")
Back to Archived Journals » Hypoxia » Volume 2
The role of PHD2 mutations in the pathogenesis of erythrocytosis
Authors Gardie B, Percy MJ, Hoogewijs D, Chowdhury R , Bento C, Arsenault P, Richard S, Almeida H, Ewing J, Lambert F, McMullin MF, Schofield CJ, Lee FS
Received 1 February 2014
Accepted for publication 20 March 2014
Published 1 July 2014 Volume 2014:2 Pages 71—90
DOI https://doi.org/10.2147/HP.S54455
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Video abstract presented by Betty Gardie et al.
Views: 1044
Betty Gardie,1,2 Melanie J Percy,3 David Hoogewijs,4 Rasheduzzaman Chowdhury,5 Celeste Bento,6 Patrick R Arsenault,7 Stéphane Richard,1,8,9 Helena Almeida,6 Joanne Ewing,10 Frédéric Lambert,11 Mary Frances McMullin,12 Christopher J Schofield,5 Frank S Lee7
1Laboratoire de Génétique Oncologique de l'Ecole Pratique des Hautes Etudes, Villejuif, 2Unité Mixte de Recherche, Institut national de la santé et de la recherche médicale U892, Centre national de la recherche scientifique 6299, Centre de Recherche en Cancérologie Nantes/Angers, Université de Nantes, Nantes, France; 3Department of Haematology, Belfast City Hospital, Belfast, UK; 4Institute of Physiology and Zürich Center for Integrative Human Physiology, University of Zürich, Zürich, Switzerland; 5Department of Chemistry and Oxford Centre for Integrative Systems Biology, Chemistry Research Laboratory, University of Oxford, Oxford, UK; 6Department of Hematology, Centro Hospitalar e Universitário de Coimbra, Coimbra, Portugal; 7Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA; 8Institut national de la santé et de la recherche médicale U753, Institut de cancérologie Gustave Roussy (IGR), Villejuif, France; 9Faculté de Médecine Paris-Sud, Le Kremlin-Bicêtre, France; 10Heart of England NHS Trust, Birmingham, UK; 11Center for Human Genetics, Pathology Institute, UniLab-Lg, Molecular Haemato-Oncology Unit, CHU of Liege, Liege, Belgium; 12Department of Haematology, Queen's University, Belfast, UK
Abstract: The transcription of the erythropoietin (EPO) gene is tightly regulated by the hypoxia response pathway to maintain oxygen homeostasis. Elevations in serum EPO level may be reflected in an augmentation in the red cell mass, thereby causing erythrocytosis. Studies on erythrocytosis have provided insights into the function of the oxygen-sensing pathway and the critical proteins involved in the regulation of EPO transcription. The α subunits of the hypoxia-inducible transcription factor are hydroxylated by three prolyl hydroxylase domain (PHD) enzymes, which belong to the iron and 2-oxoglutarate-dependent oxygenase superfamily. Sequence analysis of the genes encoding the PHDs in patients with erythrocytosis has revealed heterozygous germline mutations only occurring in Egl nine homolog 1 (EGLN1, also known as PHD2), the gene that encodes PHD2. To date, 24 different EGLN1 mutations comprising missense, frameshift, and nonsense mutations have been described. The phenotypes associated with the patients carrying these mutations are fairly homogeneous and typically limited to erythrocytosis with normal to elevated EPO. However, exceptions exist; for example, there is one case with development of concurrent paraganglioma (PHD2-H374R). Analysis of the erythrocytosis-associated PHD2 missense mutations has shown heterogeneous results. Structural studies reveal that mutations can affect different domains of PHD2. Some are close to the hypoxia-inducible transcription factor α/2-oxoglutarate or the iron binding sites for PHD2. In silico studies demonstrate that the mutations do not always affect fully conserved residues. In vitro and in cellulo studies showed varying effects of the mutations, ranging from mild effects to severe loss of function. The exact mechanism of a potential tumor-suppressor role for PHD2 still needs to be elucidated. A knockin mouse model expressing the first reported PHD2-P317R mutation recapitulates the phenotype observed in humans (erythrocytosis with inappropriately normal serum EPO levels) and demonstrates that haploinsufficiency and partial deregulation of PHD2 is sufficient to cause erythrocytosis.
Keywords: PHD2, EGLN1, HIF, hypoxia, erythropoietin, erythrocytosis
Introduction
An erythrocytosis is present when the red cell mass is increased to greater than 125% of the predicted value for the age and body mass of the subject1 and may be reflected in an increased hemoglobin level, with or without an increased hematocrit.2 It may be necessary to formally access the red cell mass to demonstrate that it is truly increased.
There can be myriad reasons for the development of an erythrocytosis; a complete list of known possibilities is listed in Table 1.3–6 Briefly, a primary erythrocytosis arises when there is increased production of red cells in the bone marrow because of an intrinsic bone marrow defect. In these cases, the individual’s erythropoietin (EPO) levels will be below the normal range. Secondary erythrocytosis arises when the red cell production is driven by EPO production. Elevated EPO levels can arise for different reasons, as summarized in Table 1. In individuals with secondary erythrocytosis, EPO levels are elevated or inappropriately normal for the associated hemoglobin. There remains a group of individuals in whom the reason for the erythrocytosis is not clear, a category that is referred to as idiopathic erythrocytosis.
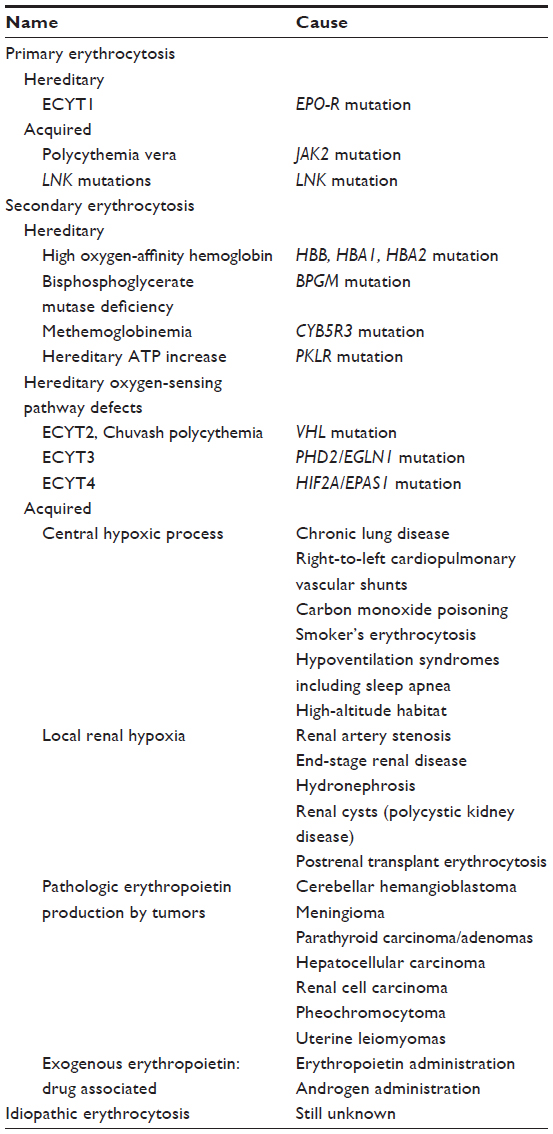 | Table 1 Classification of erythrocytoses |
Erythrocytosis can be hereditary (associated with germline mutation) or acquired, depending on whether it is present from birth or develops later in life. In hereditary cases, the erythrocytosis may not often be tested for or detected at birth, but these individuals usually present at a young age. This review focuses on secondary hereditary erythrocytosis caused by defects in the oxygen-sensing pathway that lead to EPO overexpression and, in particular, germline defects in the Egl nine homolog 1 (EGLN1) gene encoding the dioxygenase prolyl hydroxylase domain-containing protein 2 (PHD2 or EGLN1), a major player in the oxygen-sensing pathway.
Regulation of the oxygen-sensing pathway
Accurate oxygen homeostasis regulation is essential for the survival of all organisms. The transcription factor hypoxia-inducible factor (HIF), which is conserved during evolution from basal metazoans to primates, plays an essential role in oxygen homeostasis during embryonic development and postnatal life. Under limiting O2 (hypoxic) conditions, HIF upregulates the expression of a number of target genes, including that encoding the glycoprotein hormone EPO, which regulates the proliferation and differentiation of erythroid progenitors, thereby linking decreased tissue oxygenation with an adequate erythropoietic response.
HIF is an α/β hetero dimer consisting of a tightly regulated oxygen-labile α-subunit and a constitutive β-subunit. Three HIF-α isoforms exist: HIF-1α and HIF-2α, which are closely related and extensively studied, and HIF-3α, a more distantly related isoform that possibly plays an antagonistic role.
