")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 9 » Issue 1
The role of microparticles in chronic obstructive pulmonary disease
Authors Takahashi T, Kubo H
Received 25 December 2013
Accepted for publication 3 February 2014
Published 27 March 2014 Volume 2014:9(1) Pages 303—314
DOI https://doi.org/10.2147/COPD.S38931
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Toru Takahashi,1–3 Hiroshi Kubo1
1Department of Advanced Preventive Medicine for Infectious Disease, Tohoku University Graduate School of Medicine, Sendai, Japan; 2Cellular and molecular lung biology research units, Institut de Recherches Cliniques de Montréal (IRCM), Montreal, Quebec, Canada; 3Department of Anesthesiology, Tohoku University Hospital, Sendai, Japan
Abstract: Accumulating evidence suggests that cell injury in lung tissues is closely connected to disease progression in chronic obstructive pulmonary disease (COPD). Microparticles (MPs) are shed membrane vesicles that are released from platelets, leukocytes, red blood cells, and endothelial cells when these cells are activated or undergo apoptosis under inflammatory conditions. Based on increasing evidence that endothelial injury in the pulmonary capillary vasculature leads to lung destruction, and because cardiovascular diseases are the main cause of death among individuals with COPD, endothelial MPs (EMPs) are now receiving attention as potential biomarkers for COPD. There are eight types of EMPs which are defined by the presence of different endothelial markers on the cell membrane: vascular endothelial-cadherin; platelet endothelial cell adhesion molecule; melanoma cell adhesion molecule; E-selectin; CD51; CD105; von Willebrand factor; and CD143 EMPs. Vascular endothelial-cadherin, platelet endothelial cell adhesion molecule, and E-selectin EMPs are increased in patients with stable COPD and are further increased in patients with exacerbated COPD compared to non-COPD patients. In addition, the levels of these three EMPs in patients with stable COPD are significantly correlated with lung destruction and airflow limitation. These results indicate that endothelial injury is closely connected to the pathophysiology of COPD. Interestingly, the variations in the levels of the eight EMP subtypes were not identical with changes in patient condition. Although the clinical significance of the differences in these eight EMP subtypes remains unclear, evaluating the expression pattern of endothelial antigens on circulating MPs might predict the presence and degree of endothelial injury in COPD patients. In addition, circulating MPs are proposed to have several physiological functions in vivo, such as intercellular crosstalk; the increase in EMPs in COPD seems to play a role in the pathophysiology of this disease.
Keywords: COPD, exacerbation, apoptosis, endothelial activation, EMPs
Introduction
Chronic obstructive pulmonary disease (COPD) is a lung disease characterized by nearly irreversible lung destruction which results in airflow limitation.1 The severity of the disease is determined according to the degree of airflow limitation, which is measured using forced expiratory volume in one second (FEV1). However, there are several limitations in the use of FEV1 to evaluate the daily condition and disease progression of COPD patients. First, the sensitivity of FEV1 in evaluating daily condition is not high. Second, although both frequent exacerbation2–5 and bronchial hyperresponsiveness6 are associated with rapid FEV1 decline, FEV1 does not reflect these conditions. Third, clinical manifestations and radiological observations are variable among COPD patients even when the degree of airflow limitation is the same.7 For these reasons, new biomarkers for COPD are being sought.8
Accumulating evidence indicates that injured cells in the lung tissue are closely involved in the pathophysiology of COPD.9,10 In animal models, the administration of a vascular endothelial growth factor receptor inhibitor induced apoptosis of pulmonary capillary endothelial cells, leading to emphysematous changes.11 The number of apoptotic epithelial and endothelial cells is increased in emphysematous lungs compared with normal lungs.12 In addition, senescence of alveolar epithelial and endothelial cells is accelerated in patients with emphysema.13 Greater numbers of apoptotic lung cells are observed in lung tissues from COPD patients than in those from smokers without COPD.9,14,15 Furthermore, morphological and biochemical markers of autophagy are increased in the lungs of patients with COPD compared with normal lung tissue.16 These results indicate the importance of injured cells in the pathophysiology of lung destruction and COPD.
Activated or injured cells release chemical mediators, such as alarmins and microvesicles, into the circulation, and these mediators can potently modulate systemic immune responses.17–20 There are three major types of microvesicles, which are primarily distinguished by size: exosomes (40–100 nm), microparticles (MPs) (0.1–1.0 μm), and apoptotic bodies (1–3 μm). In this review, we focus on endothelium-derived MPs, which can be detected by their endothelial-specific antigens using fluorescence-activated cell sorting (FACS),18–23and discuss their potential as biomarkers for COPD.
MPs
MPs are membrane vesicles that are released by budding or shedding from the plasma membrane of activated or injured cells in response to inflammation (Figure 1).18–23 MP release from platelets,24 red blood cells,25 endothelial cells, tumor cells,26,27 and leucocytes28 has been reported. Because MPs are shed membrane vesicles, the antigens expressed on the original cells are expressed on the membrane of the MPs. For example, CD144, CD31, and CD62E are expressed on endothelial cell-derived MPs, whereas CD31 and CD41 are expressed on platelet-derived MPs. Various stimuli, such as increased intracellular Ca2+29 and the activation of protein kinase C (PKC)30 and purinergic receptors of adenosine triphosphate (ATP) such as the P2X7 and P2Y receptors,31,32 increase the rate of MP release. The contents of MPs are also variable and can include nucleic acids, particularly messenger ribonucleic acid (mRNA) and microRNA,22 proteolytic enzymes such as met alloproteinases,33 tissue factors,34 and alarmins.35 Released MPs interact with their target cells by binding to the surface of the target cell via specific receptors, followed by direct fusion of the shedding vesicle with the plasma membrane of the target cell, endocytic uptake of the vesicle, and horizontal transfer of molecules and RNA to the target cell.20 MPs play many roles, including modulating the biological functions of cells.36 For example, during leukocyte activation and arrest, activated mast cells and platelets deliver chemoattractants to endothelial cells through circulating MPs.37 MPs also display proinflammatory and prothrombotic activities through the activation of Toll-like receptors and other signaling pathways.17,21 MPs are released from cells in response to various conditions or stimuli and may be heterogeneous.38
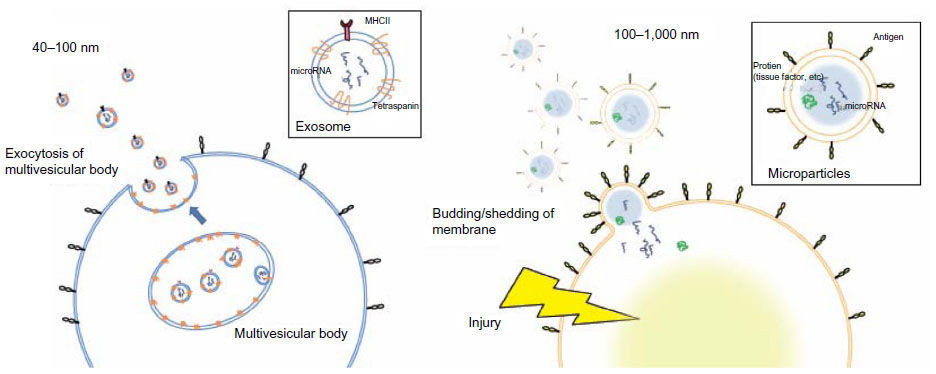 | Figure 1 Comparison of the size and release mechanism of exosomes and MPs. |
MPs and smoking
Smoking is the most important risk factor for COPD. Cigarette smoke increases autophagy16,39 and induces apoptosis and necrosis in lung cells.40–42 Smoking increases the levels of circulating MPs. In vitro studies have demonstrated that cigarette smoke extract induces the release of tissue factor-positive MPs as well as MPs with proteolytic activity attributed to matrix met alloproteinase-14 (MMP-14) from human macrophages in vitro.43,44 Incubating pulmonary microvascular endothelial cells or aortic endothelial cells with cigarette smoke extract induces the release of CD31+/CD41− MPs and CD146+ MPs in vitro.40 Secondhand smoke exposure induces the rapid release of circulating CD144+ endothelial microparticles (EMPs), within a few hours.45 Other groups have reported that CD31+/CD42b− EMPs are increased in healthy active smokers and further increased in active smokers with emphysema compared with healthy non-smokers.46
MPs and oxidative stress
Oxidative stress induced by cigarette smoke exposure or activated neutrophils and/or macrophages in response to infection is also involved in the pathophysiology of COPD.47,48 Oxidative stress induces MP release. H2O2 induces apoptosis in endothelial cells and the release of CD31+/CD41− or CD146+ MPs from aortic and pulmonary microvascular endothelial cells in vitro.40 Plasma levels of glutathione peroxidase, a marker of oxidative stress, are correlated with increased levels of platelet-, erythrocyte-, and endothelial-derived MPs independent of other factors involved in endothelial injury in patients with metabolic disease.49
MPs and infection
Viral and/or bacterial infection was reported to be detected in up to 60% of COPD exacerbation and has been found to be associated with a rapid decline in lung function.50,51 Several articles reported the impact of infections on circulating MPs,52–55 and the number of circulating MPs is significantly increased by infection. Therefore, the presence of airway infection may induce MP release in patients with COPD exacerbation.
