")
Back to Journals » Integrated Pharmacy Research and Practice » Volume 4
The role of medications and their management in acute kidney injury
Authors McDaniel B, Bentley M
Received 3 January 2015
Accepted for publication 13 March 2015
Published 18 May 2015 Volume 2015:4 Pages 21—29
DOI https://doi.org/10.2147/IPRP.S52930
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jonathan Ling
Bradford L McDaniel,1 Michael L Bentley1,2
1Department of Pharmacy, Carilion Clinic, Roanoke, VA, USA; 2Department of Biomedical Science, Virginia Tech Carilion School of Medicine, Roanoke, VA, USA
Abstract: Prior to 2002, the incidence of acute renal failure (ARF) varied as there was no standard definition. To better understand its incidence and etiology and to develop treatment and prevention strategies, while moving research forward, the Acute Dialysis Quality Initiative workgroup developed the RIFLE (risk, injury, failure, loss, end-stage kidney disease) classification. After continued data suggesting that even small increases in serum creatinine lead to worse outcomes, the Acute Kidney Injury Network (AKIN) modified the RIFLE criteria and used the term acute kidney injury (AKI) instead of ARF. These classification and staging systems provide the clinician and researcher a starting point for refining the understanding and treatment of AKI. An important initial step in evaluating AKI is determining the likely location of injury, generally classified as prerenal, renal, or postrenal. There is no single biomarker or test that definitively defines the mechanism of the injury. Identifying the insult(s) requires a thorough assessment of the patient and their medical and medication histories. Prerenal injuries arise primarily due to renal hypoperfusion. This may be the result of systemic or focal conditions or secondary to the effects of drugs such as nonsteroidal anti-inflammatory drugs, calcineurin inhibitors (CIs), and modulators of the renin–angiotensin–aldosterone system. Renal, or intrinsic, injury is an overarching term that represents complex conditions leading to considerable damage to a component of the intrinsic renal system (renal tubules, glomerulus, vascular structures, interstitium, or renal tubule obstruction). Acute tubular necrosis and acute interstitial nephritis are the more common types of intrinsic renal injury. Each type of injury has several drugs that are implicated as a possible cause, with antiinfectives being the most common. Postrenal injuries that result from obstruction block the flow of urine, leading to hydronephrosis and subsequent damage to the renal parenchyma. Drugs associated with tubular obstruction include acyclovir, methotrexate, and several antiretrovirals. Renal recovery from drug-induced AKI begins once the offending agent has been removed, if clinically possible, and is complete in most cases. It is uncommon that renal replacement therapy will be needed while recovery occurs. Pharmacists can play a pivotal role in identifying possible causes of drug-induced AKI and limit their toxic effect by identifying those most likely to cause or contribute to injury. Dose adjustment is critical during changes in renal function, and the pharmacist can ensure that optimal therapy is provided during this critical time.
Keywords: acute kidney injury, acute renal failure, acute tubular necrosis, drug-induced kidney injury, renal insufficiency
Introduction
Decreasing renal function can result in the accumulation of metabolic waste products (eg, urea, creatinine), electrolyte and acid–base abnormalities, and fluid retention. If not corrected in a timely fashion, the kidney will continue to fail and renal replacement therapy may be needed. Determining the epidemiology and etiology of acute renal failure (ARF) has been problematic. In 2002, the Acute Dialysis Quality Initiative (ADQI) workgroup met and proposed a consensus definition. The RIFLE (risk, injury, failure, loss, end-stage kidney disease) classification categorizes ARF into three grades of increasing severity and two outcome variables.1 Since that time, additional evidence has emerged suggesting that even small increases in serum creatinine (Scr) (≥0.3 mg/dL) lead to increased morbidity and mortality. With this understanding and a desire to include the entire spectrum of acute dysfunction, the Acute Kidney Injury Network (AKIN) modified the RIFLE classification and suggested the term acute kidney injury (AKI) be used.2–4 The new staging system, AKIN, is not meant to replace RIFLE but should be considered complementary.