The HIF-α subunits contain an oxygen-dependent degradation (ODD) domain that confers oxygen-dependent instability and two independent transcriptional activation domains (HIF-1α N-TAD: amino acids 531–575; C-TAD: amino acids 786–826). Hydroxylation is the main posttranslational modification regulating the oxygen-dependent stability of HIF-α subunits. In humans, HIF-α hydroxylation is catalyzed by three prolyl hydroxylase domain proteins (PHD 1–3) and an asparaginyl hydroxylase, factor-inhibiting HIF (FIH), collectively called the HIF hydroxylases. The HIF hydroxylases are dioxygenases, which use 2-oxoglutarate (2-OG) and oxygen as cosubstrates, providing a molecular basis for the oxygen-sensing function of these enzymes.7,8 The PHD proteins control the stability of HIF-α subunits, and FIH regulates their transcriptional activity. PHDs hydroxylate prolyl residues located within the HIF-α ODD (P402 and P564 for HIF-1α and P405 and P531 for HIF-2α), which allows binding of HIF-α to the von Hippel–Lindau (VHL) protein, a recognition component of an E3 ubiquitin ligase multiprotein complex. Binding of VHL to HIF-α isoforms induces ubiquitination that targets them for degradation by the proteasome (Figure 1). Of the three PHD isoforms (1–3) in mammalian cells, PHD2 appears to be a particularly critical oxygen sensor controlling HIF-1α stability in vivo.8–10 FIH hydroxylates the asparagine in the C-TAD (Asn 803/HIF-1α, Asn 847/HIF-2α), a modification that inhibits the binding of HIF-α to the p300/CBP transcriptional coactivator proteins.11
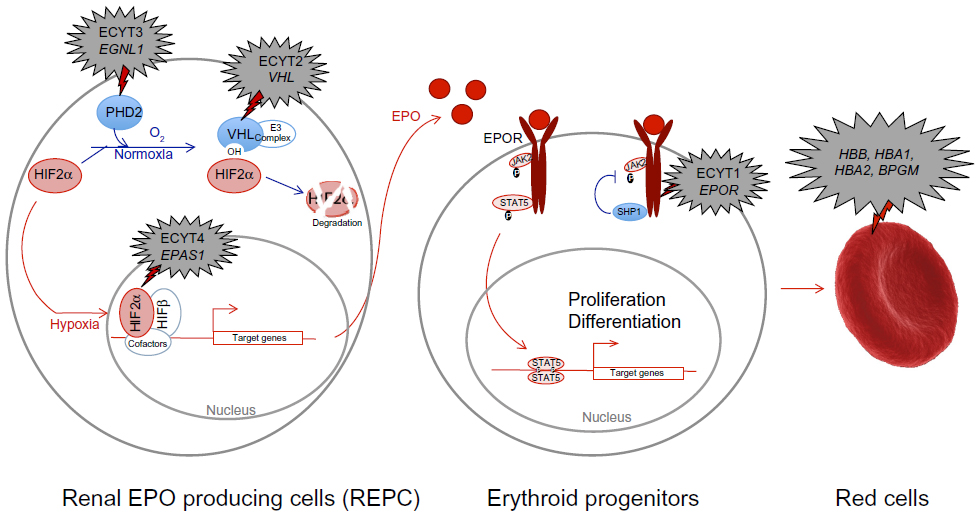 | Figure 1 Hypoxia and red cell production signalization pathway with associated mutations responsible for erythrocytoses. |
Under hypoxic conditions, when the cofactor oxygen is limiting, hydroxylation of HIF-α subunits slows down, resulting in HIF-α stabilization. HIF-α then translocates to the nucleus, associates with the HIF-1β subunit, and forms an active heterodimer. On recruiting appropriate coactivators such as p300/CBP, the HIF-α/β heterodimer binds to hypoxia response elements [HREs; pentanucleotide sequence 5′-(A/G)CGTG-3′] within DNA and activates expression of HIF target genes.12,13 More than 200 genes are transcriptionally regulated by HIF, and many genes are involved in cell survival, including erythropoiesis (via the synthesis of EPO14). The regulation of HIF target genes is very complex and not yet fully understood. It depends on the cell type, the specific HIF-α subunit, the N- or C-terminal transactivation domain involved,15 and the oxygen concentration, as well as other context-dependent factors. Moreover, the PHDs and FIH have distinct and different affinities for oxygen, as reflected by the Km values for oxygen for PHD2 and FIH. PHDs have a lower affinity, and hence are inhibited more rapidly than FIH when the concentration of oxygen decreases (other factors being equal). Therefore, with moderate hypoxic conditions, the HIF-α subunits are stabilized (PHDs are inhibited), but the C-TAD is still inhibited (FIH is active); as a consequence, only genes regulated by the N-terminal transactivation domain are induced.16 Furthermore, PHDs have been shown to manifest a certain degree of selectivity between HIF-1α and HIF-2α.9
Regulation of the EPO and red cell production
The major HIF-α isoform involved in the regulation of the EPO gene is HIF-2α,17–20 which also regulates genes required for cell survival under low oxygen tension, such as heme synthesis (ALAS2) and iron regulation (TRF2, TF).12,21–24 During development, EPO production switches in a species-specific manner from the liver to the kidney, the latter of which accounts for ~90% of total EPO synthesis in the adult.25 Hypoxia-inducible EPO transcription is controlled by distinct regulatory sequences in liver and kidney. These regulatory regions flank the EPO coding sequence on both sides: the enigmatic kidney-inducible element located far upstream in the 5′-regulatory region and the well-established liver-inducible element in the proximal 3′-region. Although the liver-inducible element has been extensively characterized26,27 and confirmed to be both essential and sufficient for liver-specific EPO expression in mice beyond embryonic day 14.5,28 only very recently was the first functional analysis of a highly conserved distal 5′-HRE, presumably representing the kidney-inducible element, reported.29 This distal 5′-HRE is located 9,248 bp upstream of the human EPO transcriptional start site, is composed of a consensus 5′-ACGTG-3′ HIF binding site and an adjacent 5′-CACA-3′ sequence very similar to the 3′-HRE, and was shown to confer oxygen-dependent EPO transcriptional regulation by HIF-2.
Extensive research efforts have been undertaken to identify the renal EPO-producing cells. In rodents, EPO production is mainly restricted to interstitial peritubular fibroblast-like cells localized in deep cortex and outer medulla.19,30–36 In addition, genetic deletion of Vhl in renin-expressing cells converted these cells in EPO-producing cells.37 In humans, the precise localization of EPO-producing cells remains unclear. In situ hybridization studies showed EPO mRNA expression in cells of renal tubules;38 a recent study has isolated peritubular cells with mesenchymal phenotype as a possible source of EPO production.39
After renal production, EPO is secreted into the circulation and enters the bone marrow, where it stimulates the proliferation of red blood progenitors (Figure 1). EPO binds its receptor EPOR on the surface of erythroid cells and induces a cascade of phosphorylation. The protein Janus kinase 2 (JAK2) is autophosphorylated and subsequently phosphorylates tyrosines located in the cytoplasmic region of the EPOR. These tyrosines act as an anchor point to many positive regulatory proteins, such as signal transducer and activator of transcription factor 5 (STAT5). After its attachment to phosphorylated EPOR, STAT5 homodimerizes and translocates into the nucleus, where it activates the transcription of various genes involved in the proliferation of erythroid precursors and the production of red blood cells. The phosphorylated tyrosines in the C-terminal domain of EPOR also serve as an anchor point to inhibitory proteins such as Src homology 2 domain-containing phosphatase 1 region (SHP1). About 30 minutes after EPO binding, SHP1 protein is recruited by EPOR and dephosphorylates it, as well as JAK2. This negative feedback loop results in the ubiquitination and degradation of EPOR.
Germline defects in some of the genes encoding proteins in red blood cells, including ones in the EPO signal transduction pathway, have been identified in hereditary erythrocytosis.40 Mutations in genes encoding hemoglobin or bisphosphoglycerate mutase induce increased affinity for oxygen by hemoglobin reducing available oxygen to the tissues. This apparent hypoxia then increases EPO synthesis to drive red cell production (Table 1 and Figure 1). Mutations in the receptor of EPO (EPOR) (responsible for ECYT1 [erythrocytosis type1]) induce the loss of the negative feedback control by SHP and the constitutive activation of the EPO signaling pathway. As discussed in more detail later, germline mutations in genes encoding regulators of the hypoxia pathway (VHL [ECYT2], EGLN1/PHD2 [ECYT3], and EPAS1/HIF2A [ECYT4]) lead to HIF-α stabilization and, ultimately, increased (or inappropriately normal) EPO levels, which would cause a secondary erythrocytosis.
Erythrocytoses described in patients with EGLN1 mutations
The first molecular defect to be associated with erythrocytosis was reported in 1993 by de la Chapelle et al.41,42 By studying a large family with erythrocytosis (33 affected patients over five generations), they uncovered significant linkage of the phenotype with the EPOR. Subsequent analysis of the EPOR gene revealed a nonsense mutation that truncated the receptor and resulted in dysregulation of the EPO signaling pathway and hypersensitivity to EPO. The first EPOR mutation was detected in cases with reduced EPO levels, and subsequently, many other EPOR truncation mutations have been described in similar erythrocytosis patients.40 However, none of the numerous erythrocytosis cases that exhibited elevated or inappropriately normal EPO levels were found to possess EPOR mutations, and their molecular basis remained uncharacterized.
It was not until a decade later that the next major discovery in the erythrocytosis field was made by Ang et al.43 Linkage studies revealed a region on chromosome 3 that cosegregated with the erythrocytosis phenotype in multiple families. This region contains the VHL gene, and sequencing uncovered a homozygous mutation of arginine to tryptophan at amino acid 200 (p.Arg200Trp, VHL-R200W). Defects in VHL were already known to cause a hereditary cancer syndrome, von Hippel–Lindau disease; very occasionally, erythrocytosis is concurrent with this syndrome because of EPO production by the tumor.44 This erythrocytosis phenotype could be explained by the disruption of VHL-mediated ubiquitination of the α subunit of the HIF transcription complex, resulting in upregulation of HIF target genes, which include EPO. The VHL mutation was initially detected in individuals from the Chuvash region of Russia, where erythrocytosis was endemic,45 but was later found to have a global distribution.46 This shone the spotlight on the oxygen-sensing pathway as a potential cause of the erythrocytosis in individuals with elevated or inappropriately normal EPO levels. Although many cases of VHL-associated erythrocytosis were characterized, once again there was a cohort of individuals with erythrocytosis who had a similar phenotype but were negative for VHL mutations.
At this time, the first characterized isoform of the HIF-α subunit, HIF-1α, was assumed to be the main HIF-α isoform that controlled EPO synthesis. Knowing that hydroxylation plays a major role in HIF stabilization and subsequent activity, Percy et al47 reasoned that diminished hydroxylation of HIF-1α could be a potential mechanism to allow the α subunit to escape proteasomal degradation and cause elevated EPO synthesis. As a consequence, they envisaged that disruption of the ODD domain structure could influence the association of HIF-1α with VHL and, hence, reduce ubiquitination of the α subunit. Screening the ODD domain in erythrocytosis patients detected one change, the p.Pro582Ser (P582S) polymorphism, at a higher frequency than in non-erythrocytosis controls.47 However, although the affected amino acid Pro582 is located in the ODD domain, functional analysis did not reveal an impairment of the binding of HIF-1α to VHL, and hence, this mutation could not account for erythrocytosis in individuals with the polymorphism. Nonetheless, the possibility remained that the HIF pathway was somehow implicated in VHL-negative erythrocytosis.
It had been established previously that the VHL-R200W mutation allows HIF-α to escape proteasomal regulation by inefficient ubiquitin modification. Before VHL can bind to HIF-α, there must be prolyl hydroxylation in the ODD, and if this was diminished, then the α subunit would be less efficiently ubiquitinated by VHL. Therefore, one may hypothesize that disruption of prolyl hydroxylation by an impaired PHD enzyme could result in erythrocytosis. As a consequence, a screening program of the three PHDs (1–3) was initiated in individuals who were previously negative for VHL mutations. This approach revealed heterozygous defects in the EGLN1 gene, but not in the EGLN2 or EGLN3 genes. The first reported PHD2 mutation, p.Pro317Arg (PHD2-P317R), was present in three members of the same family from two generations (Table 2).48 Residue Pro317 is located in a conserved region of PHD2 within the catalytic domain. It is close in primary structure to two of the three iron-chelating residues at 313 and 315, and as revealed by crystallographic studies, in the three-dimensional PHD2 structure, Pro317 is in the vicinity of the active site (also, see the “Structural insights into clinical mutations” section). In contrast to the VHL-R200W mutation, which is usually associated with substantially elevated serum EPO and raised hemoglobin, the PHD2-P317R mutation results in an EPO level in the normal range, and subtly raised hemoglobin. Functional studies confirmed that this mutation diminishes the ability of PHD2 to hydroxylate the HIF-α subunit as compared to the normal wild type hydroxylase.