EMPs
EMPs are released from injured endothelial cells during apoptosis and cellular activation.19,56,57 EMPs are defined by the surface expression of endothelial antigens, such as CD144 (vascular endothelial [VE]-cadherin), CD31 (platelet endothelial cell adhesion molecule [PECAM]-1), CD62e (E-selectin), CD146 (melanoma cell adhesion molecule [MCAM]), and CD51 (vitronectin) (Table 1 and Figure 2). Circulating EMP numbers are increased in patients with vascular disorders such as cardiovascular diseases,58–60 vasculitis,61,62 thrombosis,63,64 renal failure,65–67 hyperlipidemia,68 and metabolic syndrome.69,70 The increase in circulating EMPs reflects the degree of endothelial injury in such conditions (Table 2).
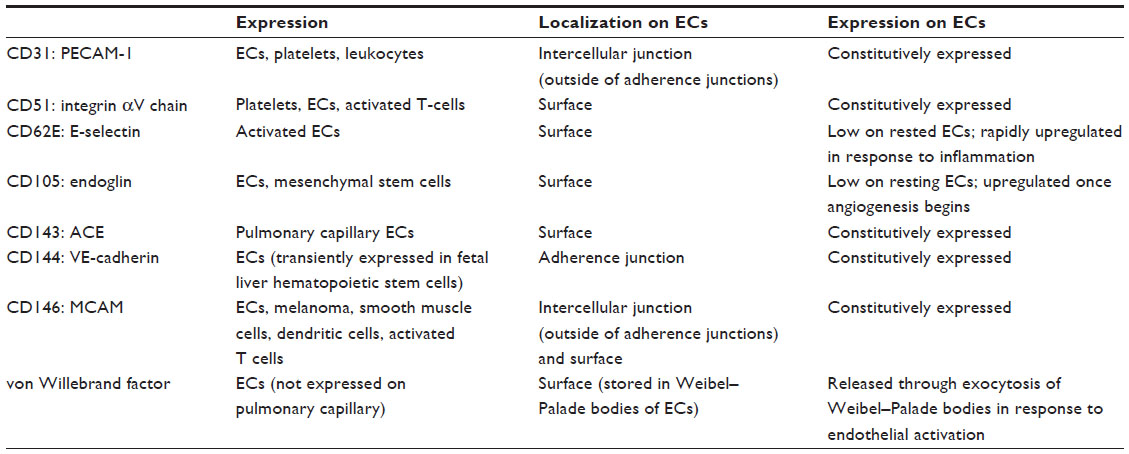 | Table 1 Endothelial markers used for detecting EMPs |
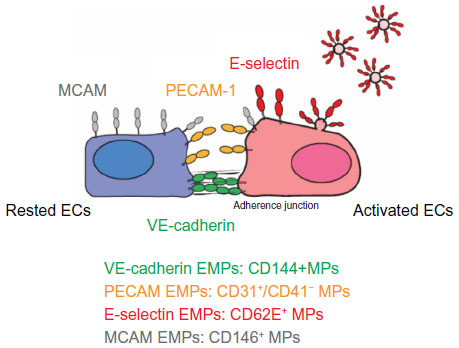 | Figure 2 Location of four EMP markers on ECs. |
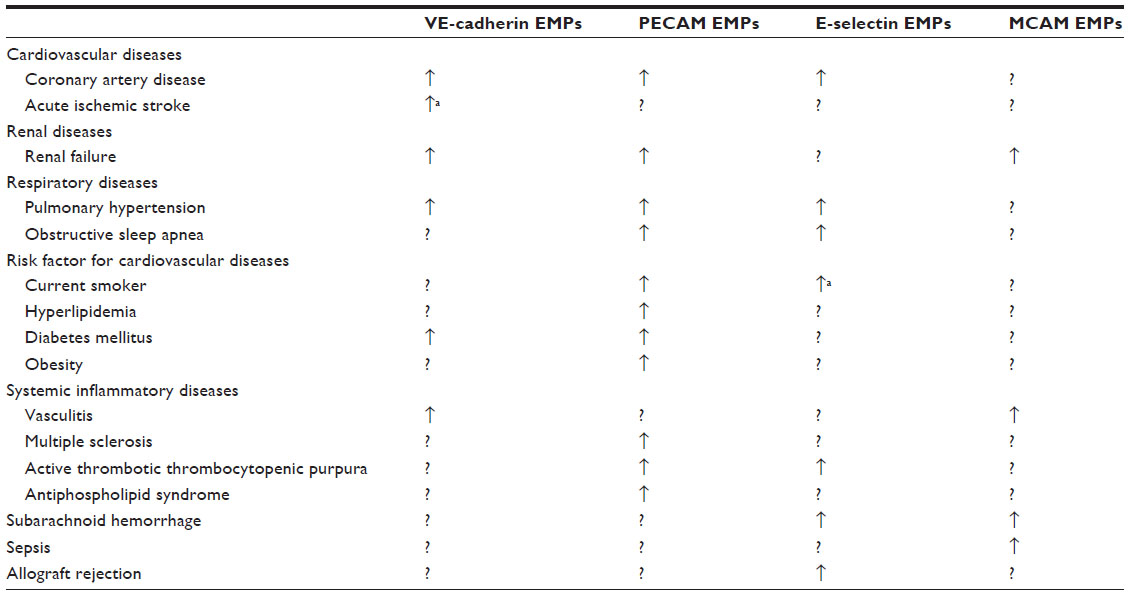 | Table 2 Numbers of circulating EMPs in various diseases |
In vitro studies have demonstrated that different stimuli, such as tumor necrosis factor α, H2O2, and cigarette smoke extract, induce the release of different types of EMPs from cultured endothelial cells. The pattern of EMP release in response to a specific stimulus differs depending on the type of endothelial cell.40 These observations suggest that different types of EMPs are released depending on the site of inflammation and the type of stimulus. Some clinical studies have compared two or more subtypes of EMPs in patients with specific diseases and observed differences in the types of released EMPs (Table 3). Although the clinical significance of these differences in EMP release remains unclear, the pattern of increased EMPs may reflect differences in inflammatory stimuli and the activated site in vivo.
