")
Back to Journals » ImmunoTargets and Therapy » Volume 4
The role of regulatory T cells in cancer immunology
Authors Whiteside T
Received 17 January 2015
Accepted for publication 14 March 2015
Published 5 August 2015 Volume 2015:4 Pages 159—171
DOI https://doi.org/10.2147/ITT.S55415
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Michael Shurin
Theresa L Whiteside
University of Pittsburgh Cancer Institute, Pittsburgh, PA, US
Abstract: Regulatory T cells (Treg) are generally considered to be significant contributors to tumor escape from the host immune system. Emerging evidence suggests, however, that in some human cancers, Treg are necessary to control chronic inflammation, prevent tissue damage, and limit inflammation-associated cancer development. The dual role of Treg in cancer and underpinnings of Treg diversity are not well understood. This review attempts to provide insights into the importance of Treg subsets in cancer development and its progression. It also considers the role of Treg as potential biomarkers of clinical outcome in cancer. The strategies for monitoring Treg in cancer patients are discussed as is the need for caution in the use of therapies which indiscriminately ablate Treg. A greater understanding of molecular pathways operating in various tumor microenvironments is necessary for defining the Treg impact on cancer and for selecting immunotherapies targeting Treg.
Keywords: cancer, regulatory T cells, tumor microenvironment, immune suppression, anti-Treg therapies
Introduction
In cancer, regulatory T cells (Treg) appear to play an important, although somewhat controversial, role. In many human cancers and in most mouse models of tumor growth, the frequency of Treg and their suppressor functions are increased as compared to those reported for healthy subjects.1–3 Despite the general perception that Treg accumulations in cancer predict poor outcome,4–6 several reports have indicated that Treg numbers and activity are associated with improved prognosis.7–11 While the role of Treg in tumor growth, progression to metastasis, and the disease outcome continues to be debated, there is considerable experimental and clinical evidence in favor of Treg being engaged in suppression of antitumor immune responses and thus contributing to tumor escape from the host immune system.11,12
Treg are called upon to mediate suppression when immune cells activated by endogenous or exogenous agents threaten to destroy tissues or when a progressing tumor actively recruits and programs Treg to downregulate antitumor immune responses.13,14 The potential of Treg utilization either for protection from tissue damage by activated T cells or for aggression against antitumor effector immune cells has led to a more extensive consideration of mechanisms underpinning Treg recruitment to tissue sites. It is known, for example, that Treg express Toll-like receptors (TLRs), and that TLR ligands can regulate functions of Treg, presumably including their migration.15 Treg recruitment to tumor sites is regulated by chemokines produced in the tumor microenvironment (TME) such as, for example, CCL22, a ligand for CCR4.16 Activated Treg express several chemokine receptors (ie, CCR4, CCR5, CCR6, CCR7, and CCR10), which can mediate Treg trafficking to tissue sites.17 In the presence of tumor-derived chemokines, Treg accumulate in the tumor, and once in place, proceed to prevent or blunt antitumor responses of immune cells infiltrating the TME. Thus, Treg which accumulate in situ and in the peripheral circulation of cancer patients can be viewed as one of multiple attempts by the tumor to promote its own escape from the host immune system by silencing antitumor immune effector cells. On the other hand, it seems equally likely that in tumors characterized by extensive inflammatory infiltrates, such as colon or breast cancers, Treg are necessary for control of chronic inflammation, prevention of tissue damage, and limiting of tumor development associated with inflammation.18,19 Interestingly, in patients with colon or breast carcinomas, the presence and frequency of Treg in the tumor are associated with improved prognosis.11,18,20,21 As a result of this potential dual role of Treg in limiting the process of chronic inflammation on the one hand and in promoting tumor escape from immune control on the other, a number of questions have emerged about the mechanisms that regulate these Treg activities. It could be surmised that the frequency and role of Treg in disease outcome depend on the tumor type and “immune signature” the tumor establishes in a given host. Today, the origin and phenotypic characteristics of Treg infiltrating human tumors are not entirely clear, and neither is the mechanism responsible for the apparent “division of labor” among these cells. This ambiguity is fueled by the rapidly emerging evidence for tremendous plasticity and phenotypic as well functional heterogeneity of Treg in man.22,23
Despite rapid progress made in our understanding of how Treg work, many aspects of their interactions with the tumor and other immune or nonimmune cells remain obscured. It is not clear, for example, that Treg found in the TME are the same cells that circulate in the periphery or that their functional repertoire is similar to or different from that of the cells in the peripheral circulation. Overexpression of multiple checkpoint receptors on Treg in the TME suggests that these cells acquire significantly different phenotype and functions once they enter the tumor.24,25 Because of their enhanced capability to suppress antitumor functions of effector T cells (Teff), Treg have been perceived as mediators of tumor escape that need to be unequivocally silenced or eliminated if antitumor functions are to be restored.26,27
The objective of this review is to address those aspects of the Treg biology that provide insights into the importance of Treg in cancer development and its progression. Another objective is to impress upon the reader a degree of caution for the use of Treg as biomarkers of cancer progression and for the use of therapies which indiscriminately ablate Treg. The review provides a rationale for exercising this caution and discusses alternative strategies for monitoring activities of human Treg that are based on the current understanding of their diversity.
Phenotypic characteristics of human Treg
Treg were first described by Sakaguchi et al as a circulating subset of murine CD4+ T cells expressing high levels of CD25 (the interleukin [IL]-2 receptor α chain), which upon adoptive transfers could prevent development of autoimmune disease.28 Today, a high level of CD25 expression still remains one of the defining surface markers of Treg, although CD25 is also expressed by activated conventional T cells (Tconv) with non-regulatory properties. Therefore, it has been suggested that in man, only CD4+CD25hi Treg, which represent 2%–5% of CD25-expressing CD4+ T cells, represent genuine Treg.29 Since the first description of Treg in 1995,28 a number of additional Treg-specifying markers have been proposed, as discussed below, although today, a marker specific for human Treg is yet to be defined.