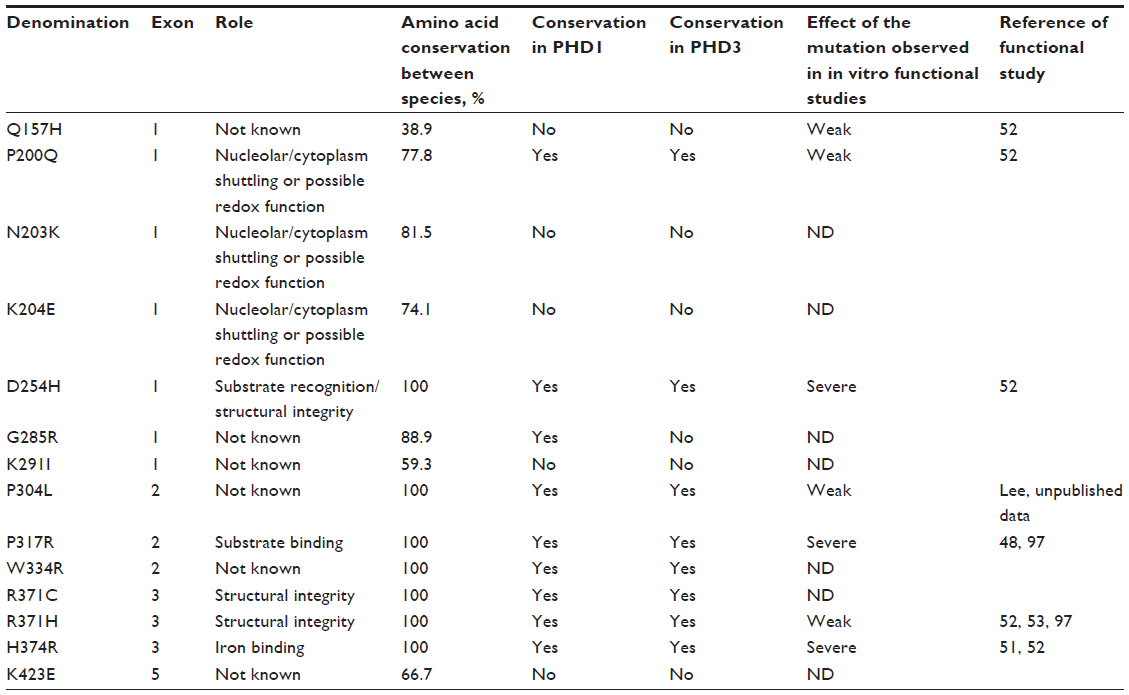
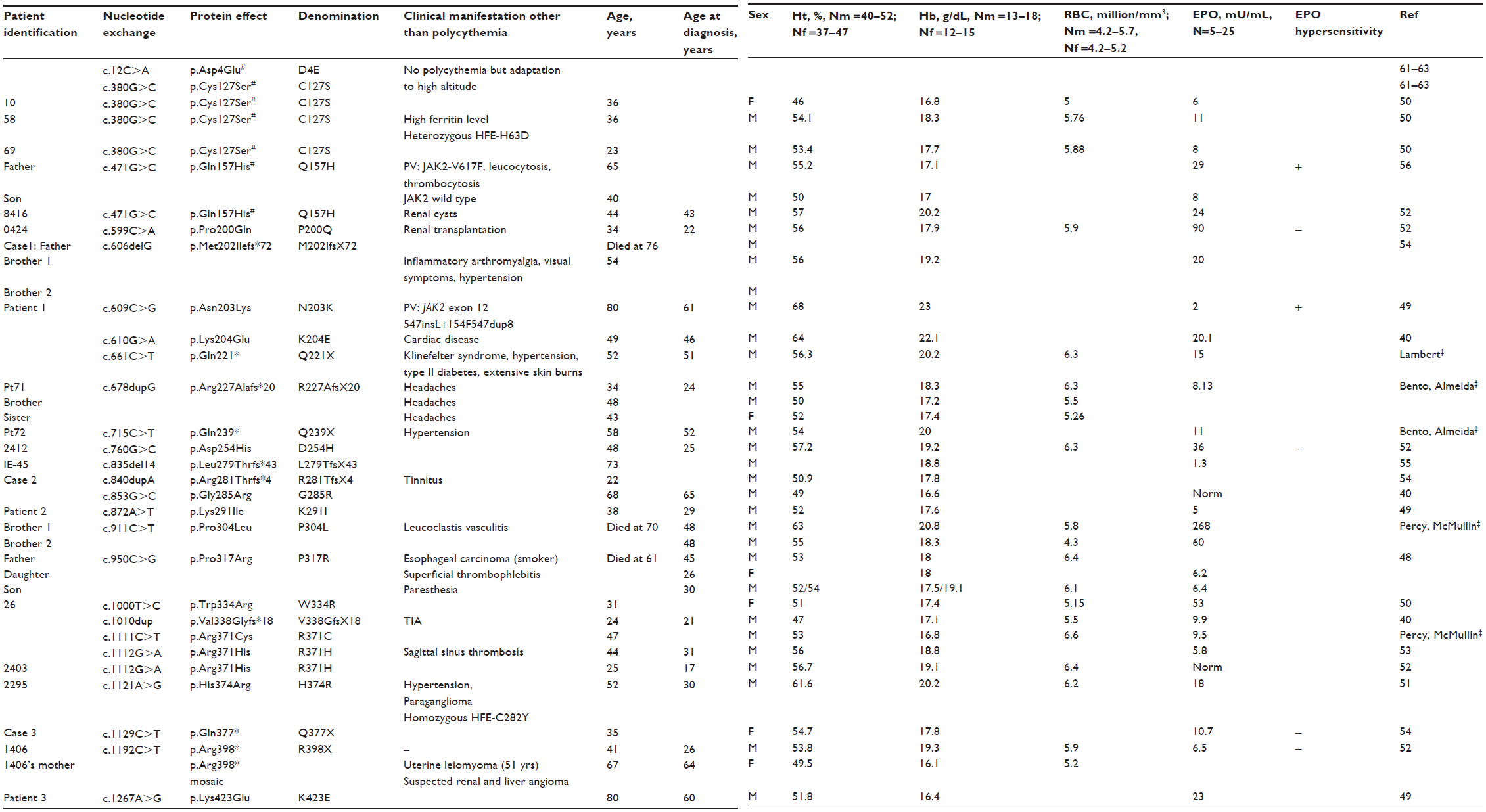 | Table 2 Description of PHD2 mutations identified in patients with erythrocytosis |
Subsequent studies of patients with erythrocytosis have identified a range of germline mutations in the EGLN1 gene, leading to PHD2-P200Q, N203K, K204E, D254H, G285R, K291I, P304L (Percy and McMullin, unpublished data, 2004), W334R, two independent cases of R371H, H374R (associated with recurrent paraganglioma), and K423E mutations (Table 2).40,49–53 More recently, a new mutation has been identified at the Arg371 residue, but with a different amino acid change (R371C) (Percy and McMullin, unpublished data, 2013), which strengthen the proposed importance of this residue in the PHD2 function. In addition, frame-shift (M202IfsX72, R227AfsX20 [Bento and Almeida, unpublished data, 2014], L279TfsX43, R281TfsX3, p.V338GfsX18) and nonsense mutations producing predicted C-terminally truncated proteins (Q221X [Lambert, unpublished data, 2013], Q239X [Bento and Almeida, unpublished data, 2014], Q377X, R398X) have been identified,40,52,54,55 and two polymorphisms described in the normal population have been identified in patients with erythrocytosis (PHD2-C127S and PHD2-Q157H).50,52,56 These studies identify a total of 37 patients with erythrocytosis who carry 24 different EGLN1 mutations. The sex ratio is unbalanced, with only six women versus 31 men. There is no hot spot of mutations (with the exception of 3 cases targeting the residue Arg371; otherwise, mutations are dispersed all through the protein). The mode of transmission is autosomal dominant but only six cases have been reported with a familial history, including one (R398X) with a mosaicism. In the majority of cases, the genotyping of the patient’s relatives was not possible (deceased parents, for example), which reduces the possibility of an accurate and enlarged phenotypes analysis. With the exception of erythrocytosis, which is modest in some cases, patients do not present other common clinical signs. Several cases have been described with hypertension (in one case with an H374R mutation related to catecholamine secretion by a paraganglioma), thrombotic events or benign renal cysts, or angioma. Two cases of erythrocytosis have been associated with hemochromatosis as a result of mutations in the high iron Fe (HFE) gene. The EPO level, when measured, is typically normal and rarely elevated (only in five cases). Two cases (patients with Q157H and N203K mutations) have been described with a polycythemia vera associated with a JAK2 mutation (the only cases with erythroid progenitors hypersensitive to EPO). Defining the contribution of the germline EGLN1 mutations to the occurrence of the acquired JAK2 mutation and the myeloproliferative neoplasms requires more extensive studies.49,56
The absence of mutations in both the EGLN2 and EGLN3 genes (which encode PHD1 and PHD3, respectively) in individuals with erythrocytosis and the detection of an increasing number of heterozygous EGLN1 mutations pointed to the potential involvement of PHD2 in the regulation of the HIF-α subunit and the development of erythrocytosis. Furthermore, this indicates there is little redundancy in the oxygen-sensing pathway regulating EPO, in that neither PHD1 nor PHD3 alone was able to, at least completely, compensate for the loss of PHD2 function.