VE-cadherin EMPs
VE-cadherin EMPs are defined as CD144+ MPs. VE-cadherin is expressed only on endothelial cells and is therefore the most specific marker for endothelial cells.71 In contrast to PECAM-1 and MCAM, VE-cadherin is specifically located at adherence junctions.72 VE-cadherin has been proposed to function as a gatekeeper of endothelial junctions.37,73 VE-cadherin does not support leukocyte migration but instead might function as an obstacle to migrating cells. Changes in the localization of VE-cadherin are associated with neutrophil migration and increased vascular permeability.74 The release of VE-cadherin EMPs may reflect the structural destruction of the endothelium rather than the inflammatory condition of the lung.
PECAM EMPs
PECAM-1 (CD31) is concentrated at endothelial junctions and is also expressed on the surfaces of platelets, neutrophils, and subsets of lymphocytes. In contrast to VE-cadherin, PECAM-1 is located outside of the adherence junctions on endothelial cells.72,75 Platelet-specific antigens such as CD41 or CD42b are used to distinguish PECAM EMPs from platelet-derived MPs; PECAM EMPs are defined as CD31+/CD41− or CD42b− MPs. PECAM-1 is not used to detect leukocyte-derived MPs because its expression on leukocytes is too low.76 PECAM-1 is a signaling molecule that plays diverse roles in vascular biology, including the regulation of platelet function, angiogenesis, T-cell and B-cell activation, endothelial cell permeability, and transmigration across the endothelium.37,77–81 Our in vitro study indicated that PECAM EMPs are released from pulmonary microvascular endothelial cells mainly in response to apoptosis induced by stimulation by H2O2 or cigarette smoke extract.40 In addition, a clinical study indicated that approximately 90% of PECAM EMPs also express annexin V in COPD82 and 60% of PECAM EMPs express annexin V in active smokers.46 Therefore, the released EMPs likely reflect the apoptosis of injured endothelial cells.
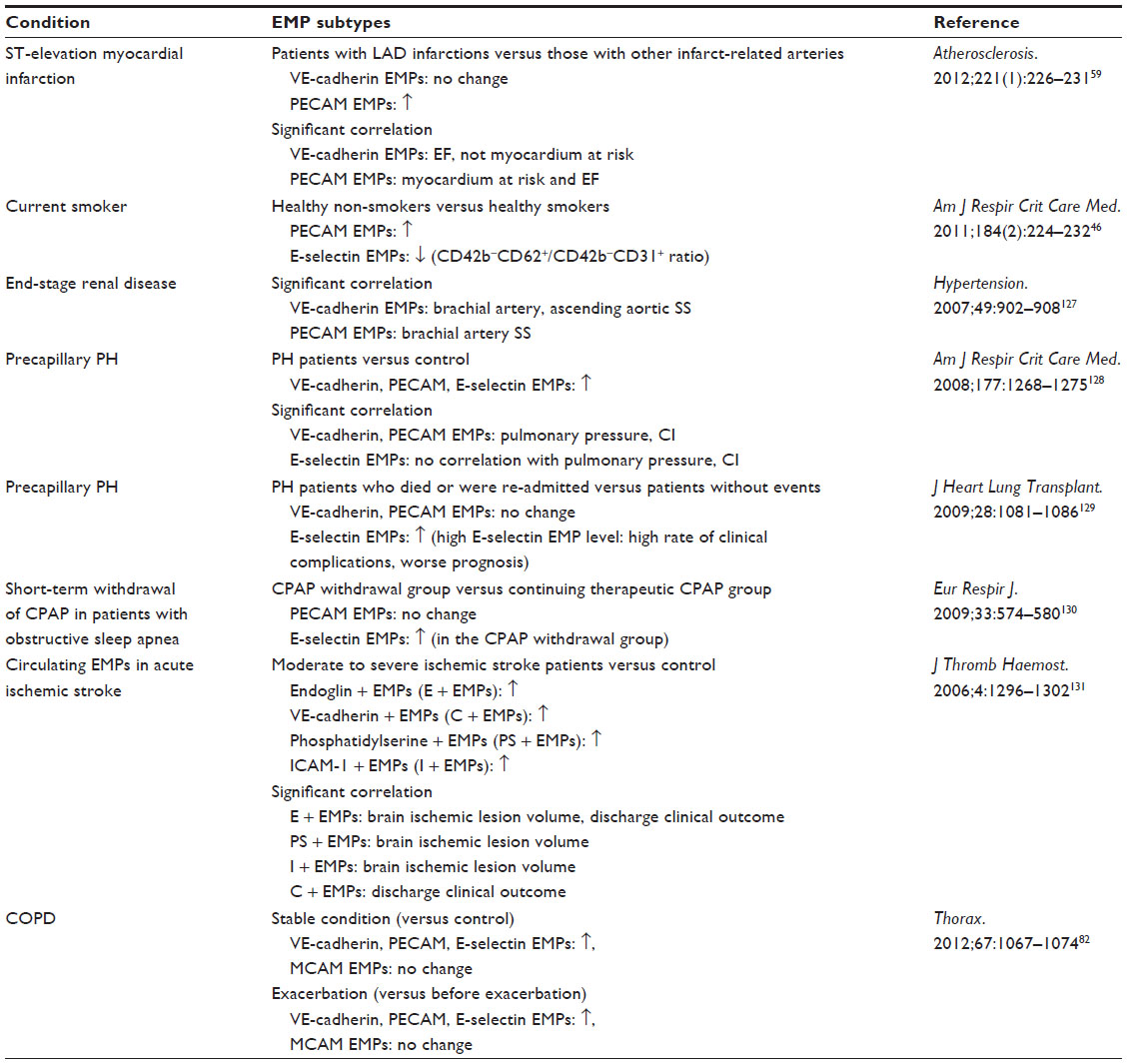 | Table 3 Articles comparing two or more EMP subtypes in subjects with pathological conditions |
MCAM EMPs
MCAM EMPs are defined as CD146+ MPs. MCAM is an adhesion molecule found on endothelial cells that is involved in processes such as endothelial permeability, signaling transduction, cell migration, angiogenesis, and the immune response.83 MCAM is located outside of the adherence junctions; however, MCAM expression is not restricted to cell junctions and is detected on the apical side of cultured endothelial cells.72 MCAM has been detected not only on endothelial cells but also on other cell types such as melanoma cells,84 a subset of T and B lymphocytes and natural killer (NK) cells under pathological conditions,85 and pericytes.86 Thus, CD146+ MPs can be released from other cell types as well as endothelial cells. In addition, the expression of MCAM, another marker of endothelial injury, is also used to detect circulating endothelial cells.85,87 Thus, circulating endothelial cells can be confounded with MCAM EMPs in FACS analysis.88
E-selectin EMPs
E-selectin is expressed only on endothelial cells and E-selectin EMPs are defined as CD62e+ MPs. E-selectin is rapidly induced on activated endothelial cells a few hours after inflammatory stimulation, whereas VE-cadherin, PECAM-1, and MCAM are constitutively expressed on endothelial cells.89 E-selectin plays an important role in recruiting leukocytes to the site of injury during inflammation.37,77–81 In vitro studies have indicated that tumor necrosis factor α upregulates E-selectin expression on endothelial cells within a few hours, resulting in the increased release of E-selectin EMPs.40,90 Thus, the presence of E-selectin EMPs might reflect the degree of ongoing endothelial inflammation.