In general, human Treg have been difficult to study for two reasons. First, they represent only a minor subset of CD4+ T cells (about 5%) and thus are only available in a limited number for extensive examinations.2,29 Second, they lack a specific phenotypic marker that could confirm their identity and could facilitate their isolation and characterization. The transcription factor, FOXP3, a reliable Treg marker in mice, is not so reliable in man, for it may be absent from some Treg subsets and present on non-Treg, as recently discussed.30,31 Further, FOXP3 is an intracellular protein32 that is not expressed on the cell surface, and thus cannot be used for Treg isolation. In the absence of a single marker that defines Treg, various panels of markers have been used to phenotypically distinguish the two major subsets of human Treg, namely naïve (nTreg) or thymus-derived Treg (tTreg) and inducible (iTreg) or peripheral Treg (pTreg). The nomenclature of Treg recently recommended by Abbas et al attempts to classify Treg based on their origin as tTreg or Treg that arise in the periphery by conversion of CD4+ Tconv to T cells mediating suppression.33 While these two Treg subsets share several phenotypic markers in common, they are not phenotypically identical. Specifically, the expression of surface markers such as CD25hi on the cell surface and intracellular FOXP3 has been used to differentiate between these two Treg subsets by flow cytometry, with pTreg exhibiting a much greater heterogeneity in levels of expression of these two markers,34–36 as indicated in Figure 1. Upregulation on the pTreg surface of checkpoint inhibitory receptors, cytotoxic lymphocyte antigen-4 (CTLA-4), programmed death-1 (PD-1), T cell immunoglobulin mucin-3 (TIM-3), and lymphocyte activation gene-3 (LAG-3), and of transforming growth factor-beta (TGF-β)-associated molecules, latency-associated protein (LAP) and glycoprotein A repetitions predominant (GARP) or co-expression of ectonucleotidases, CD39 and CD73, is a characteristic feature that helps in distinguishing pTreg from tTreg.25,35,37 These features of pTreg are especially evident at tumor sites and are interpreted as evidence of greater ability to mediate suppression.37,38 Further, these phenotypic features appear to emphasize that there is a division of labor among these cell subsets, with tTreg responsible for maintaining tolerance to self and pTreg regulating responses to non-self.27 The absence on the Treg surface of markers such as CD127 or CD26 has often been useful for differentiating Treg from CD4+ Teff.39,40 However, due to Treg plasticity and the possibility that co-expression of certain phenotypic markers distinguishes subsets of Treg with quantitatively different suppressive functions, this differentiation is not simple or easy. In the absence of a single specific, stable marker for Treg, combinations of markers are often used to define Treg.37 This need for the use of multiple marker panels further emphasizes the existence of considerable heterogeneity among human Treg populations.37
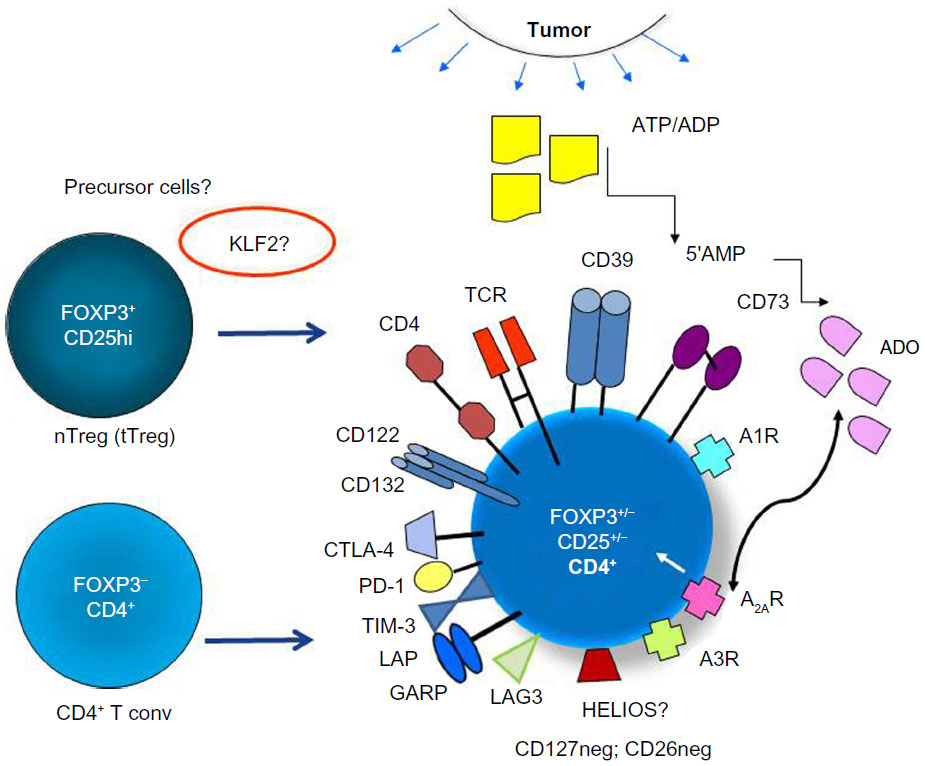 | Figure 1 The phenotypic profile and potential cellular origins of induced (i)Treg present in the tumor microenvironment. |
Immunohistochemistry broadly used for Treg detection in formaldehyde-fixed paraffin-embedded human tissues depends entirely on selection of antibodies (Abs) that work well with such specimens, and as discussed elsewhere,12 should not depend on expression of FOXP3 alone, as pTreg present in tumors may be negative for this marker.
Attempting to bring some measure of consensus to the field, a recent international workshop on Treg was organized by the Collaborative Immunoguiding Program.41 The workshop made the following (soon to be published) suggestions regarding the flow cytometry panels to be used for human Treg assessments: a) a minimal definition of human Treg should include CD3, CD4, CD25, CD127, and FOXP3 markers, with an addition of Ki67 and CD45RA to clarify the Treg activation status; b) the sole use of any of the three most commonly used flow panels for the Treg phenotypic definition – 1) [CD25+CD127loFOXP3+ Treg], 2) [FOXP3+HELIOS+ Treg], and 3) [FOXP3hi CD45RAneg vs FOXP3intCD45RA+ to distinguish activated vs nTreg, respectively] – leads to underestimation of the Treg frequency ranging between 25% and 65%.41 The same workshop concluded that CD39 and CTLA-4, which have been described as functional markers on Treg,42,43 denote activated or iTreg and thus are considered as “optional” markers.15
However, a somewhat different view of identifying Treg subsets in cancer patients could be taken based on the observed differences between the phenotype of Treg in healthy donors vs that in patients with cancer34 or between the phenotype of Treg at the tumor site vs that in the patients’ peripheral blood.25–38 Specifically, it appears that in cancer patients, the frequency of pTreg in the blood and tumor tissues is often elevated,2,34 and these accumulating Treg have high expression levels of surface markers associated with suppression such as CD39, CD73, LAP, GARP, COX-2, and others.44,45 These Treg also have intracytoplasmic expression of perforin, granzyme B, and/or IL-10, molecules associated with immune suppression.25,46 Expression by Treg of these “functional” markers is counterbalanced by the presence at tumor sites of Treg co-expressing inhibitory receptors, CTLA-4, PD-1, TIM-3, or LAG-3.23–25,47,48 In aggregate, these observations suggest that pTreg present in the TME may be phenotypically and functionally distinct from tTreg. Therefore, a broader Treg definition, one that allows for the more precise discrimination of tTreg from pTreg in patients with cancer, is needed. As pTreg populations, which likely include subsets of heterogeneous suppressor cells, predominate in cancer,34,45 their localization, numbers, phenotypic signatures, and suppressor functions are of utmost importance. While none of these markers are specific for Treg, when combined with surface CD25hi and/or intracytoplasmic FOXP3, they are useful because they allow for the assessment of the functional potential of Treg by flow cytometry without the need for Treg isolation required for conventional CFSE-based suppressor assays.2,34
More recently, efforts to identify a specific Treg marker that might distinguish tTreg from pTreg have focused on Kruppel-like factor 2 (KLF2), a transcription factor that regulates chronic inflammation and that is necessary for the development of pTreg but not of tTreg.49 This finding not only emphasizes the phenotypic and functional distinction between these Treg subsets but also suggests that discrimination between them may have critical therapeutic implications for selective rather than “global” Treg depletion.