PHD2 and other adaptations/ pathologies
The HIF pathway plays a crucial role in regulation of the normal oxygen homeostasis, and it can be activated in pathophysiological processes such as erythrocytosis, as described earlier. Moreover, genomewide association studies of populations living at high altitude identified single-nucleotide polymorphisms in regions that contain HIF pathway genes.57,58 Indeed, adaptation to high altitude includes protection from erythrocytosis. Noteworthy, multiple single-nucleotide polymorphisms located in the noncoding regulatory regions of EGLN1 have been associated with the decreased hemoglobin that is observed in the Tibetan population adapted to living at high altitude.59 Different studies on altitude-specific habitants (highland Tibetans and Andeans, moderate-altitude Eurasians) identified EGLN1 as a shared locus with genetic signatures in all of these populations, emphasizing a major role of EGLN1 in physiological hypoxia adaptation.58,60 Xiang et al resequenced the complete genomic region of EGLN1 in a series of Tibetans and discovered one nonsynonymous mutation in the coding sequence (PHD2-D4E) that is highly prevalent in Tibetans but extremely rare in lowlander Han Chinese (Table 2).61 Another polymorphism has been identified (PHD2-C127S), but the allelic divergence between Tibetans and non-Tibetans at this site was reported to be much smaller. Functional study of the PHD2-D4E-C127S mutant showed a slightly increased activity compared with the wild-type protein.62,63 A recent publication reports that the Tibetan PHD2 haplotype (D4E/C127S) strikingly diminishes the interaction of PHD2 with p23, a HSP90 cochaperone, resulting in impaired PHD2 downregulation of the HIF pathway.64 These results suggest that EGLN1 has been under Darwinian positive selection and may contribute to high-altitude hypoxic adaptation in Tibetans.65
It has long been recognized that HIF plays a major role in tumor development, angiogenesis, and dissemination.66 Indeed, many HIF target genes are involved in cell proliferation and survival, such as angiogenesis (vascular endothelial growth factor and platelet derivative growth factor), cell proliferation (transforming growth factor alpha), regulation of glucose uptake and metabolism (glucose transporter), autophagy, and cell cycle regulation. Many tumors overexpress HIF, which is associated with aggressiveness and adverse prognosis. In hereditary cancer syndromes, the HIF pathway is also targeted. Germline mutations in the VHL tumor suppressor gene, which plays a major role in the direct downregulation of HIF, predispose patients to von Hippel–Lindau disease. This rare condition predisposes to the development of very vascularized tumors from the central nervous system and retina (hemangioblastomas), pheochromocytomas, pancreatic endocrine tumors and cysts, and clear cell renal cell carcinoma.67 Of note, HIF-2α is the isoform that plays a major role in clear cell renal cell carcinoma development.68–71
Other, more indirect genetic alterations induce stabilization of the HIF pathway. For example, mutations in the Krebs cycle enzymes fumarate hydratase (FH) and succinate dehydrogenase (SDH) apparently activate HIF.72–76 In these cases, HIF activation could be a consequence of inhibition of the PHD by accumulated fumarate/or succinate, which are proposed as 2-oxoglutarate analogs and competitors.77,78 Mutations to isocitrate dehydrogenase lead to substantially increased levels of 2-hydroxyglutarate, which is also proposed to inhibit the PHDs,79 although biochemical studies have revealed it is a weak PHD inhibitor80 and, in some studies, even a PHD activator.81 Germline mutations in FH predispose to the development of papillary type II renal cancers and leiomyomas; germline mutations in SDH predispose to hereditary pheochromocytoma and paraganglioma (extra-adrenal pheochromocytoma) syndrome and rare renal cancers. Therefore, even though the PHD/VHL/HIF system is expressed ubiquitously in mammalian cells, associated cancers are highly tissue-specific, being largely confined to renal cancer and pheochromocytoma/paraganglioma. Indeed, paragangliomas have already been described in patients with an erythrocytosis carrying a particular germline EGLN1 mutation (PHD2-H374R) and a EPAS1 mutation (HIF-2α-F374Y).51,82 Examination of the paraganglioma from the patient with the PHD2-H374R mutation indicated a loss of the remaining EGLN1 wild-type allele in the tumor.51 EGLN1 may therefore be a potential tumor suppressor gene, as was previously suggested.83,84 The exact mechanism of the paraganglioma development in this patient is not elucidated, but a quantitative effect may be involved, as has been suggested in patients with the Chuvash VHL-R200W mutation who develop erythrocytosis but no tumor.45,85 Indeed, functional study of the PHD2-H374R mutation indicated a severe loss of function of this mutant with respect to HIF regulation compared with other mutations associated with erythrocytosis.52
To date, none of the EGLN1 or EPAS1 germline mutations that have been described predispose patients to renal cancer.86 A high HIF level may be necessary to cancer development.87–89 Mutations in EGLN1 or EPAS1 may not be sufficiently deleterious to induce high levels of HIF. Indeed, mutations in EGLN1 may be compensated for by other PHD isoforms (PHD1 or PHD3) not sufficiently active to override erythrocytosis but sufficient to avoid tumor transformation. Regarding EPAS1 mutations, none of the germline mutations identified in patients with erythrocytosis target the sites of prolyl hydroxylation (Pro405 and Pro531), although Pro531 mutations have been reported in sporadic pheochromocytomas.90,91 Conceivably, high-level activation of HIF-2α may be necessary for renal tumorigenesis but perhaps may not be compatible with life when constitutive and global.
Functional studies
Structural insights into clinical mutations
PHD2 is a 46-kDa enzyme that contains of an N-terminal MYND zinc finger and a C-terminal catalytic domain, the latter with a double-stranded β-helix core-fold, which is archetypical of non-heme Fe(II)/2-OG dioxygenases. The active site iron is located within the double-stranded β-helix core and is octahedrally coordinated, including by a HisXAsp/Glu...His facial triad. Binding of the His facial triad residues from the double-stranded β-helix92,93 leave two coordination sites for the 2-OG oxalyl group and one for H2O/O2 binding. During the catalytic cycle, the enzyme forms a reactive oxidizing species (Fe[IV]=O) that oxidizes its substrate(s) to give the hydroxylated product(s).94 The active site is lined by hydrophobic residues that likely prevent any potential oxidative damage by the reactive oxidizing species via Fenton-type chemistry.92,93 Biochemical studies have demonstrated that because of its high affinity, the enzyme forms a long-lived complex with Fe(II) and 2-OG.95 Further studies have revealed that PHD2 has a high Km for oxygen slightly above the atmospheric concentration, a property that marks a direct link between molecular dioxygen and PHD catalysis and renders PHD2 as an important oxygen sensor in cells.96
Depending on their location on the PHD2 domains, mutations do not necessarily have, of course, the same impact on PHD2 catalysis.97 The possible functional consequences of identified mutations to PHD2 are discussed here in the light of structural analyses, principally by crystallography, on PHD2.92,93 It is perhaps important to state that with the HIF system, as with other transcriptional regulatory systems, detailed chemical understanding is only at an early stage of development. Thus, other than situations in which there is a complete loss of active protein, in only a few cases can (predicted) protein modifications be directly linked with functional outcomes, especially in cells. Nonetheless, for reasons including the fact that HIF-α is highly inducible and HIF activity is related to clear phenotypes, the HIF system would appear to be an excellent model system for developing a molecular understanding of how pathophysiological consequences arise from mutations. In the following paragraph, we outline early-stage studies showing how mutations to PHD2 and HIF-2α may have consequences for PHD-catalyzed HIF hydroxylation.
Biochemical studies reveal a significant loss of PHD2 activity in isolated protein form when it is truncated in its C-terminal region (residues 395–399).98 The shortest isolated PHD2 that retained near full activity when compared with wild-type PHD2 was truncated at residue 402, which is located immediately after the C-terminal α-helix. Significant sequence variation occurs to the C-terminal side of residue 402 in the three human different PHD isoforms.98 Together with insights from a PHD2.CODD556–574 complex crystal structure,92 these biochemical data demonstrate an important role of C-terminal helix α4 in substrate binding. Structurally informed sequence comparisons indicate that the frameshift and nonsense mutations will produce PHD2 proteins lacking a competent catalytic domain (if expressed) and, thereby, prevent PHD2-catalyzed HIF degradation.
In contrast, the likely molecular consequences of the reported mutations leading to residue substitutions are less clear. HIF binding involves multiple residues from the PHD catalytic domain in addition to the C-terminal domain (Figure S1). On the basis of the available structural and biochemical information on the interaction of HIF and PHD2, five identified substituted residues, Asp254, Lys291, Pro317, Arg371, and His374, are located near the HIF/2-OG or ferrous iron binding sites of PHD2 (Figure 2).
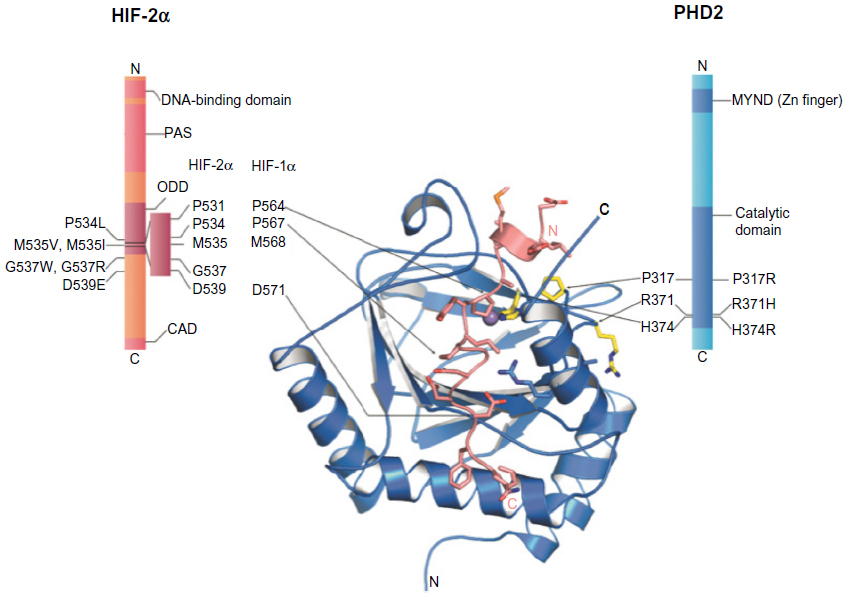 | Figure 2 Erythrocytosis-associated mutation sites in human prolyl hydroxylase domain 2 (PHD2) and HIF-2α. |
Mutations to Asp254 lead to (near) complete loss of hydroxylation activity, likely because this residue is crucial for maintaining a substrate recognition loop in the active enzyme state (Table 3).52,92 PHD2-P317R (full-length) displays a kcat/Km value toward a HIF-2α (467–572) peptide that is 18% of that of wild-type PHD2.97 In the PHD2 crystal structures, Pro317 is close to the iron coordinating HXD motif (His313 and Asp315 are two of the three iron ligating residues), whereas Arg371 is close to the other metal coordinating residue, His374 (β9).93 In a PHD2.CODD(HIF-1α) complex crystal structure, Pro317 forms part of a hydrophobic region that interacts with the 310-helix of the hydroxylated LXXLAP motif (Figure 2). Lys291 and Arg371 are relatively remote from the substrate-binding region; Arg371 is involved in making an internal salt-bridge interaction with Asp369 (Asp369 Oδ1-Arg371 Nε, 3.2 Å; Asp369 Oδ2-Arg371 NH2, 3.0 Å) and thus likely has a structural role. The presence of two different mutations at Arg371 highlights the importance of this residue for the function of PHD2, even though it is not at the iron binding site. Because His374 is one of the three iron binding residues (‘facial triad’), arginine substitution at this position will likely substantially hinder iron binding and thereby ablate, or substantially reduce, activity. In vitro functional studies described a total loss of function of the PHD2-H374R mutant.51
 | Table 3 Summary of functional studies data |
Ten heterozygous mutations have been identified in exon 12 of the EPAS1 gene, leading to HIF-2α variants.82,99–104 These variants have been linked with idiopathic erythrocytosis, and in all cases, patients were diagnosed with elevated EPO levels. Interestingly, the majority of these missense mutations (I533V, P534L, M535T, M535V, M535I, G537W, G537R, D539E, and F540L) cluster close/C-terminal to one of the two hydroxylated proline residues (Pro531) in HIF-2α. The combined biochemical and cellular assays suggest that most of these mutations may reduce both hydroxylation of HIF-2α by the PHDs (full-length) and subsequent recognition of (likely hydroxylated) HIF-2α by pVHL, except for mutations at Met535, which may only impair interactions with PHD2.4,105 Although detailed insights into the molecular mechanism behind the predicted reduced binding of the HIF-2α variants with PHD2 are unclear, close inspection of a PHD2.CODD (HIF-1α) crystal structure suggests these mutated residues bind in a region with some conformational flexibility, suggesting they may reduce, but not completely block, HIF-2α substrate binding to PHD2.