CD51 EMPs
CD51 is the integrin α chain and is also known as the vitronectin receptor α chain. CD51 forms a heterodimer with an integrin β3 chain, such as glycoprotein IIIa (GPIIIa) or the CD61 molecule, and binds to vitronectin, von Willebrand factor (vWF), and fibronectin.91 CD51 is found on endothelial cells, B lymphocytes, monocytes, macrophages, and platelets.92 CD51 plays important roles in leukocyte homing and rolling and angiogenesis.37,93 Although the specificity of endothelial origin is less than the other four EMP subtypes, CD51 MPs are defined as EMPs in some articles.94–96
COPD and EMPs
We examined the levels of VE-cadherin, PECAM, MCAM, and E-selectin EMPs in 80 patients with stable COPD and 27 patients with exacerbated COPD.97 The MESA group examined the numbers of PECAM, E-selectin, and CD51 EMPs in 104 patients with stable COPD and 76 non-COPD patients.96 These are the only two reported studies of EMP numbers in COPD patients.
Circulating EMPs in patients with stable COPD
Recent studies have indicated that the main causes of death in COPD are cardiovascular events such as ischemic heart diseases and stroke, not respiratory events.98 Vascular abnormalities in the endothelium in both the pulmonary99 and systemic vasculatures100 have been reported in COPD patients. Impaired endothelial function as assessed by flow-mediated dilation of the brachial artery is associated with low FEV1 in COPD.101 Subclinical arteriosclerosis, as evaluated by carotid intima–media thickness and focal athermanous plaque, is exaggerated early in the disease process of COPD.102 Reduced white matter integrity throughout the brain has been detected by magnetic resonance imaging in COPD patients, suggesting cerebral small vessel injury caused by COPD-associated conditions.103,104 The repair capacity of endothelial progenitor cells is also significantly impaired in the early stage of COPD.97,105 Furthermore, some animal studies have demonstrated apoptosis of pulmonary capillary endothelial cells due to emphysematous changes.11 These reports indicate that endothelial injury is closely connected to the pathophysiology of COPD.
We reported that VE-cadherin EMPs, PECAM EMPs, and E-selectin EMPs were significantly increased in patients with stable COPD compared to ex-smoker controls independent of smoking history, sex, body mass index, and age.82 This increase in PECAM EMPs was confirmed in a larger study performed by the MESA group.96 However, when adjusted for other factors, there were no significant differences in the levels of CD51 EMPs and E-selectin EMPs, although both EMPs tended to increase in COPD in the MESA group’s study. According to the Global initiative for chronic Obstructive Lung Disease (GOLD) stage progression, VE-cadherin EMPs, PECAM EMPs, and E-selectin EMPs were significantly increased in our study, although we could not analyze differences between each stage due to the small sample sizes. The MESA group reported that PECAM EMPs were significantly increased in mild and moderate COPD compared to control subjects in analyses adjusted for other factors; in contrast CD51 EMPs were not significantly elevated and E-selectin EMPs were only elevated in severe COPD. These results indicate that PECAM, VE-cadherin, and E-selectin EMPs are increased in patients with stable COPD compared to healthy controls independent of other factors associated with endothelial injury.
These two studies also examined the relationships between the degree of lung destruction and the numbers of EMPs. PECAM EMPs, E-selectin EMPs, and VE-cadherin EMPs were inversely correlated with parameters of lung functions such as the percent predicted FEV1. The MESA group also reported that PECAM EMPs were significantly associated with parameters of lung destruction such as percent emphysema (low attenuation area; LAA), pulmonary microvascular perfusion as assessed by magnetic resonance imaging, and diffusing capacity of the lung for carbon monoxide (DLCO). However, there was no significant correlation between E-selectin EMP levels and these parameters. By contrast, E-selectin EMPs were significantly related to hyperinflation characterized by both increased RV and a higher RV/TLC ratio; no such relationship was observed for PECAM EMPs. These results indicate that endothelial injury combined with impaired mobilization capacity of endothelial progenitor cells97,105 might be involved in the pathogenesis of emphysema and COPD.
Endothelial injury occurs in both the pulmonary and systemic vasculatures in COPD. Thus, EMPs can be released from both the pulmonary vasculatures and systemic vasculatures. To distinguish between EMPs originating from pulmonary and systemic circulation, the absence of vWF82 and the presence of angiotensin-converting enzyme (ACE; CD143)46 on EMPs were examined. vWF is a marker of endothelial cells that is not expressed on pulmonary capillary cells.106,107 Thus, vWF-negative EMPs are of pulmonary capillary origin. The pulmonary capillary endothelium contains abundant ACE. Thus, ACE-positive EMPs are of pulmonary capillary origin. In active smokers with normal spirometry and low DLCO, approximately 75% of PECAM EMPs were ACE positive, indicating pulmonary capillary origin.46 In stage II COPD patients, approximately 60% of the increased EMPs were vWF negative (Figure 3). In addition, the numbers of EMPs in stage IV COPD patients with severe emphysema were lower than those with mild emphysema, reflecting the reduction of pulmonary capillary vascular beds in emphysema. These results confirmed two facts. First, the increased EMPs in COPD mainly originate from the pulmonary capillary vasculature. Second, the numbers of vWF-negative or ACE-positive EMPs are sensitive markers for the detection of endothelial injury in the pulmonary capillary vasculature.
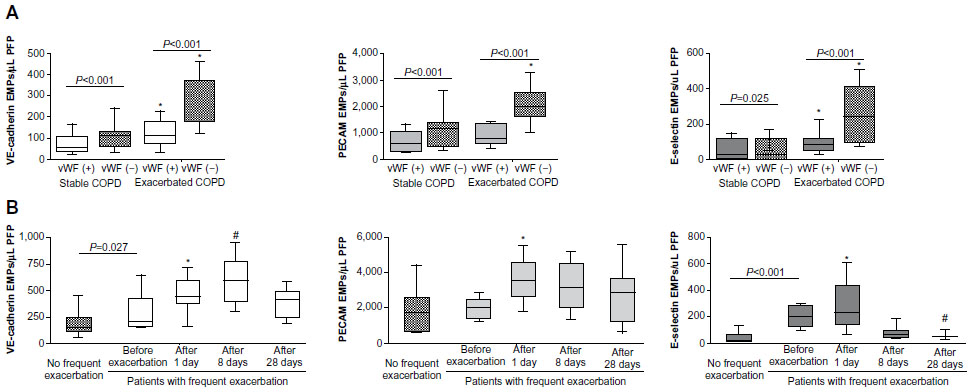 | Figure 3 EMP levels in patients with exacerbated COPD. |
Multivariable analysis revealed that E-selectin levels were higher in patients with stable COPD with a history of frequent exacerbation (two or more episodes of exacerbation each year) than in COPD patients without frequent exacerbation.82 Unfortunately, the relationship between the number of EMPs and the rate of exacerbation was not analyzed in the MESA study.96 E-selectin EMP levels may reflect ongoing endothelial inflammation. Because exacerbations are associated with accelerated loss of lung function,2–5 we hypothesize that high E-selectin EMP levels led to a rapid loss of lung function in patients with stable COPD with a history of frequent exacerbation.