Functional attributes of Treg in patients with cancer
We and many others have commented on elevated suppressor functions mediated by Treg in the peripheral circulation of patients with solid or hematological malignancies.2,26,50,51 In the context of Treg, “suppressor functions” are generally defined as significant inhibition in responder cells of activation (including signaling via activating receptors), proliferation, cytokine/soluble factor production, or of gene expression levels. A number of in vitro assays have been developed to measure suppression mediated by Treg.38,52,53 Some are based on multicolor flow cytometry to measure surface or intracytoplasmic expression levels by Treg of suppression-associated molecules (LAP, GARP, CD39, CD73), as discussed above. These flow-based assays do not require isolation or culture of Treg and thus are commonly used for monitoring of Treg in human specimens. Other assays require coculture of isolated Treg and CFSE-labeled responder T cells, to quantitate levels of suppression mediated by Treg.52,53 In such cocultures, suppression of responder cell activation, proliferation, cytokine production, or gene expression can be quantitatively determined.2,53–55 Still other assays measure FOXP3 demethylation in Treg by MS-qRT-PCR to estimate Treg-specific demethylation region.56 Methodological details for these functional Treg assays can be found in the references.53,54
With a recent more reliable detection and discrimination of Treg in tissues and the peripheral circulation of patients with cancer,57,58 it has become apparent that Treg accumulating in at the tumor site are phenotypically and functionally altered relative to circulating Treg.25,37,48 We have recently reported that expression levels of inhibitory receptors, PD-1, CTLA-4, and TIM-3, as well as of CD39, an enzyme which participates in conversion of adenosine-5′-triphosphate (ATP) to immunosuppressive adenosine (ADO), were significantly elevated in Treg within TIL isolated from human head and neck squamous cell carcinomas (HNSCCs) relative to expression of these markers in paired peripheral blood Treg.25 Others have observed similar upregulated expression of inhibitory receptors on TIL, including Treg, in various other solid tumors. For example, in human non-small-cell lung cancer, a majority of CD4+FOXP3+ TIL also expressed TIM-3,47 and TIM-3+FOXP3+CD4+ Treg preferentially accumulated in the tumor nests in hepatocellular carcinoma.48 Camisaschi et al reported that in melanoma and in colorectal carcinoma, LAG-3+CD4+CD25hiFOXP3+ Treg were preferentially expanded in PBMC as well as in TIL and mediated strong suppressor activity.24 In aggregate, these data suggest that the tumor can induce changes in the receptor profile of Treg thus altering their functions. This may lead to Treg “activation” and upregulation of their suppressor functions or to downregulation of suppression by signaling of the inhibitory receptor expressed on Treg. The implication of these data is that highly suppressive Treg accumulating in the TME may need to be “restrained” via the upregulation of inhibitory checkpoint receptors from excessive suppression that might interfere with immunologic homeostasis.59 Alternatively, it has been suggested that, in contrast to functional blockade induced by signals delivered via checkpoint receptors to all other immune cells, iTreg induction, proliferation, and suppressive functions are promoted by checkpoint receptor engagement.23 Thus, co-expression of “activation” markers and inhibitory receptors on pTreg in the peripheral circulation of cancer patients, and especially at tumor sites,60,61 emerges as an important surrogate marker for Treg functions, and as such, should be included in monitoring of Treg.
The emerging evidence further suggests that in the presence of tumor-derived signals, Treg might be regulated to preferentially use specific inhibitory molecular pathways.37 As previously discussed37 and illustrated in Figure 2, Treg are known to utilize a variety of mechanisms for mediating suppression.46,55,62–64 However, it is unclear whether all Treg are capable of perusing these different mechanisms or whether Treg subsets specializing in one type of suppression exist. Therefore, it may be reasonable to envision the scenario where different solid tumors create microenvironments in which Treg are instructed to preferentially adopt the suppression pathway that best fits with environmental programming in situ. For example, HNSCCs are known to express COX-2 and secrete PGE2, which signals via four prostaglandin E receptors expressed on immune (as well as other) cells, upregulating 3′,5′-cyclic adenosine monophosphate (3′,5′-cAMP) levels in responder cells and thus inducing immune suppression.45,65 We have shown that human CD4+CD25neg T cells cocultured in the presence of COX-2+ HNSCC cells differentiated into highly suppressive FOXP3+COX-2+ Treg, which produced PGE2 and other suppressive factors.45 In contrast, COX-2neg HNSCC induced Treg with a significantly lower expression of inhibitory receptors and lower output of immunosuppressive factors.45 The TME of most tumors is enriched in ATP, and the ability of accumulating CD4+CD39+ Treg to hydrolyze ATP to AMP and then, upon upregulation of CD73, to ADO likely represents one of the most common mechanisms of tumor-induced immunosuppression. Expression on the Treg surface of markers such as LAP and GARP suggests the involvement of the TGF-β pathway in tumor-induced suppression by Treg, which is common for cancers producing this cytokine, for example, HNSCCs66 (our unpublished data). These examples illustrate how human tumors regulate suppressive functions of Treg that are recruited to the TME.
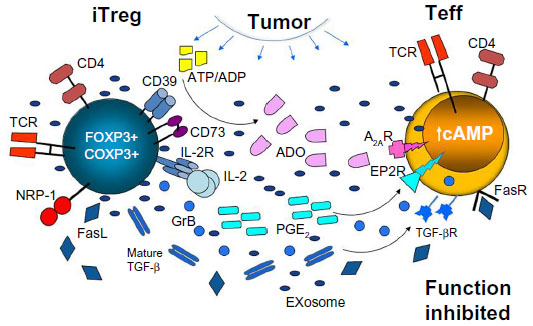 | Figure 2 Treg accumulating in the TME (activated iTreg) utilize various suppressive mechanisms to inhibit functions of Teff. |
The ADO-PGE2 pathway and Treg-mediated suppression
ADO is a well-known mediator of diverse regulatory processes in the endocrine, vascular, neurological, renal, pulmonary, and immunological systems.67,68 It plays a key role in various diseases, including cancer, chronic inflammation, infections, and autoimmune disorders.45,67 Exogenous ADO is a product of ATP hydrolysis by two ectoenzymes acting in sequence: CD39, an ectonucleoside triphosphate diphospho-hydrolase-1, which hydrolyzes ATP to ADP and AMP, and CD73, an ecto-5′-nucleotidase which catalyzes AMP conversion to ADO. Signaling via its four surface G-protein-coupled receptors, A1, A2A, A2B, and A3 which are widely distributed throughout tissues, ADO mediates regulatory effects via up- or downregulation of intracellular levels of 3′,5′-cAMP. Expression of CD39 and CD73 on Treg was first reported by Borsellino et al69 and Deaglio et al70 in 2007. Since then, we have studied the ability of human Treg to produce ADO and showed that in vitro-generated, cultured iTreg (the so-called Tr1 cells) upregulated surface expression of CD73, co-expressed CD39 and CD73, efficiently hydrolyzed ATP to 5′-AMP and ADO, secreted copious levels of ADO, and mediated suppression of Teff functions via the A2AR engagement.55 In contrast, nTreg, which expressed CD39 but not CD73 on the cell surface, mainly produced 5′-AMP and produced ADO only when co-incubated with CD4+CD73+ T cells, CD73+ B cells, or tumor-derived exosomes, which carried membrane-tethered CD39 and CD73.71 In contrast to CD4+ Teff, human Treg express little if any CD26, which is linked to ADO deaminase at the cell surface, and thus are inefficient in converting ADO to inosine.72 Increased pericellular levels of ADO in Treg might facilitate autocrine signaling, potentially augmenting their suppressor activity. As the A2AR is expressed on Treg, ADO generated by Treg could signal via this ADO receptor to promote Treg functions.72 These data suggest that the ADO pathway may not only be important for Treg proliferation and Treg-mediated suppression of other immune cells expressing ADO but, via its autocrine activity, might also play a key role in the upregulation of Treg suppressor functions.72
In the immune system, ADO inhibits functions of immune cells and is considered to be a powerful anti-inflammatory factor.65,67,68 In cancer, however, in addition to promoting migration of immune cells to the tumor and inhibiting antitumor functions of accumulating Teff, ADO promotes differentiation, expansion, and suppressor activity of Treg and myeloid-derived suppressor cells (MDSCs).73,74 As recently discussed, the ADO pathway in the TME ceases to be a protective pathway guarding against tissue damage by activated immune cells and becomes a tool for suppressing antitumor immune functions, and through its effects on the vasculature, for promoting metastasis.37,65 Importantly, as indicated above, these pro-tumor activities of Treg occur in cooperation with the tumor-driven PGE2 pathway,72 as the ADO and PGE2 pathways converge at the adenylate cyclase, upregulating its activity and thus 3′,5′-cAMP levels in responder cells. Together, these two factors deliver powerful immunoinhibitory signals to antitumor responder cells.