In silico studies
Conservation of sequences and residues can provide information on functional significance. To assess the conservation of all PHD2 mutated residues described in Table 2, a multiple sequence alignment was generated covering a broad range of taxa from different phyla. Vertebrate homologs across 100 species including primates, mammals, birds, and fish were extracted, using the MULTIZ whole-genome multiple alignment algorithm106 implemented in the University of California, Santa Cruz, Genome Browser.107 Invertebrate homologs were obtained using the FlyBase implemented InParanoid algorithm.108 Individual BLASTP (protein-protein Basic Local Alignment Search Tool) searches were performed to find homologous PHD2 sequences in representative species of various protostome and deuterostome taxa, including Urochordata, Cephalochordata, Echinodermata, Hemichordata, Gastropoda, Annelida, Crustacea, Hexapoda, Nematoda, and Placozoa. Trichoplax adhaerens, part of the latter phylum, has been described as the most basal multicellular metazoan organism containing a functional HIF system.109 Human PHD1 and PHD3 sequences were added to the PHD2 ortholog sequence set. Species with large gaps were manually omitted. Subsequently, all sequences were realigned using the MAFFT version 7 multiple alignment tool with an iterative refinement option incorporating local pairwise alignment information (L-INS-i) and default parameters.110 Gaps introduced in mammalian PHD2 homologs by alignment to distantly related invertebrate sequences were deleted.
The multiple sequence alignment clearly reflects more conservation within the C-terminal catalytic domain (Figure 3). Interestingly, the mutation in the fourth codon of PHD2 (D4E), recently identified independently by various groups, affects a strongly conserved amino acid in mammals, but not in lower vertebrates or in human PHD1 or PHD3. The PHD2-C127S mutation, in contrast, occurs in a residue that is not conserved at all in any primate species. The Gln157 amino acid is only moderately conserved between mammals, and not within the PHD protein family (PHD1 and PHD3). The residues affected by the PHD2-P200Q and the PHD2-M202I mutations are conserved in all vertebrates, but only Pro200 is conserved in both PHD1 and PHD3. Both the PHD2-P200Q and M202I mutations affect residues that immediately flank C201, which has been shown to chelate zinc and cadmium ions, providing evidence for the existence of a second metal binding site on PHD2.111,112 In contrast, the PHD2-N203K, K204E, G285R, and K291I mutations affect residues not strictly conserved in all vertebrates. The active site residue D254 (the target of the PHD2-D245H mutation) is strictly conserved in all eukaryotes. Other strictly conserved residues in metazoans and both PHD1 and PHD3 have been targeted by mutations PHD2-P304L, P317R, W334R, R371C, R371H, and H374R, with the latter mutation in one of the three invariant iron coordination sites and being associated with paraganglioma.51 Although not part of the catalytic domain, PHD2-K423E occurs in a residue that is highly conserved down to the coelacanth Latimeria chalumnae.
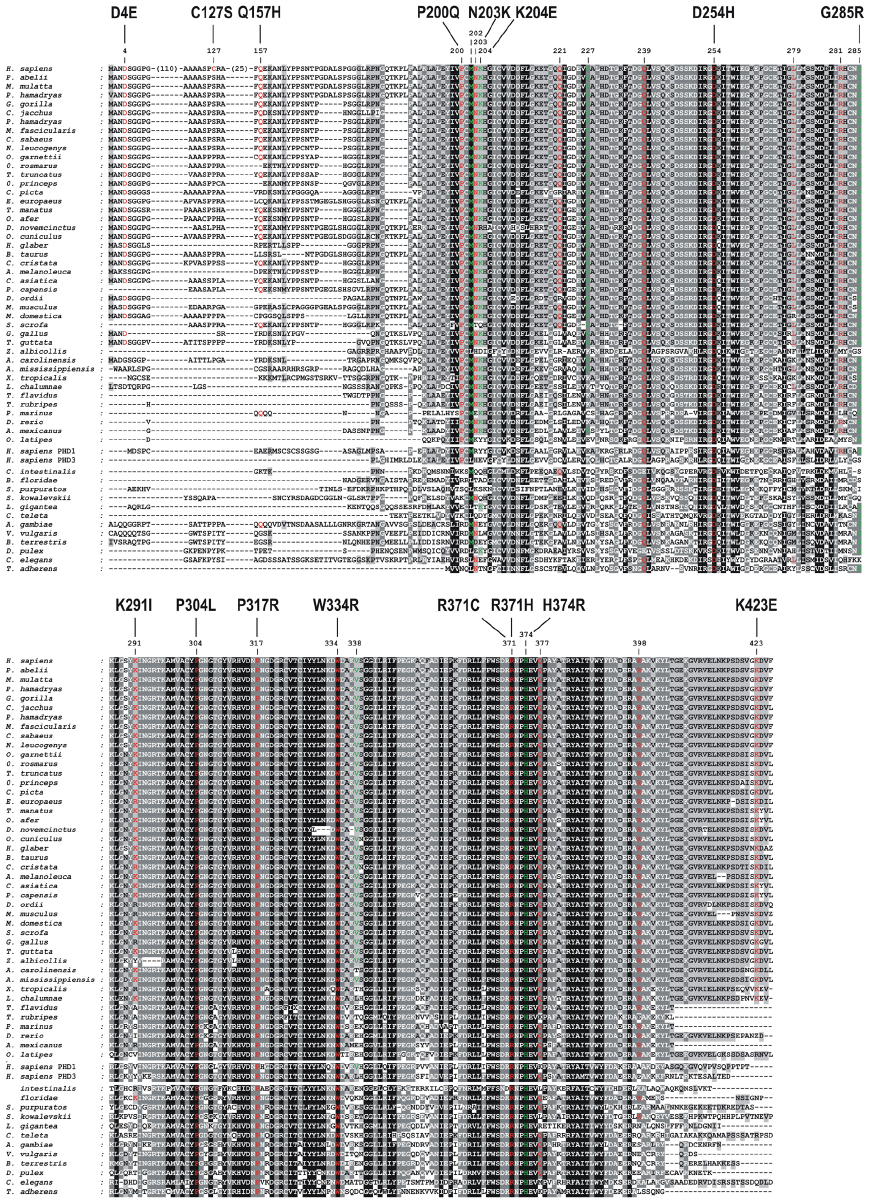 | Figure 3 Multiple sequence alignment of PHD2 homologs. |
In vitro functional studies
Different assay methods have been described to test the prolyl hydroxylase activity of PHD2.113 PHD2 is a member of the Fe(II) and 2-OG-dependent oxygenase superfamily in which almost all members appear to follow the same overall reaction mechanism: in the presence of cosubstrates (2-OG, dioxygen) and cofactors (Fe(II) and, sometimes, ascorbic acid), the substrate (HIF-α) is hydroxylated and the hydroxylation reaction is accompanied by oxidative decarboxylation of 2-OG to give CO2 and succinate. Indirect measurement of PHD2 activity can therefore be inferred, for example, by quantification of the amount of dioxygen consumed or by the release of 14CO2 from 142-OG,114 although direct measurement of hydroxylation is more reliable. These assays are generic to the dioxygenase family. A direct quantification of PHD2 activity can be measured by the amount of hydroxylated HIF-α produced after reaction with PHD2. A peptide containing the proline of C-terminal ODD domain of HIF-1α (P564) is normally used (amino acids 556–574 or 549–575) and is amenable to mass spectrometric analyses. It is also noticeable that hydroxylation induces a mobility shift of a GAL4 fusion protein containing a HIF-1α peptide (amino acids, 531–652) after electrophoresis on an sodium dodecyl sulfate-polyacrylamide gel electrophoresis.115 Another direct quantification of HIF hydroxylation can also be performed by using antibodies specific to the hydroxylated proline.52,116 Indirect measurement of HIF hydroxylation is frequently performed by using VHL capture assays.48,53 In vitro assays to measure PHD2 binding to HIF have also been performed.48,53 These reactions used recombinant proteins that are either produced in bacteria or are in vitro translated (with wheat germ extract to avoid contamination with endogenous hydroxylases). In cellulo functional study of PHD2 mutants has used reporter gene assays.48,51,52 The inhibitory effect of PHD2 on HIF activity (endogenous52 or transfected48) has been performed with luciferase reporter genes under the regulation of HRE. PHD2 mutants have also been studied using a one-hybrid reporter assay on the basis of the capacity of PHD2 to induce HIF-α protein instability.52,117
Several missense PHD2 mutations have been tested (results are summarized in Table 3). In the case of the P317R mutation, functional studies supported a loss of function of the mutants instead of a dominant negative inhibitory effect.48 The mutations substantially decrease, although do not abolish completely, PHD2 activity, with the exception of two mutations with a severe loss of function (PHD2-H374R and PHD2-D254H).52 Some of the mutants even present with a very subtle loss of activity, close to the wild-type protein (PHD2-P200Q, PHD2-R371H in one study52 and P304L [Lee, unpublished data, 2012]). More-sensitive assays should be developed to determine subtle effects of these PHD2 mutations (and polymorphisms such as PHD2-Q157H) that are difficult to study with current in vitro and in cellulo assays based on overexpressed proteins. The studies, in some instances, showed the same effect of the mutants on different HIF-α isoforms (HIF-1α and HIF-2α) and under various oxygen concentrations.
The hydroxylase activity of PHD2 may not necessarily be the only functionally relevant target of the mutations. PHD2 binds a number of proteins, and the effect of the mutations on this binding could conceivably be affected.118–120 Notably, PHD2 binds FKPB38, which plays a major role in PHD2 stability.121 PHD2 also regulates angiogenesis and vasculogenesis via the regulation of nuclear factor kappa-B122 in a likely HIF-independent pathway that may also be affected by the mutations. It has also been shown that PHD2 undergoes nuclear-cytoplasmic shuttling and that the deletion of PHD2 sequences required for nuclear entry is associated with a greatly impaired capacity to downregulate HIF.123,124 PHD2 residues 196–205 have been identified as crucial for nuclear import. Some PHD2 mutants (including the PHD2-P200Q mutant) have been tested for the capability to shuttle between the cytoplasm and the nucleus, but so far, no effect of the mutations has been observed (Ladroue et al;52 Gardie and Ladroue, unpublished data, 2010).
Mouse models of PHD2 inactivation
Two implications that followed the initial identification of the family with a heterozygous PHD2-P317R mutation and erythrocytosis48 are that in the EPO pathway, the PHDs are not redundant, such that loss of PHD2 function could not be compensated for by PHD1 or PHD3, and the EPO pathway is sensitive to modest changes in PHD2 activity, such that even a heterozygous PHD2 mutation was sufficient to induce erythrocytosis. Both implications have been borne out by subsequent mouse models examining Phd2 and erythrocytosis, which have provided additional insights into the role of PHD2 in the control of red cell mass. In such mouse models, one has the capacity to genetically introduce knockout of the gene of interest (in this case, Phd2) in a manner that can be global, tissue-specific, and/or inducible. Moreover, one also has the capacity to introduce single-amino acid substitutions to precisely model human disease.