Circulating EMPs in patients with exacerbated COPD
Recent studies have indicated that an exacerbation episode is connected to the progression of COPD. A history of frequent exacerbation and/or numbers of exacerbation episodes is associated with future rapid FEV1 decline, increased morbidity, readmission rates, and mortality.108–112 Exacerbation episodes are also associated with emphysema progression, as evaluated by chest computed tomography scans in patients with COPD.113 Although the importance of exacerbation in COPD management is becoming clearer, the diagnosis and evaluation of exacerbation are mainly based on clinical symptoms.
Accumulating evidence suggests that further endothelial injury occurs during exacerbation. For example, flow and nitroglycerin-mediated peripheral vascular dilation are impaired during acute exacerbation.114 In addition, inflammatory responses, particularly platelet activation, are upregulated during exacerbation. Platelet aggregation is exaggerated,115 mean platelet volume is increased,116 and plasma vWF and fibrinogen levels are increased during exacerbation.117 To clarify the influence of COPD exacerbation on the endothelium, we examined the numbers of EMPs during exacerbation.
VE-cadherin, PECAM, and E-selectin EMPs were significantly higher in patients during COPD exacerbation than in stable patients. Interestingly, the majority of the EMPs that were increased during COPD exacerbation were vWF negative (Figure 3), indicating that pulmonary capillary endothelial cells were the main targets of injury during exacerbation. In addition, exacerbation is associated with the progression of emphysema,113 which is connected to endothelial injury in the pulmonary vasculature.11 These reports confirmed that endothelial injury, primarily in pulmonary capillary endothelial cells, occurred during exacerbation.
The trends in the levels of circulating VE-cadherin EMPs, PECAM EMPs, and E-selectin EMPs differed after exacerbation (Figure 3). All EMP levels increased significantly one day after the onset of exacerbation, but after 28 days, the levels of VE-cadherin EMPs and PECAM EMPs remained high, while the level of E-selectin EMPs declined to less than the baseline level. The persistent high VE-cadherin and PECAM EMP levels after 28 days indicate that endothelial injury induced by exacerbation continues even after clinical symptoms disappear. Similar to E-selectin EMP levels, plasma fibrinogen levels also increase during exacerbation and decrease significantly in 4–6 weeks.118–120 Although there are no data to indicate a significant correlation between E-selectin EMP levels and plasma fibrinogen levels, these reports provide additional evidence that E-selectin EMP levels reflect ongoing endothelial inflammation and that VE-cadherin and PECAM EMP levels reflect endothelial injury as a consequence of endothelial inflammation. The decrease in the high E-selectin EMP levels during the early phase of exacerbation to levels lower than the baseline after treatment indicates that inflammation is present on the endothelium even during the stable phase before the onset of exacerbation. Most of the patients who underwent exacerbation received a systemic corticosteroid therapy, however, the role of steroid in the EMP levels is not defined yet. Further analyses are needed to clarify the impact of the COPD drugs on the kinetics of EMPs. Furthermore, drug therapies that ameliorate increased EMP levels may have an effect on prognosis of COPD patients.
Effects of increased EMPs on COPD pathophysiology
Increased numbers of EMPs are observed in patients with stable COPD. The release of EMPs increases further during exacerbation. In addition, COPD patients with a history of frequent exacerbation exhibit high levels of circulating EMPs not only during exacerbation but also in the stable phase. However, the effects of this increase in EMPs on COPD pathophysiology remain unclear. In patients with severe systemic inflammatory syndrome, EMPs are produced and actively bind to leukocytes.121 MPs from human atherosclerotic plaques promote transendothelial migration of monocytes.122 Circulating MPs in patients with myocardial infarction induce endothelial dysfunction.123 Based on these reports, the increased EMPs released from injured pulmonary endothelium might induce further endothelial injury in both the pulmonary vasculature and distant systemic vasculature; this injury might be involved in further lung destruction as well as the increased incidence of cardiovascular diseases in COPD patients in both the stable phase and after exacerbation.108 Because the MPs released in response to various stimuli are heterogeneous, future studies should examine the effects of MPs on the endothelium using MPs isolated from COPD patients. Prospective studies are also needed to clarify the relationship between increased EMP levels and the incidence of cardiovascular diseases.
Technical difficulties with measuring EMPs
There are several technical difficulties reported in the measurement of MPs. For example, there is no clear definition of MPs. The differences between exosomes and MPs remain unclear. Thus, there is no standard protocol for isolating and detecting circulating MPs from the plasma, and the results of microparticle studies are often inconsistent.124 Differences in flow cytometers influence the sensitivity of MP detection. In addition, differences in centrifugation protocols influence the number of MPs. Clear definitions and standardization of protocols are essential.
Other remaining questions
Although much evidence for a link between COPD and circulating EMPs has been documented, there are several issues to clarify in the future.
Firstly, the effects of increased EMPs on the pathophysiological condition or progression of exacerbation are not clear. MPs are not the passive parameter induced from activated or injured cells but rather active modulators that promote both pro-inflammatory and anti-inflammatory signals.125 MPs contain proteins and microRNAs and have a capacity to deliver those components to distant endothelial cells.22 Therefore, increased EMPs may influence vascular function and systemic inflammation under COPD exacerbation.
Secondly, the release of MPs originating from other cell types is not clearly evaluated in COPD patients. Other MPs, such as platelet-derived and leukocyte-derived MPs, play different roles in endothelial phenotypes; in particular, platelet-derived MPs are known to increase in cardiovascular diseases, including myocardial infarction.126 Therefore, the role of other MPs in the comorbidity and the prognosis of COPD would be a great interest.
Lastly, epithelial-derived MPs in airways are not elucidated in COPD patients. Tissue factor-bearing MPs, which demonstrate procoagulant activity, are elevated in the pulmonary edema fluid of acute respiratory distress syndrome patients;127 these MPs appear to be derived from alveolar epithelial cells. Similarly, epithelial injury present in COPD airways may produce the epithelial-derived MPs.
Conclusion
In this review, we proposed the potential use of circulating MPs, particularly EMPs, as novel biomarkers for COPD (Figure 4). In addition, by comparing subtypes of MPs that are increased in COPD patients, we may be able to reclassify heterogeneous COPD. Furthermore, the relationship between COPD and other MPs, such as platelet-derived MPs, could be an intriguing area of investigation in the future.
 | Figure 4 EMPs as potential biomarkers for COPD. |
Disclosure
The authors report no conflicts of interest in this work.