Treg as potential biomarkers in cancer
The ADO/PGE2 pathway operating in the TME and discussed above is but one example of strategies that tumors employ in an attempt to utilize Treg for silencing of antitumor responses. Other mechanisms of suppression exercised by Treg include production of inhibitory cytokines (IL-10, TGF-β),66,75,76 Fas/FasL-dependent apoptosis of activated CD8+ T cells,46 or the engagement of the Neuropilin/semaphorin-4a pathway.76,77 Accumulations of Treg as well as MDSC in human tumors and their increased frequency in the circulation of cancer patients have been widely reported.1–3,74 Many reports, but not all, link these accumulations of CD4+FOXP3+CD25hi Treg to poor prognosis due to suppression of antitumor responses by the accumulating Treg.4–11 Notably, in human colorectal cancer and in breast cancer, the presence and density of FOXP3+ Treg have been reported to predict favorable outcome and a better locoregional control of the tumor.18,20 Also, in human lymphomas, elevated circulating Treg predict better outcome.8–10 Thus, Treg frequency in the tumor or in the periphery is a potentially important prognostic biomarker in cancer. Treg enumeration and characterization in situ could provide important clues about the tumor’s immune signature, which currently is becoming recognized as an important prognostic factor in various human solid tumors.79 As the TME is created and maintained by the tumor, phenotypic and functional characterization of Treg among TIL and in the peripheral circulation of patients with cancer might inform us about tumor itself, especially its aggressiveness or propensity to metastasize. Given recent emphasis on the tumor immune signature and emerging correlations of immunohistochemistry data to cancer patients’ survival,79,80 the phenotypic, and especially functional, characterization of Treg in situ assumes a new and potentially important role in establishing the prognostic significance of Treg. While Treg significance as a prognostic marker is best established in colorectal carcinoma,81 investigations are in progress to extend and confirm these findings to other solid tumors.
Based on measures of the magnitude of immune response silencing by Treg in cancer (ie, Treg suppressor functions) rather than their phenotype in situ or in the peripheral circulation, it might be possible to arrive at an even better estimate of Treg prognostic significance. Similarly, measures of Treg functions might correlate better with responses to oncological therapies than does their phenotypic enumeration. However, because Treg are heterogeneous, consisting of many subsets of functionally distinct cells, and because no universal distinguishing marker for Treg is currently available, their use as a biomarker of prognosis is limited and has to be taken with caution. Furthermore, current attempts to therapeutically deplete Treg might enhance tumor immunity in some patients but be detrimental in others.1,7 It is necessary to remember that most of the studies examining the association of the Treg phenotype with prognosis or response to therapies were based on the use of FOXP3 as a “specific” Treg marker.20 However, a recent comprehensive review of the prognostic significance of FOXP3+ T cells in 16 nonlymphoid cancers suggested that FOXP3 by itself is not a reliable marker of human Treg and that the tumor site, that is, the TME, has a major impact on biologic effects of FOXP3+ Treg.30 Overall, the prognostic value of Treg in cancer remains questionable, although it is possible that introduction in the future of more specific high-throughput assays for Treg might provide a more discriminating approach for evaluating their value as surrogate markers of prognosis, outcome, or response to therapy.
It has been reported that expression of surface markers on Treg can be altered in disease82 and in patients undergoing conventional therapies or immune therapies.82–84 Therefore, the selection of a panel of markers for measuring Treg is a critical task that will ultimately determine the Treg role as biomarkers of prognosis in cancer and other disease. As discussed earlier, there is still no consensus as to which marker panel (of several available) is best, and which subset of Treg should be monitored. Focusing on one functional subset, for example, on the CD4+CD39+CD25+ ADO-producing Treg, as is done in the author’s laboratory, may be limiting in scope but offers an advantage of following disease-associated changes in this subset of Treg and correlating these changes to disease progression.85
Therapeutic approaches to eliminating Treg-induced suppression
If Treg play a role in promoting tumor escape, as suggested by many in vitro and in vivo studies, then elimination or silencing of Treg becomes a desirable therapeutic objective. Indeed, a considerable body of recent literature deals with various methods for Treg depletion.86–91 Treg express surface molecules that can be specifically targeted by Abs or pharmacologic inhibitors. Table 1 lists the Treg-associated molecules that potentially could be targeted for Treg silencing either by Treg removal or impairment of Treg suppressor functions. To date, a variety of agents, including Abs (daclizumab: anti-CD25 Ab), IL-2 fusion toxins such as denileukin diftitox (Ontak), or drugs such as cyclophosphamide or tyrosine kinase inhibitors (sunitinib), have been tested in preclinical in vitro studies with human cells.87–91 Many in vivo studies in animal models of cancer have been performed testing for efficiency in depleting Treg.86 Advantages and disadvantages of these depletion strategies have been extensively reviewed.86
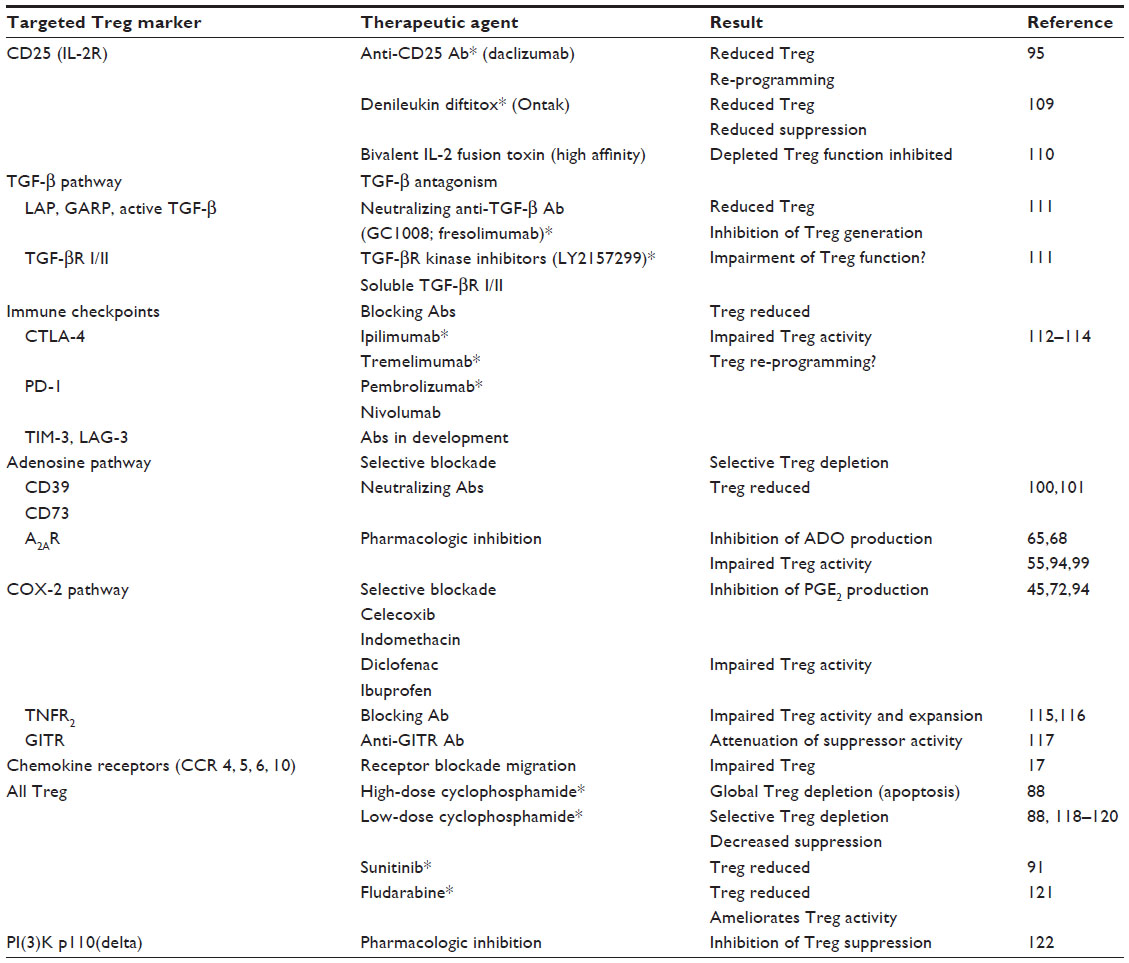 | Table 1 Potential molecular targets for therapeutic depletion or re-programming of human Treg |