Epidemiology
AKI occurring in the community (CA-AKI) is relatively uncommon. It accounts for ~1% of hospital admissions in the United States.5 However, its true incidence is likely unknown. Not until recently have investigators used standardized definitions in an attempt to better understand its incidence and underlying causes. In the hospital, AKI is seen in 5%–7% of patients,6 and critically ill patients are at the highest risk. In this group AKI is seen in 5%–20% of patients. Furthermore, ~6% of these individuals will require renal replacement therapy (RRT) during their stay in the intensive care unit.7
Classifying and staging AKI
The RIFLE classification system defines “Risk” as oliguria for more than 6 hours or a Scr increase of at least 50%, “Injury” as a 2-fold increase in Scr or oliguria for 12 hours, and “Failure” as a 3-fold increase in Scr or Scr ≥4 mg/dL (with an acute increase of at least 0.5 mg/dL) or anuria for 12 hours. The last two letters of the acronym represent outcome variables, where “L” or loss is the complete loss of function for >4 weeks and “E” or end-stage kidney disease is the complete loss of function for >3 months.1 The worse Scr or urine output defines the class.
The AKIN workgroup defined AKI using a staging system, stages 1–3. Stage 1 is met when Scr increases to 1.5- to 2-fold above baseline or by ≥0.3 mg/dL or documented oliguria (<0.5 mL/kg/h for more than 6 hours) despite adequate fluid resuscitation. Stage 2 is met when Scr increases to 2- to 3-fold above baseline or urine output is <0.5 mL/kg/h for 12 hours. Stage 3 is met when Scr increases >3-fold above baseline or ≥4 mg/dL with an acute rise of ≥0.5 mg/dL, or urine output is <0.3 mL/kg/h for 24 hours, or anuria for 12 hours, all occurring over no more than 48 hours. The AKIN definition categorizes all patients requiring RRT as Stage 3.8 Table 1 summarizes Scr and urine output for both the RIFLE classification and AKIN staging systems.
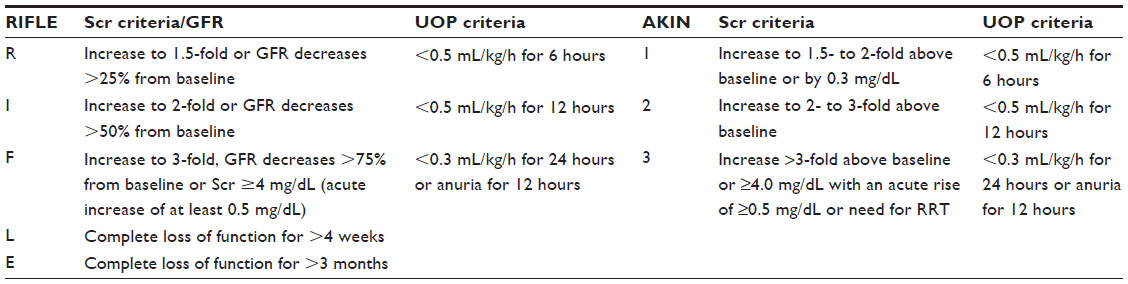 | Table 1 Classification and staging of AKI |
Etiology
AKI can be divided into categories depending on the location of injury, and more than one can contribute: prerenal, renal, and postrenal.9–12 Determining the location of the injury may assist in the search to identify the most likely cause. However, there is no one single biomarker or test that is definitive. Rather, the underlying insult or insults are most often identified after a detailed review of the patient’s medical history, recent and current medications, a complete physical examination, and assessment of laboratory values. It is important to recognize that many laboratory variables used to assist in the diagnosis of AKI can be influenced by nonrenal factors. Table 2 lists several influences of Scr and urea. In addition, in the critically ill, reaching a steady-state Scr is often difficult as there are several variables that influence its production, volume of distribution, and rate of elimination.13–15
 | Table 2 Factors affecting serum creatinine and urea |
Prerenal injury
Prerenal injury accounts for 40%–80% of AKI cases.16 The primary insult is renal hypoperfusion. Renal autoregulation is primarily governed by a combination of preglomerular (afferent) arteriolar vasodilatation and postglomerular (efferent) arteriolar vasodilation. These mechanisms enable a fairly consistent degree of renal blood flow and glomeruli filtration across a wide range of mean arterial pressures. Preglomerular arteriolar vasodilation is directed by prostaglandins and nitric oxide. Postglomerular arteriolar vasodilation is directed by angiotensin-II. Renal autoregulation becomes impaired when mean arterial pressures fall below 65 mm Hg. A decline in the rate of glomerular filtration is proportional to the level of hypoperfusion.17