References
Rabe KF, Hurd S, Anzueto A, et al; Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176(6):532–555. | |
Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57(10):847–852. | |
Vestbo J, Edwards LD, Scanlon PD, et al; ECLIPSE Investigators. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011;365(13):1184–1192. | |
Kanner RE, Anthonisen NR, Connett JE; Lung Health Study Research Group. Lower respiratory illnesses promote FEV(1) decline in current smokers but not ex-smokers with mild chronic obstructive pulmonary disease: results from the lung health study. Am J Respir Crit Care Med. 2001;164(3):358–364. | |
Calverley PM, Anderson JA, Celli B, et al; TORCH investigators. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med. 2007;356(8):775–789. | |
Brutsche MH, Downs SH, Schindler C, et al; SAPALDIA Team. Bronchial hyperresponsiveness and the development of asthma and COPD in asymptomatic individuals: SAPALDIA cohort study. Thorax. 2006;61(8):671–677. | |
Agusti A, Calverley PM, Celli B, et al; Evaluation of COPD Longitudinally to Identify Predictive Surrogate Endpoints (ECLIPSE) investigators. Characterisation of COPD heterogeneity in the ECLIPSE cohort.Respir Res. 2010;11:122. | |
Vestbo J, Rennard S. Chronic obstructive pulmonary disease biomarker(s) for disease activity needed – urgently. Am J Respir Crit Care Med. 2010;182(7):863–864. | |
Henson PM, Vandivier RW, Douglas IS. Cell death, remodeling, and repair in chronic obstructive pulmonary disease? Proc Am Thorac Soc. 2006;3(8):713–717. | |
Schmidt EP, Tuder RM. Role of Apoptosis in Amplifying Inflammatory Responses in Lung Diseases. J Cell Death. 2010;2010(3):41–53. | |
Kasahara Y, Tuder RM, Taraseviciene-Stewart L, et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106(11):1311–1319. | |
Kasahara Y, Tuder RM, Cool CD, Lynch DA, Flores SC, Voelkel NF. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med. 2001;163(3 Pt 1):737–744. | |
Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med. 2006;174(8):886–893. | |
Hodge S, Hodge G, Holmes M, Reynolds PN. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur Respir J. 2005;25(3):447–454. | |
Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129(6):1673–1682. | |
Ryter SW, Choi AM. Autophagy in the lung. Proc Am Thorac Soc. 2010;7(1):13–21. | |
Pisetsky DS, Gauley J, Ullal AJ. HMGB1 and microparticles as mediators of the immune response to cell death. Antioxid Redox Signal. 2011;15(8):2209–2219. | |
Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–593. | |
Gyorgy B, Szabo TG, Pasztoi M, et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011;68(16):2667–2688. | |
Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19(2):43–51. | |
Nomura S, Ozaki Y, Ikeda Y. Function and role of microparticles in various clinical settings. Thromb Res. 2008;123(1):8–23. | |
Martinez MC, Tual-Chalot S, Leonetti D, Andriantsitohaina R. Microparticles: targets and tools in cardiovascular disease. Trends Pharmacol Sci. 2011;32(11):659–665. | |
Rautou PE, Vion AC, Amabile N, et al. Microparticles, vascular function, and atherothrombosis. Circ Res. 2011;109(5):593–606. | |
Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327(5965):580–583. | |
Rubin O, Canellini G, Delobel J, Lion N, Tissot JD. Red blood cell microparticles: clinical relevance. Transfus Med Hemother. 2012;39(5):342–347. | |
Smalley DM, Sheman NE, Nelson K, Theodorescu D. Isolation and identification of potential urinary microparticle biomarkers of bladder cancer. J Proteome Res. 2008;7(5):2088–2096. | |
Valenti R, Huber V, Iero M, Filipazzi P, Parmiani G, Rivoltini L. Tumor-released microvesicles as vehicles of immunosuppression. Cancer Res. 2007;67(7):2912–2915. | |
Mostefai HA, Meziani F, Mastronardi ML, et al. Circulating microparticles from patients with septic shock exert protective role in vascular function. Am J Respir Crit Care Med. 2008;178(11):1148–1155. | |
Boulanger CM, Amabile N, Tedgui A. Circulating microparticles: a potential prognostic marker for atherosclerotic vascular disease. Hypertension. 2006;48(2):180–186. | |
Sidhu SS, Mengistab AT, Tauscher AN, LaVail J, Basbaum C. The microvesicle as a vehicle for EMMPRIN in tumor-stromal interactions. Oncogene. 2004;23(4):956–963. | |
MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity. 2001;15(5):825–835. | |
Wilson HL, Francis SE, Dower SK, Crossman DC. Secretion of intracellular IL-1 receptor antagonist (type 1) is dependent on P2X7 receptor activation. J Immunol. 2004;173(2):1202–1208. | |
Gasser O, Hess C, Miot S, Deon C, Sanchez JC, Schifferli JA. Characterisation and properties of ectosomes released by human polymorphonuclear neutrophils. Exp Cell Res. 2003;285(2):243–257. | |
Baroni M, Pizzirani C, Pinotti M, et al. Stimulation of P2 (P2X7) receptors in human dendritic cells induces the release of tissue factor-bearing microparticles. FASEB J. 2007;21(8):1926–1933. | |
Pisetsky DS, Ullal AJ, Gauley J, Ning TC. Microparticles as mediators and biomarkers of rheumatic disease. Rheumatology (Oxford). 2012;51(10):1737–1746. | |
Martinez MC, Tesse A, Zobairi F, Andriantsitohaina R. Shed membrane microparticles from circulating and vascular cells in regulating vascular function. Am J Physiol Heart Circ Physiol. 2005;288(3):H1004–H1009. | |
Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7(9):678–689. | |
Lovren F, Verma S. Evolving role of microparticles in the pathophysiology of endothelial dysfunction. Clin Chem. 2013;59(8):1166–1174. | |
Ryter SW, Chen ZH, Kim HP, Choi AM. Autophagy in chronic obstructive pulmonary disease: homeostatic or pathogenic mechanism? Autophagy. 2009;5(2):235–237. | |
Takahashi T, Kobayashi S, Fujino N, et al. Differences in the released endothelial microparticle subtypes between human pulmonary microvascular endothelial cells and aortic endothelial cells in vitro. Exp Lung Res. 2013;39(4–5):155–161. | |
Kosmider B, Messier EM, Chu HW, Mason RJ. Human alveolar epithelial cell injury induced by cigarette smoke. PLoS One. 2011;6(12):e26059. | |
Bernhard D, Pfister G, Huck CW, et al. Disruption of vascular endothelial homeostasis by tobacco smoke: impact on atherosclerosis. FASEB J. 2003;17(15):2302–2304. | |
Li M, Yu D, Williams KJ, Liu ML. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arterioscler Thromb Vasc Biol. 2010;30(9):1818–1824. | |
Li CJ, Liu Y, Chen Y, Yu D, Williams KJ, Liu ML. Novel proteolytic microvesicles released from human macrophages after exposure to tobacco smoke. Am J Pathol. 2013;182(5):1552–1562. | |
Heiss C, Amabile N, Lee AC, et al. Brief secondhand smoke exposure depresses endothelial progenitor cells activity and endothelial function: sustained vascular injury and blunted nitric oxide production. J Am Coll Cardiol. 2008;51(18):1760–1771. | |
Gordon C, Gudi K, Krause A, et al. Circulating endothelial microparticles as a measure of early lung destruction in cigarette smokers. Am J Respir Crit Care Med. 2011;184(2):224–232. | |
Sundar IK, Yao H, Rahman I. Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases. Antioxid Redox Signal. 2013;18(15):1956–1971. | |
Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest. 2012;122(8):2749–2755. | |
Helal O, Defoort C, Robert S, et al. Increased levels of microparticles originating from endothelial cells, platelets and erythrocytes in subjects with metabolic syndrome: relationship with oxidative stress. Nutr Metab Cardiovasc Dis. 2011;21(9):665–671. | |