Based on favorable preclinical results, some depletion strategies are being used alone or in combination with immunotherapies in human clinical trials (Table 2). The table lists clinical trials posted online at http://www.clinicaltrials.gov that have utilized one or more Treg-depleting strategies presented in Table 1. However, in contrast to successful and meaningful Treg depletion studies in mouse models, it has not been possible to convincingly correlate Treg depletion by these agents with clinical benefits in patients with cancer.92 This may be due to inadequate depletion efficacy of the drugs, innate resistance of Treg to certain drugs, selective sensitivity of some but not all Treg subsets to the drugs being used, or the ability of the host to rapidly re-populate the depleted Treg. Recent experiments support the notion that not all Treg are the same, and that Treg mediating antitumor responses (ie, iTreg) represent a unique subset or subsets of CD4+ T cells with properties distinct from those of Treg responsible for mediating tolerance to self-antigens.93 This argues against the concept of a “global” Treg depletion (eg, with high-dose cyclophosphamide) and for the use of more selective depletion strategies that would protect Treg-regulating autoimmunity and eliminate those that mediate suppression of antitumor immunity.86,94 However, because the phenotypic distinction between tTreg and iTreg is blurred at this time, it may be difficult to selectively target one Treg subset in preference of another. Surprisingly, there is evidence that CD4+CD25+FOXP3+CD45RA+ Treg were not altered by daclizumab administered prior to an antitumor vaccine in patients with breast cancer, while the depletion of CD4+CD25+FOXP3+CD45RAneg Treg has led to selective re-population of the partly depleted Treg compartment with re-programmed or newly minted CD4+IFN-γ+ T cells that no longer mediated suppression.95 These studies suggest that interference with the Treg compartment by the Treg-depleting therapies may profoundly alter properties of Treg subsets, leading to re-population with T-cell subsets showing unexpected characteristics. In this context, our cross-sectional studies of patients with HNSCC treated with the standard-of-care chemoradiotherapy (CRT) showed that in a substantial proportion of these patients, circulating Treg become more resistant to CRT than conventional CD4+ Teff cells, persist for prolonged time periods in the circulation, upregulate expression of pro-survival molecules Bcl-2 and Bcl-xL, and might contribute to depressed antitumor immunity potentially associated with cancer recurrence.85 Although effects of conventional or immune therapies on the Treg frequency and functions remain a subject of contention, it appears that much of this controversy arises from the tremendous plasticity of Treg, which in cancer is almost certainly orchestrated by the presence of the tumor and of various tumor-derived factors.
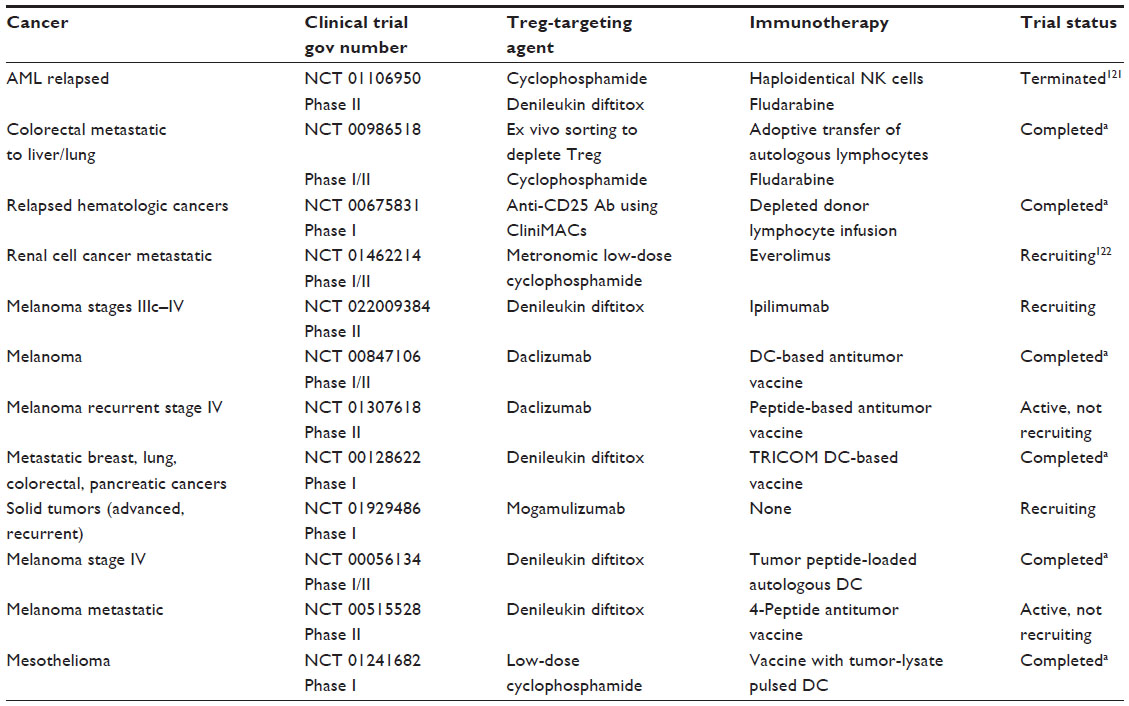 | Table 2 Immunotherapy clinical trials incorporating strategies for Treg depletion in patients with solid or hematologic malignancies |
In view of the existence of multiple inhibitory pathways in the TME, such as the ADO/PGE2, TGF-β, IL-2/IL-2R, and Neuropilin-1/semaphorin-4a pathways,44,76,77,96 and emerging evidence that Treg might selectively utilize these pathways for mediating immune suppression, the strategy of “blocking the inhibitors” with neutralizing Abs or pharmacologic inhibitors has been considered.97 The strategy is based on the inhibitor ability to antagonize negative signaling ongoing between iTreg and Teff, and it calls for the delivery to patients of Abs or pharmacologic agents specific for key components of an inhibitory pathway operating in the TME and responsible for tumor-induced suppression.97 The difficulty is to a priori discern which of the multiple inhibitory pathways plays a key role in immune escape of a given tumor. For example, in patients with tumors expressing COX-2 such as HNSCC, which are richly infiltrated with iTreg-producing PGE2, inhibitors of the PGE2 pathway (celecoxib, indomethacin, diclofenac, ibuprofen) have been clinically used with an intent to block immune suppression.45 As ATP levels are generally high in the TME of human solid tumors,98 and as tumor-associated iTreg overexpress CD39 and CD73 producing lots of ADO,71 it is safe to predict that antagonizing this pathway at the ectoenzyme or the A2AR level would effectively reduce or eliminate ADO-mediated suppression. This approach has been shown to work both in vitro with human Treg–Teff cocultures71 and in preclinical models of cancer.99,100 It is important to remember that antagonistic drugs or Abs with specificity for an antigen present on Treg and on tumor cells, as is the case with CD39 and CD73 ectonucleotidases, for example, will target not only Treg but also tumor cells, potentially amplifying their effectiveness. An additional benefit of antagonizing ADO-induced suppression may derive from the fact that it involves blocking of Treg suppressor functions without depletion of all Treg and risking the development of autoimmunity.101
Currently, the most widely used strategy for reducing tumor-induced immune suppression is the immune checkpoint blockade with Abs specific for CTLA-4, PD-1, or PD-L1.102,103 The targeted molecules are negative inhibitors of immune responses mediated by activated Teff. However, Treg, especially those present in the TME, are known to express a variety of the same regulatory molecules.24,25 Therefore, it has been suggested that in addition to blocking negative signaling in Teff, the checkpoint blockade with, for example, anti-CTLA-4 (ipilimumab) or anti-PD-1 (nivolumab) Abs also eliminates Treg by a mechanism referred to as antibody-dependent cellular cytotoxicity (ADCC).104 There is recent evidence that in vitro targeting of CTLA-4+ Treg with ipilimumab reduces suppression exerted by Treg on natural killer cells, which are now able to mediate ADCC and thus potentiate antitumor functions of ipilimumab.64 The reported antitumor efficacy of checkpoint-blocking Abs in human clinical trials may be related to inhibition of activated T cells and also to the Ab-driven elimination of iTreg. The potential of ipilimumab for elimination of CTLA-4+ Treg is especially intriguing in view of current reports that some monoclonal Abs used for cancer therapy, for example, cetuximab approved for treatment of HNSCC, actually increase the frequency and suppressor functions of Treg and that these increases can be related with poor prognosis.64 This would argue for a combination of cetuximab and ipilimumab in the future to improve antitumor effectiveness of immunotherapy.64 As these cellular mechanisms of Ab cancer immunotherapy are potentially related to the observed clinical responses and outcome in patients with cancer, they remain under intense scrutiny.