Conventional knockout of Phd2 in the mouse leads to embryonic lethality.125 To circumvent this embryonic lethality, global tamoxifen-inducible knockouts of Phd2 in the mouse have been generated and have been found to result in massive erythrocytosis with markedly elevated Epo levels.126,127 This erythrocytosis is observed not only in young mice but also in aging mice.128 In contrast, erythrocytosis is not observed with individual knockouts of either Phd1 or Phd3.126 In the globally inducible Phd2 knockout, the kidney is the primary source of the elevated Epo,126,127 and other studies have shown that cortical interstitial cells are the source of Epo within this organ.34 Consistent with the latter, conditional knockout of Phd2 that targets these renal interstitial cells, using either a CD68-cre or a Pax3-cre transgene, also results in marked erythrocytosis.129,130 Taken together, these studies identify a critical nonredundant role for Phd2 in the control of Epo in the Epo-producing renal cortical interstitial cells. This contrasts with the liver, another potential source of Epo, in which there is redundancy among the three Phd paralogs. Liver-specific knockout of Phd2 is insufficient to induce erythrocytosis, but it is observed with concurrent knockout in liver of Phd1 and Phd3.131,132
With regard to the issue of the sensitivity of the EPO pathway to PHD2 gene dosage, a knockin mouse model that bears a heterozygous amino acid substitution (Phd2-P294R) homologous to the first reported human EGLN1 mutation (PHD2-P317R) displays erythrocytosis.130 This result not only formally demonstrates that this is the cause of the human phenotype but also shows that a heterozygous mutation is sufficient to do so. The degree of erythrocytosis observed in this mouse line, which is modest, is comparable to that seen with Phd2+/− mice, consistent with haplo-insufficiency being the mechanism for this particular mutation (as opposed to a dominant negative mechanism). The erythrocytosis is reversed by heterozygous knockout of Hif2a, but not Hif1a,130 consistent with a substantial body of work that identifies HIF-2α as the key HIF-α isoform regulating EPO in adult mammals.17,20,129,133 Indeed, as mentioned previously, mutations in the EPAS1 gene are another cause of erythrocytosis.102
The Epo levels in the Phd2P294R/+ mice, as in the Phd2+/− mice, are normal, recapitulating the findings in the human patients that bear the homologous mutation.130 The levels should be regarded as inappropriately normal, as the elevated hematocrit would ordinarily depress Epo production from the kidney. At the same time, it should be recognized that Phd2 control over red cell mass extends beyond the Hif-2α:Epo axis. For example, deletion of Phd2 in hematopoietic precursors using a Vav-cre transgene induces erythrocytosis, and bone marrow cells derived from these as well as those from an induced globally deleted Phd2 mouse line display hypersensitivity to Epo, as assessed by erythroid burst forming unit assays.128,130 This is reminiscent of Chuvash polycythemia due to the homozygous hypomorphic mutation in the Vhl gene. Both patients and mice that bear this mutation (VHL-R200W in humans, Vhl-R166W in mice) display erythroid precursors with hypersensitivity to Epo.45,134 Moreover, loss of Phd2 in hematopoietic stem cells induced by either a CD68-driven or globally inducible cre transgene results in increased numbers of Lineage− Sca1+ c-Kit+ cells, which are early HSCs, and subsets of these cells, including multipotent progenitors MPP1, MPP2, and MPP3.126,135
Discussion
Observations of patients carrying germline mutations in the EGLN1 gene showed a relatively homogeneous phenotype that can be presumed to be erythrocytosis. However, at the present time, the total number of patients is still too small to perform genotype/phenotype correlation studies to explain exceptional clinical manifestations, such as the tumor occurrence described in only one case. The international database on erythrocytosis, the European Congenital Erythrocytosis Consortium (http://www.erythrocytosis.org) should help solve this limitation.
Mapping erythrocytosis-associated mutations onto the structure of the PHD2 protein has indicated they are typically present in conserved regions. Several frameshift and nonsense mutations are predicted to result in loss of the C-terminal portion of PHD2 and, in some cases, the full catalytic domain, thereby resulting in a nonactive enzyme. In addition, only a minority of the erythrocytosis-associated mutations are located in the active site of PHD2, so as to directly affect either the 2-OG or iron-binding amino acids. Functional studies have revealed that many mutations have a mild effect on PHD2 activity. Whether this reflects limitations of current PHD2 assay methods or whether this subtle impairment of HIF-2α regulation is indeed sufficient to increase red cell production is not clear at the present time. All identified germline EGLN1 mutations are present in the heterozygous state, implying that only partial loss of PHD2 activity is sufficient to induce erythrocytosis. As proof of this, a mouse model expressing a mutation homologous to the first reported heterozygous mutation, PHD2-P317R, exhibits erythrocytosis with inappropriately normal serum EPO levels. Similar results were obtained with Phd2+/− mice; thus, a heterozygous mutation is sufficient to cause the erythrocytosis phenotype. Mouse models also identify a role for Phd2 in the hematopoietic compartment in regulating red cell development. In addition, the presence of normal Phd1 and Phd3 in the knockin mouse indicates there is no redundancy in the oxygen-sensing pathway that regulates Epo in the kidney, and that the other two isoforms of Phd are unable to compensate for loss of Phd2 in connection with Epo synthesis.
Intriguingly, PHD2 may also have an important role in facilitating adaptation to high altitude. Genome-wide association studies have identified two coding sequence variants in PHD2, with PHD2-D4E being highly prevalent in the Tibetan population but very rare in lowlanders. The second variant, PHD2-C127S, exhibits less allelic divergence between these populations.
Although HIF plays a major role in tumor development and angiogenesis, only one germline EGLN1 mutation (PHD2-H374R) has been described as associated with tumor development (paraganglioma). The exact mechanism of the paraganglioma development in this patient is not elucidated, and the functionality of PHD2 is yet not fully understood. Other PHD2 partners and pathways may be explored to address this question. Given this, one may consider follow-up of EGLN1 germline mutation carriers to monitor potential tumor occurrence.
In conclusion, study of the inherited red cell disorder of erythrocytosis has been exemplary in providing insight into the hypoxia response pathway and defining the major role of the HIF prolyl hydroxylase PHD2 in the regulation of EPO gene transcription. Further investigations need to be performed to fully understand the complex and subtle pathways regulated by PHD2. Moreover, in many cases of familial secondary erythrocytosis, the cause remains unknown. Future research implementing next-generation sequencing technology will hopefully shed more light on additional causal mutations, not only in the coding sequence of the PHD2 gene, but also in its regulatory regions, as well as potentially even other genes that affect the HIF pathway.
Acknowledgments
The authors are grateful to the European networks that made collaboration possible: HypoxiaNet, Cooperation in Science and Technology (COST) Action TD0901, and Molecular Diagnosis of Myeloproliferative Neoplasms and MPN-Related Congenital Diseases-EuroNet, COST Action BM0902. Research in Dr Gardie’s lab has been supported by grants from the Ligue Nationale contre le Cancer (Comités du Cher, de l’Indre, de la Loire Atlantique et des Côtes d’Armor) and the Association pour la Recherche sur le Cancer. Research in Dr Lee’s lab has been supported by grants from the National Institutes of Health, Roche Foundation for Anemia Research, and Penn Institute of Aging. Dr Hoogewijs acknowledges support from the National Centre of Competence in Research Kidney.CH, funded by the Swiss National Science Foundation. We thank Dr Aguado-Diaz and Dr Francesca Pierdomenico for their precious help in recruiting patients and sharing data.
Disclosure
The authors report no conflicts of interest in this work.
References
Pearson TC, Guthrie DL, Simpson J, et al. Interpretation of measured red cell mass and plasma volume in adults: Expert Panel on Radionuclides of the International Council for Standardization in Haematology. Br J Haematol. 1995;89(4):748–756. | |
Johansson PL, Safai-Kutti S, Kutti J. An elevated venous haemoglobin concentration cannot be used as a surrogate marker for absolute erythrocytosis: a study of patients with polycythaemia vera and apparent polycythaemia. Br J Haematol. 2005;129(5):701–705. | |
Franke K, Gassmann M, Wielockx B. Erythrocytosis: the HIF pathway in control. Blood. 2013;122(7):1122–1128. | |
Lee FS, Percy MJ. The HIF pathway and erythrocytosis. Annu Rev Pathol. 2011:165–192. | |
McMullin MF. Idiopathic erythrocytosis: a disappearing entity. Hematology Am Soc Hematol Educ Program. 2009;629–635. | |
Semenza GL. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood. 2009;114(10):2015–2019. | |
Berra E, Ginouvès A, Pouysségur J. The hypoxia-inducible-factor hydroxylases bring fresh air into hypoxia signalling. EMBO Rep. 2006;7(1):41–45. | |
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402. | |
Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–38465. | |
Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22(16):4082–4090. | |
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295(5556):858–861. | |
Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005(306):re12. | |
Wenger RH, Hoogewijs D. Regulated oxygen sensing by protein hydroxylation in renal erythropoietin-producing cells. Am J Physiol Renal Physiol. 2010;298(6):F1287–F1296. | |
Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88(13):5680–5684. | |
Pugh CW, O’Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ. Activation of hypoxia-inducible factor-1; definition of regulatory domains within the alpha subunit. J Biol Chem. 1997;272(17):11205–11214. | |
Dayan F, Roux D, Brahimi-Horn MC, Pouyssegur J, Mazure NM. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res. 2006;66(7):3688–3698. | |
Gruber M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal ablation of Hif-2alpha results in anemia. Proc Natl Acad Sci U S A. 2007;104(7):2301–2306. | |
Rankin EB, Biju MP, Liu Q, et al. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest. 2007;117(4):1068–1077. | |
Paliege A, Rosenberger C, Bondke A, et al. Hypoxia-inducible factor-2alpha-expressing interstitial fibroblasts are the only renal cells that express erythropoietin under hypoxia-inducible factor stabilization. Kidney Int. 2010;77(4):312–318. | |
Kapitsinou PP, Liu Q, Unger TL, et al. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood. 2010;116(16):3039–3048. | |
Haase VH. Hypoxic regulation of erythropoiesis and iron metabolism. Am J Physiol Renal Physiol. 2010;299(1):F1–F13. | |
Lok CN, Ponka P. Identification of a hypoxia response element in the transferrin receptor gene. J Biol Chem. 1999;274(34):24147–152. | |
Zhang FL, Shen GM, Liu XL, et al. Hypoxic induction of human erythroid-specific δ-aminolevulinate synthase mediated by hypoxia-inducible factor 1. Biochemistry. 2011;50(7):1194–1202. | |
Zhang FL, Shen GM, Liu XL, Wang F, Zhao YZ, Zhang JW. Hypoxia-inducible factor 1-mediated human GATA1 induction promotes erythroid differentiation under hypoxic conditions. J Cell Mol Med. 2012;16(8):1889–1899. | |
Dame C, Fahnenstich H, Freitag P, et al. Erythropoietin mRNA expression in human fetal and neonatal tissue. Blood. 1998;92(9):3218–3225. | |
Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12(12):5447–5454. | |
Blanchard KL, Acquaviva AM, Galson DL, Bunn HF. Hypoxic induction of the human erythropoietin gene: cooperation between the promoter and enhancer, each of which contains steroid receptor response elements. Mol Cell Biol. 1992;12(12):5373–5385. | |
Suzuki N, Obara N, Pan X, et al. Specific contribution of the erythropoietin gene 3’ enhancer to hepatic erythropoiesis after late embryonic stages. Mol Cell Biol. 2011;31(18):3896–3905. | |
Storti F, Santambrogio S, Crowther L, et al. A novel distal upstream hypoxia response element regulating oxygen-dependent erythropoietin gene expression. Haematologica. 2014;99(4):e45–e48. | |
Bachmann S, Le Hir M, Eckardt KU. Co-localization of erythropoietin mRNA and ecto-5’-nucleotidase immunoreactivity in peritubular cells of rat renal cortex indicates that fibroblasts produce erythropoietin. J Histochem Cytochem. 1993;41(3):335–341. | |
Koury ST, Bondurant MC, Koury MJ. Localization of erythropoietin synthesizing cells in murine kidneys by in situ hybridization. Blood. 1988;71(2):524–527. | |
Lacombe C, Da Silva JL, Bruneval P, et al. Peritubular cells are the site of erythropoietin synthesis in the murine hypoxic kidney. J Clin Invest. 1988;81(2):620–623. | |
Maxwell PH, Osmond MK, Pugh CW, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int. 1993;44(5):1149–1162. | |
Obara N, Suzuki N, Kim K, Nagasawa T, Imagawa S, Yamamoto M. Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood. 2008;111(10):5223–5232. | |
Pan X, Suzuki N, Hirano I, Yamazaki S, Minegishi N, Yamamoto M. Isolation and characterization of renal erythropoietin-producing cells from genetically produced anemia mice. PLoS One. 2011;6(10):e25839. | |
Yamazaki S, Souma T, Hirano I, et al. A mouse model of adult-onset anaemia due to erythropoietin deficiency. Nat Commun. 2013;4:1950. | |
Kurt B, Paliege A, Willam C, et al. Deletion of von Hippel-Lindau protein converts renin-producing cells into erythropoietin-producing cells. J Am Soc Nephrol. 2013;24(3):433–444. | |
Beirão I, Moreira L, Barandela T, et al. Erythropoietin production by distal nephron in normal and familial amyloidotic adult human kidneys. Clin Nephrol. 2010;74(5):327–335. | |
Bussolati B, Lauritano C, Moggio A, Collino F, Mazzone M, Camussi G. Renal CD133(+)/CD73(+) progenitors produce erythropoietin under hypoxia and prolyl hydroxylase inhibition. J Am Soc Nephrol. 2013;24(8):1234–1241. | |
Bento C, Percy MJ, Gardie B, et al; ECE-Consortium. Genetic basis of congenital erythrocytosis: mutation update and online databases. Hum Mutat. 2014;35(1):15–26. | |
de la Chapelle A, Sistonen P, Lehväslaiho H, Ikkala E, Juvonen E. Familial erythrocytosis genetically linked to erythropoietin receptor gene. Lancet. 1993;341(8837):82–84. | |
de la Chapelle A, Träskelin AL, Juvonen E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc Natl Acad Sci U S A. 1993;90(10):4495–4499. | |
Ang SO, Chen H, Gordeuk VR, et al. Endemic polycythemia in Russia: mutation in the VHL gene. Blood Cells Mol Dis. 2002;28(1):57–62. | |
Horton JC, Harsh GR 4th, Fisher JW, Hoyt WF. Von Hippel-Lindau disease and erythrocytosis: radioimmunoassay of erythropoietin in cyst fluid from a brainstem hemangioblastoma. Neurology. 1991;41(5):753–754. | |
Ang SO, Chen H, Hirota K, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32(4):614–621. | |
Liu E, Percy MJ, Amos CI, et al. The worldwide distribution of the VHL 598C>T mutation indicates a single founding event. Blood. 2004;103(5):1937–1940. | |
Percy MJ, Mooney SM, McMullin MF, Flores A, Lappin TR, Lee FS. A common polymorphism in the oxygen-dependent degradation (ODD) domain of hypoxia inducible factor-1alpha (HIF-1alpha) does not impair Pro-564 hydroxylation. Mol Cancer. 2003;2:31. | |
Percy MJ, Zhao Q, Flores A, et al. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci U S A. 2006;103(3):654–659. | |
Albiero E, Ruggeri M, Fortuna S, et al. Isolated erythrocytosis: study of 67 patients and identification of three novel germ-line mutations in the prolyl hydroxylase domain protein 2 (PHD2) gene. Haematologica. 2012;97(1):123–127. | |
Bento C, Almeida H, Maia TM, et al. Molecular study of congenital erythrocytosis in 70 unrelated patients revealed a potential causal mutation in less than half of the cases (Where is/are the missing gene(s)?). Eur J Haematol. 2013;91(4):361–368. | |
Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359(25):2685–2692. | |
Ladroue C, Hoogewijs D, Gad S, et al. Distinct deregulation of the hypoxia inducible factor by PHD2 mutants identified in germline DNA of patients with polycythemia. Haematologica. 2012;97(1):9–14. | |
Percy MJ, Furlow PW, Beer PA, Lappin TR, McMullin MF, Lee FS. A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood. 2007;110(6):2193–2196. | |
Al-Sheikh M, Moradkhani K, Lopez M, Wajcman H, Préhu C. Disturbance in the HIF-1alpha pathway associated with erythrocytosis: further evidences brought by frameshift and nonsense mutations in the prolyl hydroxylase domain protein 2 (PHD2) gene. Blood Cells Mol Dis. 2008;40(2):160–165. | |
Jang JH, Seo JY, Jang J, et al. Hereditary gene mutations in Korean patients with isolated erythrocytosis. Ann Hematol. Epub January 31, 2014. | |
Albiero E, Ruggeri M, Fortuna S, et al. Analysis of the oxygen sensing pathway genes in familial chronic myeloproliferative neoplasms and identification of a novel EGLN1 germ-line mutation. Br J Haematol. 2011;153(3):405–408. | |
Yi X, Liang Y, Huerta-Sanchez E, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. 2010;329(5987):75–78. | |
Bigham A, Bauchet M, Pinto D, et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLoS Genet. 2010;6(9):e1001116. | |
Simonson TS, Yang Y, Huff CD, et al. Genetic evidence for high-altitude adaptation in Tibet. Science. 2010;329(5987):72–75. | |
Ji LD, Qiu YQ, Xu J, et al. Genetic adaptation of the hypoxia-inducible factor pathway to oxygen pressure among eurasian human populations. Mol Biol Evol. 2012;29(11):3359–3370. | |
Xiang K, Ouzhuluobu, Peng Y, et al. Identification of a Tibetan-specific mutation in the hypoxic gene EGLN1 and its contribution to high-altitude adaptation. Mol Biol Evol. 2013;30(8):1889–1898. | |
Petousi N, Sciesielski LK, Ratcliffe PJ, Robbins PA, Masson N. Investigation of the functional significance of two coding variants found in Tibetans. Keystone Symposia 2014. Sensing and signaling of hypoxia: interfaces with biology and medicine. 2014. | |
Lorenzo FR, Huff CD, Myllymaki M, et al. EGLN1 variant associated with the Tibetan adaptation in high altitude. Keystone Symposia 2014. Sensing and signaling of hypoxia : interfaces with biology and medicine. 2014. | |
Song DS, Li L, Arsenault PR, et al. Defective Tibetan PHD2 binding to p23 links high altitude adaptation to altered oxygen sensing. J Biol Chem. In press 2014. | |
Petousi N, Robbins PA. Human adaptation to the hypoxia of high altitude – the Tibetan paradigm from the pre-genomic to the post-genomic era. J Appl Physiol (1985). Epub November 7, 2013. | |
Semenza GL. Life with oxygen. Science. 2007;318(5847):62–64. | |
Richard S, Gardie B, Couvé S, Gad S. Von Hippel-Lindau: how a rare disease illuminates cancer biology. Semin Cancer Biol. 2013;23(1):26–37. | |
Kondo K, Kim WY, Lechpammer M, Kaelin WG Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1(3):E83. | |
Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL. Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res. 2003;63(19):6130–6134. | |
Schödel J, Bardella C, Sciesielski LK, et al. Common genetic variants at the 11q13.3 renal cancer susceptibility locus influence binding of HIF to an enhancer of cyclin D1 expression. Nat Genet. 2012;44(4):420–425. | |
Purdue MP, Johansson M, Zelenika D, et al. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat Genet. 2011;43(1):60–65. | |
Hewitson KS, Liénard BM, McDonough MA, et al. Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates. J Biol Chem. 2007;282(5):3293–3301. | |
Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8(2):143–153. | |
Kaelin WG Jr. SDH5 mutations and familial paraganglioma: somewhere Warburg is smiling. Cancer Cell. 2009;16(3):180–182. | |