Sethi S, Maloney J, Grove L, Wrona C, Berenson CS. Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173(9):991–998. | |
Seemungal T, Sykes A; ICEAD Contributors. Recent advances in exacerbations of COPD. Thorax. 2008;63(10):850–852. | |
Mayne E, Funderburg NT, Sieg SF, et al. Increased platelet and microparticle activation in HIV infection: upregulation of P-selectin and tissue factor expression. J Acquir Immune Defic Syndr. 2012;59(4):340–346. | |
Kornek M, Lynch M, Mehta SH, et al. Circulating microparticles as disease-specific biomarkers of severity of inflammation in patients with hepatitis C or nonalcoholic steatohepatitis. Gastroenterology. 2012;143(2):448–458. | |
Ge S, Hertel B, Emden SH, et al. Microparticle generation and leucocyte death in Shiga toxin-mediated HUS. Nephrol Dial Transplant. 2012;27(7):2768–2775. | |
Nieuwland R, Berckmans RJ, McGregor S, et al. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood. 2000;95(3):930–935. | |
Diamant M, Tushuizen ME, Sturk A, Nieuwland R. Cellular microparticles: new players in the field of vascular disease? Eur J Clin Invest. 2004;34(6):392–401. | |
Freyssinet JM. Cellular microparticles: what are they bad or good for? J Thromb Haemost. 2003;1(7):1655–1662. | |
Singh N, Van Craeyveld E, Tjwa M, et al. Circulating apoptotic endothelial cells and apoptotic endothelial microparticles independently predict the presence of cardiac allograft vasculopathy. J Am Coll Cardiol. 2012;60(4):324–331. | |
Jung C, Sorensson P, Saleh N, Arheden H, Ryden L, Pernow J. Circulating endothelial and platelet derived microparticles reflect the size of myocardium at risk in patients with ST-elevation myocardial infarction. Atherosclerosis. 2012;221(1):226–231. | |
Nozaki T, Sugiyama S, Koga H, et al. Significance of a multiple biomarkers strategy including endothelial dysfunction to improve risk stratification for cardiovascular events in patients at high risk for coronary heart disease. J Am Coll Cardiol. 2009;54(7):601–608. | |
Erdbruegger U, Grossheim M, Hertel B, et al. Diagnostic role of endothelial microparticles in vasculitis. Rheumatology (Oxford). 2008;47(12):1820–1825. | |
Duval A, Helley D, Capron L, et al. Endothelial dysfunction in systemic lupus patients with low disease activity: evaluation by quantification and characterization of circulating endothelial microparticles, role of anti-endothelial cell antibodies. Rheumatology (Oxford). 2010;49(6):1049–1055. | |
Jimenez JJ, Jy W, Mauro LM, Horstman LL, Soderland C, Ahn YS. Endothelial microparticles released in thrombotic thrombocytopenic purpura express von Willebrand factor and markers of endothelial activation. Br J Haematol. 2003;123(5):896–902. | |
Combes V, Simon AC, Grau GE, et al. In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant. J Clin Invest. 1999;104(1):93–102. | |
Amabile N, Guerin AP, Leroyer A, et al. Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J Am Soc Nephrol. 2005;16(11):3381–3388. | |
Faure V, Dou L, Sabatier F, et al. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J Thromb Haemost. 2006;4(3):566–573. | |
Amabile N, Guerin AP, Tedgui A, Boulanger CM, London GM. Predictive value of circulating endothelial microparticles for cardiovascular mortality in end-stage renal failure: a pilot study. Nephrol Dial Transplant. 2012;27(5):1873–1880. | |
Pirro M, Schillaci G, Paltriccia R, et al. Increased ratio of CD31+/CD42- microparticles to endothelial progenitors as a novel marker of atherosclerosis in hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2006;26(11):2530–2535. | |
Moore XL, Michell D, Lee S, et al. Increased carotid intima-media thickness and reduced distensibility in human class III obesity: independent and differential influences of adiposity and blood pressure on the vasculature. PLoS One. 2013;8(1):e53972. | |
Stepanian A, Bourguignat L, Hennou S, et al. Microparticle increase in severe obesity: Not related to metabolic syndrome and unchanged after massive weight loss. Obesity.2013;21:2236–2243. | |
Vincent PA, Xiao K, Buckley KM, Kowalczyk AP. VE-cadherin: adhesion at arm’s length. Am J Physiol Cell Physiol. 2004;286(5):C987–C997. | |
Bardin N, Anfosso F, Masse JM, et al. Identification of CD146 as a component of the endothelial junction involved in the control of cell-cell cohesion. Blood. 2001;98(13):3677–3684. | |
Shaw SK, Bamba PS, Perkins BN, Luscinskas FW. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J Immunol. 2001;167(4):2323–2330. | |
Conway D, Schwartz MA. Lessons from the endothelial junctional mechanosensory complex. F1000 Biol Rep. 2012;4:1. | |
Newman PJ, Berndt MC, Gorski J, et al. PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science. 1990;247(4947):1219–1222. | |
Angelillo-Scherrer A. Leukocyte-derived microparticles in vascular homeostasis. Circ Res. 2012;110(2):356–369. | |
Patil S, Newman DK, Newman PJ. Platelet endothelial cell adhesion molecule-1 serves as an inhibitory receptor that modulates platelet responses to collagen. Blood. 2001;97(6):1727–1732. | |
Cicmil M, Thomas JM, Leduc M, Bon C, Gibbins JM. Platelet endothelial cell adhesion molecule-1 signaling inhibits the activation of human platelets. Blood. 2002;99(1):137–144. | |
Falati S, Patil S, Gross PL, et al. Platelet PECAM-1 inhibits thrombus formation in vivo. Blood. 2006;107(2):535–541. | |
Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24(6):327–334. | |
Woodfin A, Voisin MB, Nourshargh S. PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler Thromb Vasc Biol. 2007;27(12):2514–2523. | |
Takahashi T, Kobayashi S, Fujino N, et al. Increased circulating endothelial microparticles in COPD patients: a potential biomarker for COPD exacerbation susceptibility. Thorax. 2012;67(12):1067–1074. | |
Wang Z, Yan X. CD146, a multi-functional molecule beyond adhesion. Cancer Lett. 2013;330(2):150–162. | |
Lehmann JM, Riethmuller G, Johnson JP. MUC18, a marker of tumor progression in human melanoma, shows sequence similarity to the neural cell adhesion molecules of the immunoglobulin superfamily. Proc Natl Acad Sci U S A. 1989;86(24):9891–9895. | |
Elshal MF, Khan SS, Takahashi Y, Solomon MA, McCoy JP Jr. CD146 (Mel-CAM), an adhesion marker of endothelial cells, is a novel marker of lymphocyte subset activation in normal peripheral blood. Blood. 2005;106(8):2923–2924. | |
Maier CL, Shepherd BR, Yi T, Pober JS. Explant outgrowth, propagation and characterization of human pericytes. Microcirculation. 2010;17(5):367–380. | |
Blann AD, Woywodt A, Bertolini F, et al. Circulating endothelial cells. Biomarker of vascular disease. Thromb Haemost. 2005;93(2):228–235. | |
Bertolini F, Shaked Y, Mancuso P, Kerbel RS. The multifaceted circulating endothelial cell in cancer: towards marker and target identification. Nat Rev Cancer. 2006;6(11):835–845. | |
Bevilacqua M, Butcher E, Furie B, et al. Selectins: a family of adhesion receptors. Cell. 1991;67(2):233. | |
Jimenez JJ, Jy W, Mauro LM, Soderland C, Horstman LL, Ahn YS. Endothelial cells release phenotypically and quantitatively distinct microparticles in activation and apoptosis. Thromb Res. 2003;109(4):175–180. | |
Sosnoski DM, Emanuel BS, Hawkins AL, et al. Chromosomal localization of the genes for the vitronectin and fibronectin receptors alpha subunits and for platelet glycoproteins IIb and IIIa. J Clin Invest. 1988;81(6):1993–1998. | |