Summary
The presence and functions of human Treg in cancer have been intensively investigated. Nevertheless, the role that these suppressor cells play in cancer progression remains controversial. It appears that while contributing to tumor escape from the host immune system, Treg are also involved in regulating immune responses to self and controlling inflammatory responses that threaten to disrupt tissue integrity. This small subset of CD4+ T cells is endowed with a remarkable characteristic of plasticity that allows Treg to rapidly respond to recruiting stimulatory signals by trafficking to sites requiring their interventions, rapid expansion, overexpression of surface receptors involved in their functions, and conversion to highly effective regulatory cells that can act in a paracrine as well as an autocrine manner. It appears that in cancer, expansion and activation of Treg occur in response to tumor-generated signals, leading to tumor escape. The remarkable plasticity of Treg infiltrating human tumors is reflected in their phenotypic and functional heterogeneity that may influence disease outcome. It appears that genetic and environmental factors promote variability in the expression of Treg cell signature genes,10 so that Treg gene repertoire differs between individuals. If so, then Treg involvement in human cancer and other diseases will have to be viewed in the light of personalized medicine.108
Recent insights into Treg accumulating at tumor sites and in the peripheral circulation of patients with cancer indicate that Treg responding to environmentally generated stimuli participate in already existing inhibitory molecular pathways, which characterize the TME created by a given tumor.37 At the same time, Treg entering a TME rich in activated inflammatory cells can regulate inflammation, decreasing the potential for pro-tumor effects.105 A better understanding of molecular pathways operating in the TME is needed to be able to discriminate “bad” Treg (promote tumor escape) from “good” Treg (restrict destructive chronic inflammation). This concept underlies the use of Treg as biomarkers of tumor progression and the selection of therapeutic strategies for Treg elimination to help restore antitumor immunity in cancer. The understanding of Treg diversity is critical for either of these strategies to be successful, and a sustained focus on the molecular pathways that Treg use in the TME is likely to facilitate future progress.
Disclosure
The author reports no conflicts of interest in this work.
References
Mougiakakos D, Choudhury A, Lladser A, Kiessling R, Johansson CC. Regulatory T cells in cancer. Adv Cancer Res. 2010;107:57–117. | |
Strauss L, Bergmann C, Gooding W, Johnson JT, Whiteside TL. The frequency and suppressor function of CD4+CD25highFoxp3+ T cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13:6301–6311. | |
Wolf AM, Wolf D, Steurer M, Gastl G, Gunsilius E, Grubeck-Loebenstein B. Increase in regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res. 2003;9:606–612. | |
Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. | |
Wolf D, Wolf AM, Rumpold H, et al. The expression of T cell-specific forkhead box transcription factor FOXP3 is associated with poor prognosis in ovarian carcinoma. Clin Cancer Res. 2005;11:8326–8331. | |
Deng L, Zhang H, Luan Y, et al. Accumulation of FOXP3+ T regulatory cells in draining lymph nodes correlates with disease progression and immune suppression in colorectal cancer patients. Clin Cancer Res. 2010;16:4105–4112. | |
Badoual C, Hans S, Rodriguez J, et al. Prognostic value of tumor-infiltrating CD4+ T cell subpopulations in head and neck cancers. Clin Cancer Res. 2006;12(2):465–472. | |
Tzankov A, Meier C, Hirschmann P, Went P, Pileri SA, Dirnhofer S. Correlation of high numbers of intratumoral FOXP3+ regulatory T cells with improved survival in germinal center-like diffuse large B-cell lymphoma, follicular lymphoma and classical Hodgkin’s lymphoma. Haematologica. 2008;93:193–200. | |
Carreras J, Lopez-Guillermo A, Fox BC, et al. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood. 2006;108: 2957–2964. | |
Farinha P, Al-Tourah A, Gill K, Klasa R, Connors JM, Gascoyne RD. The architectural pattern of FOXP3-positive T cells in follicular lymphoma is an independent predictor of survival and histologic transformation. Blood. 2010;115:289–295. | |
Droeser R, Zlobec I, Kilic E, et al. Differential pattern and prognostic significance of CD4+, FOXP3+ and IL-17+ tumor infiltrating lymphocytes in ductal and lobular breast cancers. BMC Cancer. 2012; 12:134. | |
Whiteside TL. What are regulatory T cells (Treg) regulating in cancer and why? Semin Cancer Biol. 2012;22:327–334. | |
Yamaguchi T, Wing JB, Sakaguchi S. Two modes of immune suppression by FoxP39(+) regulatory T cells under inflammatory or non-inflammatory conditions. Semin Immunol. 2011;23:424–430. | |
Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27:5904–5912. | |
Oberg HH, Juricke M, Kabelitz D, Wesch D. Regulation of T cell activation by TLR ligands. Eur J Cell Biol. 2011;90:582–592. | |
Yoshie O, Matsushima K. CCR4 and its ligands: from bench to bedside. Int Immunol. 2015;27(1):11–20. | |
Ondondo B, Jones E, Godkin A, Gallimore A. Home sweet home: the tumor microenvironment as a haven for regulatory T cells. Front Immunol. 2013;4:197. | |
Ladoire S, Martin F, Ghiringhelli F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother. 2011;60(7):909–918. | |
Rech AJ, Mick R, Kaplan DE, Chang KM, Domchek SM, Vonderheide RH. Homeostasis of peripheral FOXP3(+)CD4(+) regulatory T cells in patients with early and late stage breast cancer. Cancer Immunol Immunother. 2010;59(4):599–607. | |
Salama P, Phillips M, Grieu F, et al. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol. 2009;27(2):186–192. | |
Frey DM, Droeser RA, Viehl CT, et al. High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer. 2010;126:2635–2643. | |
Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood. 2012;119:4430–4440. | |
Francisco LM, Salinas VH, Brown KE, et al. PD-1 regulates the development, maintenance and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029. | |
Camisaschi C, Casati C, Rini F, et al. LAG-3 expression defines a subset of CD4(+)CD25(high) Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol. 2010;184:6545–6551. | |
Jie HB, Gildener-Leapman N, Li J, et al. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer. 2013;109:2629–2635. | |
Whiteside TL, Schuler P, Schilling B. Induced and natural regulatory T cells in human cancer. Expert Opin Biol Ther. 2012;12:1383–1397. | |
Adeegbe DO, Nishikawa H. Natural and induced T regulatory cells in cancer. Front Immunol. 2013;4:190. | |
Sakaguchi S, Sakeguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155: 1151–1164. | |
Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25 high regulatory cells in human peripheral blood. J Immunol. 2001;167: 1245–1253. | |
deLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FOXP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res. 2012;18:3022–3029. | |
