:10.1016/j.neuropharm.2015.01.009
128. Tokami H, Ago T, Sugimori H, et al. RANTES has a potential to play a neuroprotective role in an autocrine/paracrine manner after ischemic stroke. Brain Res. 2013;23:122–132. doi:10.1016/j.brainres.2013.04.022
129. Victoria ECG, de Brito Toscano EC, de Sousa Cardoso AC, et al. Knockdown of C-C chemokine receptor 5 (CCR5) is protective against cerebral ischemia and reperfusion injury. Curr Neurovasc Res. 2017;14:125–131. doi:10.2174/1567202614666170313113056
130. Fan Y, Xiong X, Zhang Y, et al. MKEY, a peptide inhibitor of CXCL4‐CCL5 heterodimer formation, protects against stroke in mice. J Am Heart Assoc. 2016;5:1–8.
131. Li P, Wang L, Zhou Y, et al. C-C chemokine receptor type 5 (CCR5)-mediated docking of transferred Tregs protects against early blood-brain. J Am Heart Assoc. 2017;6:1–17. doi:10.1161/JAHA.117.006387
132. Montecucco F, Lenglet S, Gayet-ageron A, et al. Systemic and intraplaque mediators of inflammation are increased in patients symptomatic for ischemic stroke. Stroke. 2010;41:1394–1404. doi:10.1161/STROKEAHA.110.578369
133. Zaremba J, Ilkowski J, Losy J. Serial measurements of levels of the chemokines CCL2, CCL3 and CCL5 in serum of patients with acute ischaemic stroke. Folia Neuropathol. 2006;44:282–289.
134. Canouï-Poitrine F, Luc G, Mallat Z, et al. Systemic chemokine levels, coronary heart disease, and ischemic stroke events. Neurology. 2011;77:1165–1173. doi:10.1212/WNL.0b013e31822dc7c8
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.