Kokubo T, Uchida H, Choi ET. Integrin alpha(v)beta(3) as a target in the prevention of neointimal hyperplasia. J Vasc Surg. 2007; 45 Suppl A:A33–A38. | |
Sheppard D. Roles of alphav integrins in vascular biology and pulmonary pathology. Curr Opin Cell Biol. 2004;16(5):552–557. | |
Minagar A, Jy W, Jimenez JJ, et al. Elevated plasma endothelial microparticles in multiple sclerosis. Neurology. 2001;56(10):1319–1324. | |
Bernal-Mizrachi L, Jy W, Jimenez JJ, et al. High levels of circulating endothelial microparticles in patients with acute coronary syndromes. Am Heart J. 2003;145(6):962–970. | |
Thomashow MA, Shimbo D, Parikh MA, et al. Endothelial microparticles in mild chronic obstructive pulmonary disease and emphysema. The Multi-Ethnic Study of Atherosclerosis Chronic Obstructive Pulmonary Disease study. Am J Respir Crit Care Med. 2013;188(1):60–68. | |
Takahashi T, Suzuki S, Kubo H, Yamaya M, Kurosawa S, Kato M. Impaired endothelial progenitor cell mobilization and colony-forming capacity in chronic obstructive pulmonary disease. Respirology. 2011;16(4):680–687. | |
Young RP, Hopkins R, Eaton TE. Forced expiratory volume in one second: not just a lung function test but a marker of premature death from all causes. Eur Respir J. 2007;30(4):616–622. | |
Peinado VI, Pizarro S, Barbera JA. Pulmonary vascular involvement in COPD. Chest. 2008;134(4):808–814. | |
Hunninghake DB. Cardiovascular disease in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2(1):44–49. | |
Barr RG, Mesia-Vela S, Austin JH, et al. Impaired flow-mediated dilation is associated with low pulmonary function and emphysema in ex-smokers: the Emphysema and Cancer Action Project (EMCAP) Study. Am J Respir Crit Care Med. 2007;176(12):1200–1207. | |
Iwamoto H, Yokoyama A, Kitahara Y, et al. Airflow limitation in smokers is associated with subclinical atherosclerosis. Am J Respir Crit Care Med. 2009;179(1):35–40. | |
Ryu CW, Jahng GH, Choi CW, et al. Microstructural change of the brain in chronic obstructive pulmonary disease: a voxel-based investigation by MRI. COPD. 2013;10(3):357–366. | |
Dodd JW, Chung AW, van den Broek MD, Barrick TR, Charlton RA, Jones PW. Brain structure and function in chronic obstructive pulmonary disease: a multimodal cranial magnetic resonance imaging study. Am J Respir Crit Care Med. 2012;186(3):240–245. | |
Palange P, Testa U, Huertas A, et al. Circulating haemopoietic and endothelial progenitor cells are decreased in COPD. Eur Respir J. 2006;27(3):529–541. | |
Muller AM, Hermanns MI, Skrzynski C, Nesslinger M, Muller KM, Kirkpatrick CJ. Expression of the endothelial markers PECAM-1, vWf, and CD34 in vivo and in vitro. Exp Mol Pathol. 2002;72(3):221–229. | |
Kawanami O, Jin E, Ghazizadeh M, et al. Mosaic-like distribution of endothelial cell antigens in capillaries and juxta-alveolar microvessels in the normal human lung. Pathol Int. 2000;50(2):136–141. | |
Donaldson GC, Hurst JR, Smith CJ, Hubbard RB, Wedzicha JA. Increased risk of myocardial infarction and stroke following exacerbation of COPD. Chest. 2010;137(5):1091–1097. | |
Mannino DM, Thorn D, Swensen A, Holguin F. Prevalence and outcomes of diabetes, hypertension and cardiovascular disease in COPD. Eur Respir J. 2008;32(4):962–969. | |
Soler-Cataluna JJ, Martinez-Garcia MA, Roman Sanchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax. 2005;60(11):925–931. | |
Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1418–1422. | |
Patel AR, Kowlessar BS, Donaldson GC, et al. Cardiovascular risk, myocardial injury, and exacerbations of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(9):1091–1099. | |
Tanabe N, Muro S, Hirai T, et al. Impact of exacerbations on emphysema progression in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(12):1653–1659. | |
Marchetti N, Ciccolella DE, Jacobs MR, et al. Hospitalized acute exacerbation of COPD impairs flow and nitroglycerin-mediated peripheral vascular dilation. COPD. 2011;8(2):60–65. | |
Maclay JD, McAllister DA, Johnston S, et al. Increased platelet activation in patients with stable and acute exacerbation of COPD. Thorax. 2011;66(9):769–774. | |
Wang RT, Li JY, Cao ZG, Li Y. Mean platelet volume is decreased during an acute exacerbation of chronic obstructive pulmonary disease. Respirology. 2013;18(8):1244–1248. | |
Polatli M, Cakir A, Cildag O, Bolaman AZ, Yenisey C, Yenicerioglu Y. Microalbuminuria, von Willebrand factor and fibrinogen levels as markers of the severity in COPD exacerbation. J Thromb Thrombolysis. 2008;26(2):97–102. | |
Wedzicha JA, Seemungal TA, MacCallum PK, et al. Acute exacerbations of chronic obstructive pulmonary disease are accompanied by elevations of plasma fibrinogen and serum IL-6 levels. Thromb Haemost. 2000;84(2):210–215. | |
Koutsokera A, Kiropoulos TS, Nikoulis DJ, et al. Clinical, functional and biochemical changes during recovery from COPD exacerbations. Respir Med. 2009;103(6):919–926. | |
Valipour A, Schreder M, Wolzt M, et al. Circulating vascular endothelial growth factor and systemic inflammatory markers in patients with stable and exacerbated chronic obstructive pulmonary disease. Clin Sci (Lond). 2008;115(7):225–232. | |
Ogura H, Tanaka H, Koh T, et al. Enhanced production of endothelial microparticles with increased binding to leukocytes in patients with severe systemic inflammatory response syndrome. J Trauma. 2004;56(4):823–830; discussion 830–821. | |
Rautou PE, Leroyer AS, Ramkhelawon B, et al. Microparticles from human atherosclerotic plaques promote endothelial ICAM-1-dependent monocyte adhesion and transendothelial migration. Circ Res. 2011;108(3):335–343. | |
Boulanger CM, Scoazec A, Ebrahimian T, et al. Circulating microparticles from patients with myocardial infarction cause endothelial dysfunction. Circulation. 2001;104(22):2649–2652. | |
Witwer KW, Buzas EI, Bemis LT, et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2. | |
Morel O, Toti F, Morel N, Freyssinet JM. Microparticles in endothelial cell and vascular homeostasis: are they really noxious? Haematologica. 2009;94(3):313–317. | |
van der Zee PM, Biro E, Ko Y, et al. P-selectin- and CD63-exposing platelet microparticles reflect platelet activation in peripheral arterial disease and myocardial infarction. Clin Chem. 2006;52(4):657–664. | |
Bastarache JA, Fremont RD, Kropski JA, Bossert FR, Ware LB. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2009;297(6):L1035–L1041. | |
Amabile N, Heiss C, Real WM, et al. Circulating endothelial microparticle levels predict hemodynamic severity of pulmonary hypertension. Am J Respir Crit Care Med. 2008;177(11):1268–1275. | |
Amabile N, Heiss C, Chang V, et al. Increased CD62e(+) endothelial microparticle levels predict poor outcome in pulmonary hypertension patients. J Heart Lung Transplant. 2009;28(10):1081–1086. | |
Ayers L, Ferry B, Craig S, Nicoll D, Stradling JR, Kohler M. Circulating cell-derived microparticles in patients with minimally symptomatic obstructive sleep apnoea. Eur Respir J. 2009;33(3):574–580. | |
Simak J, Gelderman MP, Yu H, Wright V, Baird AE. Circulating endothelial microparticles in acute ischemic stroke: a link to severity, lesion volume and outcome. J Thromb Haemost. 2006;4(6):1296–1302. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.