Devaud C, Darcy PK, Kershaw MH. Foxp3 expression in T regulatory cells and other cell lineages. Cancer Immunol Immunother. 2014;63: 869–876. | |
Allan SE, Passerini L, Bacchetta R, et al. The role of 2FOXP3 isoforms in the generation of human CD4+ Tregs. J Clin Invest. 2005;115: 3276–3284. | |
Abbas AK, Benoist C, Bluestone JA, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. 2013;14: 307–308. | |
Bergmann C, Strauss L, Wang Y, et al. T regulatory type 1 cells in squamous cell carcinoma of the head and neck: mechanisms of suppression and expansion in advanced disease. Clin Cancer Res. 2008;14:3706–3715. | |
Schuler PJ, Schilling B, Harasymczuk M, et al. Phenotypic and functional characteristics of CD4+ CD39+ FOXP3+ and CD4+ CD39+ FOXP3neg T-cell subsets in cancer patients. Eur J Immunol. 2012;42:1876–1885. | |
deLeeuw RJ, Kroeger DR, Kost SE, Chang PP, Webb JR, Nelson BH. CD25 identifies a subset of CD4+FoxP3- TIL that are exhausted yet prognostically favorable in human ovarian cancer. Cancer Immunol Res. 2014;3:1–9. | |
Whiteside TL. Induced regulatory T cells in inhibitory microenvironments created by cancer. Expert Opin Biol Ther. 2014;14(10):1411–1425. | |
Strauss L, Bergmann C, Szczepanski MJ, Lang S, Kirkwood JM, Whiteside TL. Expression of ICOS on human melanoma-infiltrating CD4+CD25highFoxp3+ T regulatory cells: implications and impact on tumor-mediated immune suppression. J Immunol. 2008;180(5):2967–2980. | |
Mandapathil M, Szczepanski M, Harasymczuk M, et al. CD26 expression and adenosine deaminase activity in regulatory T cells (Treg) and CD4(+) T effector cells in patients with head and neck squamous cell carcinoma. Oncoimmunology. 2012;1(5):659–669. | |
Liu W, Putnam AL, Xu-Yu Z, et al. CD127 expression inversely correlates with FOXP3 and suppressive function of human CD4+ Treg cells. J Exp Med. 2006;203:1701–1711. | |
Santegoets S, Dijkgraaf E, Battaglia A, et al. Monitoring regulatory T cells in clinical samples: consensus on an essential marker set and gating strategy for regulatory T cell analysis by flow cytometry. In press 2015. | |
Salomon B, Lenschow DJ, Rhee L, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. | |
Mandapathil M, Hilldorfer B, Szczepanski MJ, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem. 2010;285: 7176–7186. | |
Mandapathil M, Szczepanski MJ, Szajnik M, et al. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem. 2010;285:27571–27580. | |
Bergmann C, Strauss L, Zeidler R, Lang S, Whiteside TL. Expansion of human T regulatory type 1 cells in the microenvironment of cyclooxygenase 2 overexpressing head and neck squamous cell carcinoma. Cancer Res. 2007;67:8865–8873. | |
Strauss L, Bergmann C, Whiteside TL. Human circulating CD4+CD25highFoxp3+ regulatory T cells kill autologous CD8+ but not CD4+ responder cells by Fas-mediated apoptosis. J Immunol. 2009;182: 1469–1480. | |
Gao X, Zhu Y, Li G, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One. 2012;7:e30676. | |
Yan J, Zhang Y, Zhang JP, Liang J, Li L, Zheng L. Tim-3 expression defines regulatory T cells in human tumors. PLoS One. 2013;8: e58006. | |
Pabbisetty SK, Rabacal W, Maseda D, et al. KLF2 is a rate-limiting transcription factor that can be targeted to enhance regulatory T-cell production. Proc Natl Acad Sci U S A. 2014;111(26):9579–9584. | |
Szczepanski MJ, Szajnik M, Czystowska M, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res. 2009;15:3325–3332. | |
Schmitt EG, Williams CB. Generation and function of induced regulatory T cells. Front Immunol. 2013;4:152. | |
Ruitenberg JJ, Boyce C, Hingorani R, Putnam A, Ghanekar SA. Rapid assessment of in vitro expanded human regulatory T cells function. J Immunol Methods. 2011;372:95–106. | |
Canavan JB, Afzali B, Scottà C, et al. A rapid diagnostic test for human regulatory T cell function to enable regulatory T cell therapy. Blood. 2012;119:e57–e66. | |
Whiteside TL. Regulatory T cell (Treg) assays: repertoire, functions and clinical importance of human Treg. In: Detrich B, Hamilton RG, Folds JD, editors. Manual of Molecular and Clinical Laboratory Immunology. 8th ed. Washington: ASM Press. In press 2015. | |
Whiteside TL, Jackson EK. Adenosine and prostaglandin e2 production by human inducible regulatory T cells in health and disease. Front Immunol. 2013;4:212. | |
Polansky JK, Schreiber L, Thelemann C, et al. Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J Mol Med (Berl). 2010;88:1029–1040. | |
Kalathil S, Lugade AA, Miller A, Iyer R, Thanavala Y. Higher frequencies of GARP(+)CTLA-4(+)Foxp3(+) T regulatory cells and myeloid-derived suppressor cells in hepatocellular carcinoma patients are associated with impaired T cell functionality. Cancer Res. 2013;73: 2435–2444. | |
Jacobs JF, Idema AJ, Bol KF, et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro Oncol. 2009;11:394–402. | |
Butt AQ, Mills KH. Immunosuppressive networks and checkpoints controlling antitumor immunity and their blockade in the development of cancer immunotherapeutics and vaccines. Oncogene. 2014;33: 4623–4631. | |
Nirschi CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunoatherapy. Clin Cancer Res. 2013;19:4917–4924. | |
Woo SR, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72:917–927. | |
Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8: 1353–1362. | |
Czystowska M, Strauss L, Bergmann C, Szajnik M, Rabinowich H, Whiteside TL. Reciprocal granzyme/perforin-mediated death of human regulatory and responder T cells is regulated by interleukin-2 (IL-2). J Mol Med (Berl). 2010;88:577–588. | |
Hyun-Bae J, Schuler PJ, Lee SC, et al. CTLA-4+ regulatory T cells are increased in cetuximab treated head and neck cancer patients, suppress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res. In press 2015. | |
Muller-Haegele S, Muller L, Whiteside TL. Immunoregulatory activity of adenosine and its role in human cancer progression. Expert Rev Clin Immunol. 2014;10(7):897–914. | |
Kumai T, Oikawa K, Aoki N, et al. Tumor-derived TGF-β and prostaglandin E2 attenuate anti-tumor immune responses in head and neck squamous cell carcinoma treated with EGFR inhibitor. J Transl Med. 2014;12:265. | |
Antonioli L, Blandizzi C, Pacher P, Hasko G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. 2013;13: 842–857. | |
Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. | |
Borsellino G, Kleinewietfeld M, Di Mitri D, et al. Expression of ectonucleotidase CD39 by FoxP3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. | |
Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. | |
Schuler PJ, Saze Z, Hong CS, et al. Human CD4(+) CD39(+) regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73(+) exosomes or CD73(+) cells. Clin Exp Immunol. 2014;177(2):531–543. | |
Mandapathil M, Whiteside TL. Targeting human inducible regulatory T cells (Tr1) in patients with cancer: blocking of adenosine-prostaglandin E2 cooperation. Expert Opin Biol Ther. 2011;11: 1203–1214. | |
Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, Whiteside TL. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS One. 2010;5: e11469. | |
Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–179. | |