Pollard PJ, Brière JJ, Alam NA, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14(15):2231–2239. | |
Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. | |
Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. 2007;282(7):4524–4532. | |
O’Flaherty L, Adam J, Heather LC, et al. Dysregulation of hypoxia pathways in fumarate hydratase-deficient cells is independent of defective mitochondrial metabolism. Hum Mol Genet. 2010;19(19):3844–3851. | |
Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. | |
Chowdhury R, Yeoh KK, Tian YM, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12(5):463–469. | |
Koivunen P, Lee S, Duncan CG, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483(7390):484–488. | |
Lorenzo FR, Yang C, Ng Tang Fui M, et al. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (Berl). 2013;91(4):507–512. | |
Kato H, Inoue T, Asanoma K, Nishimura C, Matsuda T, Wake N. Induction of human endometrial cancer cell senescence through modulation of HIF-1alpha activity by EGLN1. Int J Cancer. 2006;118(5):1144–1153. | |
Lee KA, Lynd JD, O’Reilly S, Kiupel M, McCormick JJ, LaPres JJ. The biphasic role of the hypoxia-inducible factor prolyl-4-hydroxylase, PHD2, in modulating tumor-forming potential. Mol Cancer Res. 2008;6(5):829–842. | |
Gardie B, Couvé S, Ladroue C, et al. Interacting germline mutations in a single allele of the VHL tumor suppressor gene reveal the importance of precisely tuned dysregulation in oncogenesis Hypoxianet meeting. Sensing Hypoxia in the Cell and the Organism. September 20–23, 2012, Essen, Germany. 2012. | |
Astuti D, Ricketts CJ, Chowdhury R, et al. Mutation analysis of HIF prolyl hydroxylases (PHD/EGLN) in individuals with features of phaeochromocytoma and renal cell carcinoma susceptibility. Endocr Relat Cancer. 2010;18(1):73–83. | |
Li L, Zhang L, Zhang X, et al. Hypoxia-inducible factor linked to differential kidney cancer risk seen with type 2A and type 2B VHL mutations. Mol Cell Biol. 2007;27(15):5381–5392. | |
Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WG Jr. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet. 2001;10(10):1019–1027. | |
Clifford SC, Cockman ME, Smallwood AC, et al. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet. 2001;10(10):1029–1038. | |
Comino-Méndez I, de Cubas AA, Bernal C, et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet. 2013;22(11):2169–2176. | |
Toledo RA, Qin Y, Srikantan S, et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2013;20(3):349–359. | |
Chowdhury R, McDonough MA, Mecinovic J, et al. Structural basis for binding of hypoxia-inducible factor to the oxygen-sensing prolyl hydroxylases. Structure. 2009;17(7):981–989. | |
McDonough MA, Li V, Flashman E, et al. Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc Natl Acad Sci U S A. 2006;103(26):9814–9819. | |
Hoffart LM, Barr EW, Guyer RB, Bollinger JM Jr, Krebs C. Direct spectroscopic detection of a C-H-cleaving high-spin Fe(IV) complex in a prolyl-4-hydroxylase. Proc Natl Acad Sci U S A. 2006;103(40):14738–14743. | |
McNeill LA, Flashman E, Buck MR, et al. Hypoxia-inducible factor prolyl hydroxylase 2 has a high affinity for ferrous iron and 2-oxoglutarate. Mol Biosyst. 2005;1(4):321–324. | |
Hirsilä M, Koivunen P, Günzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278(33):30772–30780. | |
Pappalardi MB, Martin JD, Jiang Y, et al. Biochemical characterization of human prolyl hydroxylase domain protein 2 variants associated with erythrocytosis. Biochemistry. 2008;47(43):11165–11167. | |
Flashman E, Bagg EA, Chowdhury R, et al. Kinetic rationale for selectivity toward N- and C-terminal oxygen-dependent degradation domain substrates mediated by a loop region of hypoxia-inducible factor prolyl hydroxylases. J Biol Chem. 2008;283(7):3808–3815. | |
Gale DP, Harten SK, Reid CD, Tuddenham EG, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood. 2008;112(3):919–921. | |
Martini M, Teofili L, Cenci T, et al. A novel heterozygous HIF2AM535I mutation reinforces the role of oxygen sensing pathway disturbances in the pathogenesis of familial erythrocytosis. Haematologica. 2008;93(7):1068–1071. | |
Percy MJ, Beer PA, Campbell G, et al. Novel exon 12 mutations in the HIF2A gene associated with erythrocytosis. Blood. 2008;111(11):5400–5402. | |
Percy MJ, Furlow PW, Lucas GS, et al. A gain-of-function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358(2):162–168. | |
Perrotta S, Della Ragione F. The HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358(18):1966. | |
van Wijk R, Sutherland S, Van Wesel AC, et al. Erythrocytosis associated with a novel missense mutation in the HIF2A gene. Haematologica. 2010;95(5):829–832. | |
Furlow PW, Percy MJ, Sutherland S, et al. Erythrocytosis-associated HIF-2alpha mutations demonstrate a critical role for residues C-terminal to the hydroxylacceptor proline. J Biol Chem. 2009;284(14):9050–9058. | |
Blanchette M, Kent WJ, Riemer C, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14(4):708–715. | |
Karolchik D, Barber GP, Casper J, et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42(Database issue):D764–D770. | |
Ostlund G, Schmitt T, Forslund K, et al. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010;38(Database issue):D196–D203. | |
Loenarz C, Coleman ML, Boleininger A, et al. The hypoxia-inducible transcription factor pathway regulates oxygen sensing in the simplest animal, Trichoplax adhaerens. EMBO Rep. 2011;12(1):63–70. | |
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. | |
Mecinovic J, Chowdhury R, Flashman E, Schofield CJ. Use of mass spectrometry to probe the nucleophilicity of cysteinyl residues of prolyl hydroxylase domain 2. Anal Biochem. 2009;393(2):215–221. | |
Mecinovic J, Chowdhury R, Liénard BM, et al. ESI-MS studies on prolyl hydroxylase domain 2 reveal a new metal binding site. ChemMedChem. 2008;3(4):569–572. | |
Hewitson KS, Schofield CJ, Ratcliffe PJ. Hypoxia-inducible factor prolyl-hydroxylase: purification and assays of PHD2. Methods Enzymol. 2007;435:25–42. | |
Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54. | |
Huang J, Zhao Q, Mooney SM, Lee FS. Sequence determinants in hypoxia-inducible factor-1alpha for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J Biol Chem. 2002;277(42):39792–39800. | |
Tian YM, Yeoh KK, Lee MK, et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J Biol Chem. 2011;286(15):13041–13051. | |
Köditz J, Nesper J, Wottawa M, et al. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110(10):3610–3617. | |
Huo Z, Ye JC, Chen J, et al. Prolyl hydroxylase domain protein 2 regulates the intracellular cyclic AMP level in cardiomyocytes through its interaction with phosphodiesterase 4D. Biochem Biophys Res Commun. 2012;427(1):73–79. | |
Song D, Li LS, Heaton-Johnson KJ, Arsenault PR, Master SR, Lee FS. Prolyl hydroxylase domain protein 2 (PHD2) binds a Pro-Xaa-Leu-Glu motif, linking it to the heat shock protein 90 pathway. J Biol Chem. 2013;288(14):9662–9674. | |
Vogel S, Wottawa M, Farhat K, et al. Prolyl hydroxylase domain (PHD) 2 affects cell migration and F-actin formation via RhoA/rho-associated kinase-dependent cofilin phosphorylation. J Biol Chem. 2010;285(44):33756–33763. | |
Barth S, Nesper J, Hasgall PA, et al. The peptidyl prolyl cis/trans isomerase FKBP38 determines hypoxia-inducible transcription factor prolyl-4-hydroxylase PHD2 protein stability. Mol Cell Biol. 2007;27(10):3758–3768. | |
Chan DA, Kawahara TL, Sutphin PD, Chang HY, Chi JT, Giaccia AJ. Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell. 2009;15(6):527–538. | |
Pientka FK, Hu J, Schindler SG, et al. Oxygen sensing by the prolyl-4-hydroxylase PHD2 within the nuclear compartment and the influence of compartmentalisation on HIF-1 signalling. J Cell Sci. 2012;125(Pt 21):5168–5176. | |
Steinhoff A, Pientka FK, Möckel S, et al. Cellular oxygen sensing: Importins and exportins are mediators of intracellular localisation of prolyl-4-hydroxylases PHD1 and PHD2. Biochem Biophys Res Commun. 2009;387(4):705–711. | |
Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26(22):8336–8346. | |
Takeda K, Aguila HL, Parikh NS, et al. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood. 2008;111(6):3229–3235. | |
Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG Jr. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111(6):3236–3244. | |
Li X, Sutherland S, Takeda K, Fong GH, Lee FS. Integrity of the prolyl hydroxylase domain protein 2:erythropoietin pathway in aging mice. Blood Cells Mol Dis. 2010;45(1):9–19. | |
Franke K, Kalucka J, Mamlouk S, et al. HIF-1α is a protective factor in conditional PHD2-deficient mice suffering from severe HIF-2α-induced excessive erythropoiesis. Blood. 2013;121(8):1436–1445. | |
Arsenault PR, Pei F, Lee R, et al. A knock-in mouse model of human PHD2 gene-associated erythrocytosis establishes a haploinsufficiency mechanism. J Biol Chem. 2013;288(47):33571–33584. | |
Minamishima YA, Kaelin WG Jr. Reactivation of hepatic EPO synthesis in mice after PHD loss. Science. 23 2010;329(5990):407. | |
Duan LJ, Takeda K, Fong GH. Hematological, hepatic, and retinal phenotypes in mice deficient for prolyl hydroxylase domain proteins in the liver. Am J Pathol. 2014;184(4):1240–1250. | |
Scortegagna M, Ding K, Zhang Q, et al. HIF-2alpha regulates murine hematopoietic development in an erythropoietin-dependent manner. Blood. 2005;105(8):3133–3140. | |
Hickey MM, Lam JC, Bezman NA, Rathmell WK, Simon MC. von Hippel-Lindau mutation in mice recapitulates Chuvash polycythemia via hypoxia-inducible factor-2alpha signaling and splenic erythropoiesis. J Clin Invest. 2007;117(12):3879–3889. | |
Singh RP, Franke K, Kalucka J, et al. HIF prolyl hydroxylase 2 (PHD2) is a critical regulator of hematopoietic stem cell maintenance during steady-state and stress. Blood. 2013;121(26):5158–5166. |
Supplementary material
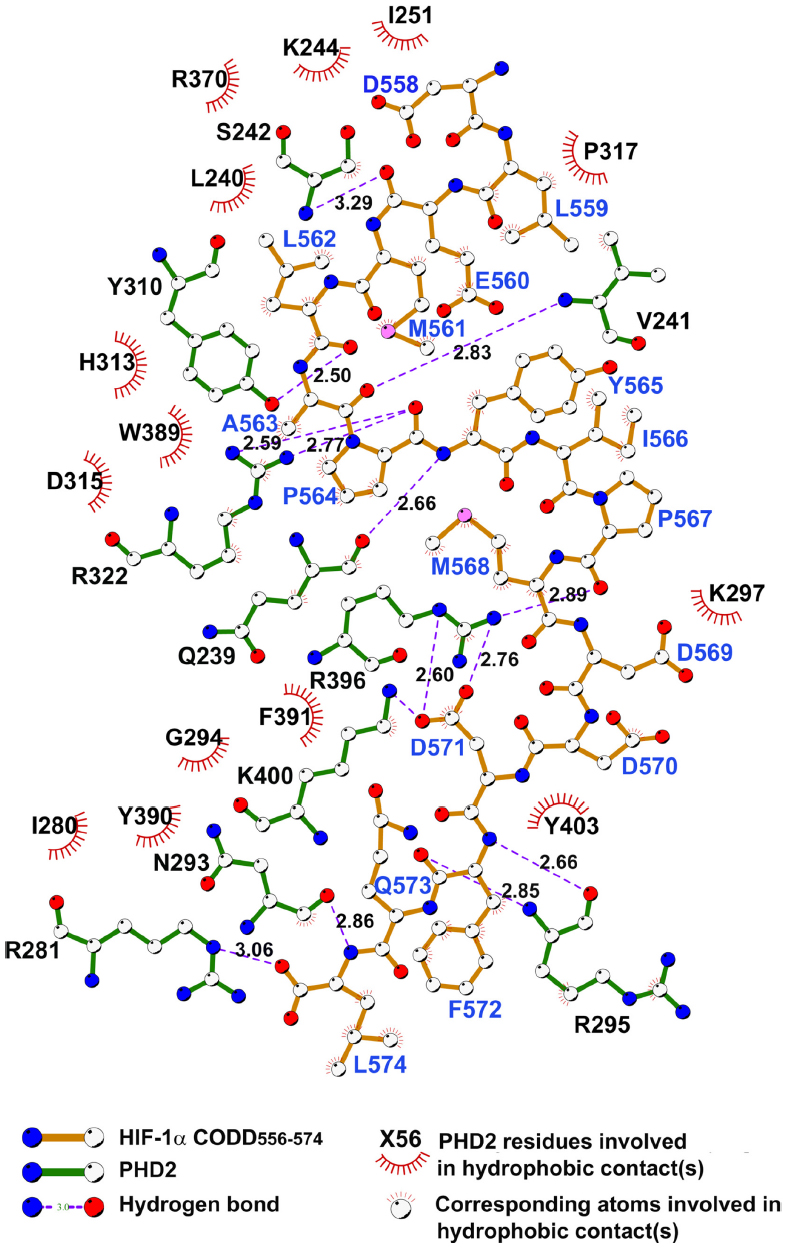 | Figure S1 Ligplot 2D figure of the HIF-PHD2 complex showing important interactions between HIF and PHD2. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.