Zhou AX, Kozhaya L, Fujii H, Unutmaz D. Garp-TGF-β complexes negatively regulate regulatory T cell development and maintenance of peripheral CD4+ T cells in vivo. J Immunol. 2013;190:5057–5064. | |
White RA, Malkoski SP, Wang XJ. TGFβ signaling in head and neck squamous cell carcinoma. Oncogene. 2010;29:5437–5446. | |
Delgoffe GM, Woo SR, Turnis ME, et al. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013;501:252–256. | |
Chaudhary B, Khaled YS, Ammori BJ, Elkord E. Neuropilin 1: function and therapeutic potential in cancer. Cancer Immunol Immunother. 2014;63:81–99. | |
Galon J, Pagès F, Marincola FM, et al. The immune score as a new possible approach for the classification of cancer. J Transl Med. 2012; 10:1. | |
Fridman WH, Galon J, Pagès F, Tartour E, Sautès-Fridman C, Kroemer G. Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res. 2011;71(17):5601–5605. | |
Nosho K, Baba Y, Tanaka N, et al. Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol. 2010;222:350–366. | |
Long SA, Buckner JH. CD4+FOXP3+ T regulatory cells in human autoimmunity: more than a numbers game. J Immunol. 2011;187: 2061–2066. | |
Kavanagh B, O’Brien S, Lee D, et al. CTLA4 blockade expands FOXP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood. 2008;112(4):1175–1183. | |
Sim GC, Martin-Orozco N, Jin L, et al. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J Clin Invest. 2014;124(1):99–110. | |
Schuler PJ, Harasymczuk M, Schilling B, et al. Effects of adjuvant chemoradiotherapy on the frequency and function of regulatory T cells in patients with head and neck cancer. Clin Cancer Res. 2013;19(23):6585–6596. | |
Colombo MP, Piconese S. Regulatory T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer. 2007;7:880–887. | |
Moltedo B, Hemmers S, Rudensky AY. Regulatory T cell ablation causes acute T cell lymphopenia. PLoS One. 2014;9:e86762. | |
Le DT, Jaffee EM. Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: a current perspective. Cancer Res. 2012;72(14):3439–3444. | |
Rech AJ, Vonderheide RH. Clinical use of anti-CD25 antibody daclizumab to enhance immune responses to tumor antigen vaccination by targeting regulatory T cells. Ann N Y Acad Sci. 2009;1174: 99–106. | |
Baur AS, Lutz MB, Schierer S, et al. Denileukin diftitox (ONTAK) induces a tolerogenic phenotype in dendritic cells and stimulates survival of resting Treg. Blood. 2013;122(13):2185–2194. | |
Finke JH, Rini B, Ireland J, et al. Sunitinib reverses type-1 immune suppression and decreases T regulatory cells in renal carcinoma patients. Clin Cancer Res. 2008;14(20):6674–6682. | |
Elkord E, Alcantar-Orozco EM, Dovedi SJ. T regulatory cells in cancer: recent advances and therapeutic potential. Exp Opin Biol Ther. 2010;10:1573–1586. | |
Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1–7. | |
Whiteside TL. Disarming suppressor cells to improve immunotherapy. Cancer Immunol Immunother. 2012;61:283–288. | |
Rech AJ, Mick R, Martin S, et al. CD25 blockade depletes and selectively reprograms regulatory T cells in concert with immunotherapy in cancer patients. Sci Transl Med. 2012;4(134):134ra62. | |
Cheng G, Yu A, Malek TR. T cell tolerance and the multi-functional role of IL-2R signaling in T regulatory cells. Immunol Rev. 2011;241: 63–76. | |
Whiteside TL. Inhibiting the inhibitors: evaluating agents targeting cancer immunosuppression. Expert Opin Biol Ther. 2010;10: 1019–1035. | |
Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;30:5346–5358. | |
Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol. 2012;3:190. | |
Stagg J, Divisekera U, McLaughlin N, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A. 2010;107:1547–1552. | |
Bastid J, Cottalorda-Regairaz A, Alberici G, Bonnefoy N, Eliaou JF, Bensussan A. ENTPD1/CD39 is a promising therapeutic target in oncology. Oncogene. 2013;32(14):1743–1751. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. | |
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. | |
Curran MA, Montalvo W, Yahita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells with B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280. | |
Whiteside TL. Regulatory T cell subsets in human cancer: are they regulating for or against tumor progression? Cancer Immunol Immunother. 2014;63:67–72. | |
Whiteside TL. Immune modulation of T-cell and NK-cell (natural killer) cell activities by TEX (tumor derived exosomes). Biochem Soc Trans. 2013;41:245–251. | |
Elkord E, Al-Ramadi BK. Helios expression in FoxP3(+) T regulatory cells. Expert Opin Biol Ther. 2012;12:1423–1425. | |
Ferraro A, D’Alise AM, Raj T, et al. Interindividual variation in human T regulatory cells. Proc Natl Acad Sci U S A. 2014;111: E1111–E1120. | |
Litzinger MT, Fernando R, Curiel TJ, Grosenbach DW, Schlom J, Palena C. IL-2 immunotoxin denileukin diftitox reduces regulatory T cells and enhances vaccine-mediated T-cell immunity. Blood. 2007;110:3192–3201. | |
Peraino JS, Zhang H, Rajasekera PV, et al. Diphtheria toxin-based bivalent human IL-2 fusion toxin with improved efficacy for targeting human CD25(+) cells. J Immunol Methods. 2014;405:57–66. | |
Smith AL, Robin TP, Ford HL. Molecular pathways: targeting the TGF-β pathway for cancer therapy. Clin Cancer Res. 2012;18:4514–4521. | |
Weber J. Immune checkpoint proteins: a new therapeutic paradigm for cancer – preclinical background: CTLA-4 and PD-1 blockade. Semin Oncol. 2010;37:430–439. | |
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. | |
Callahan MK, Wolchok JD. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol. 2013;94: 41–53. | |
Chen X, Bäumel M, Männel DN, Howard OM, Oppenheim JJ. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol. 2007;179:154–161. | |
Nagar M, Jacob-Hirsch J, Vernitsky H, et al. TNF activates a NF-kappaB-regulated cellular program in human CD45RA- regulatory T cells that modulates their suppressive function. J Immunol. 2010;184: 3570–3581. | |
Cohen AD, Diab A, Perales MA, et al. Agonist anti-GITR antibody enhances vaccine-induced CD8(+) T-cell responses and tumor immunity. Cancer Res. 2006;66:4904–4912. | |
Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood. 2005;105:2862–2868. | |
van der Most RG, Currie AJ, Mahendran S, et al. Tumor eradication after cyclophosphamide depends on concurrent depletion of regulatory T cells: a role for cycling TNFR2-expressing effector-suppressor T cells in limiting effective chemotherapy. Cancer Immunol Immunother. 2009;58:1219–1228. | |
Zhao J, Cao Y, Lei Z, Yang Z, Zhang B, Huang B. Selective depletion of CD4+CD25+Foxp3+ regulatory T cells by low-dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer Res. 2010;70:4850–4858. | |
Beyer M, Kochanek M, Darabi K, et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. 2005;106:2018–2025. | |
Ali K, Soond DR, Piñeiro R, et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510:407–411. | |
Bachanova V, Cooley S, Defor TE, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123:3855–3863. | |
Huijts CM, Santegoets SJ, van den Eertwegh AJ, et al. Phase I-II study of everolimus and low-dose oral cyclophosphamide in patients with metastatic renal cell cancer. BMC Cancer. 2011;11:505. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.