Diminished renal perfusion can be a complication of systemic or focal conditions resulting from decreased intravascular volume, a change in vascular resistance, or low cardiac output.16 Nonsteroidal anti-inflammatory drugs (NSAIDs) have been implicated in several forms of kidney injury, including hemodynamically-mediated (prerenal) AKI, electrolyte and acid–base disorders, acute interstitial nephritis (AIN), and papillary necrosis.18 Prostaglandin synthesis is usually increased in the conditions of hypotension and afferent arteriolar vasodilation. Prostaglandins act to vasodilate the afferent arteriole and thus help to increase blood flow into the glomerulus.19,20 NSAIDs, by inhibiting the synthesis of prostaglandins, can worsen renal failure by further preventing afferent arteriole dilatation.16–18,20 AKI associated with NSAID administration is usually reversible once the offending agent has been discontinued. However, full recovery may take weeks to months.
Calcineurin inhibitors (CIs) can compromise renal perfusion by causing vasodilation of the afferent renal arteriole. When first introduced in the 1980s, their use in solid organ allogeneic transplants helped to improve 1-year graft survival, shortened postoperative hospital length of stay, and resulted in fewer rejection crises, among other advantages.9,21 CI-induced nephrotoxicity was initially thought to be only dose-dependent, which led to routine therapeutic drug monitoring. However, it is now recognized that they can cause nephrotoxicity at low dosages.22,23 It is thought that changes in renal hemodynamics are the product of increased endothelin and thromboxane A2 production, which vasoconstrict, and decreased prostaglandins production, which have renal vasodilatory effects. Other proposed mechanisms are the inhibition of nitric oxide and the activation of the sympathetic nervous system.23–26 Acute injury caused by CIs is typically alleviated with a dose reduction or withdrawal of the drug.27 Kidney injury corresponding with chronic use is associated with an interstitial nephritis pathology and is generally not reversible. It has been seen with low doses and is more commonly associated with mild-to-moderate dysfunction.9 Close monitoring of renal function and drug levels during the early period of therapy (days to weeks) can assist in preemptively managing deterioration of renal perfusion.
Drugs that impact the renin–angiotensin–aldosterone system may increase the risk of a prerenal injury by their actions on the efferent arteriole. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers reduce the synthesis or the activity of angiotensin-II, which results in the dilation of the efferent arteriole, consequently decreasing the intrarenal perfusion pressure (glomerular filtration rate).9,28 In the early use of these medications, those with bilateral renal artery stenosis were identified to be at the highest risk. With much broader use in the last couple of decades, AKI has been documented in those without renal artery stenosis.28,29 Monitoring is most important following the initiation of therapy. A rise in Scr may be expected and is typically not of concern unless it exceeds 30% of baseline. A rise above this threshold or one that does not stabilize in the 6–12 weeks following initiation may be considered an induced kidney injury. Patients also at risk include those who may have altered renal blood flow at baseline (heart failure) and induced states of dehydration (overdiuresis, excessive gastrointestinal losses). Clinicians may also choose to start at very low doses, use short-acting agents, and increase monitoring in those who are at an increased risk. As patients become stable following initiation of therapy, doses may be increased as tolerated and long-acting versions can be substituted.9,30
Renal (intrinsic) injury
Renal injury to the kidney results from damage to the renal tubules, glomerulus, vascular structures, or interstitium, or from obstruction of the renal tubules. It has been implicated in 10%–50% of AKI cases.17,31,32 Of these, necrosis to renal tubules is the most common (acute tubular necrosis [ATN]). Intrinsic renal failure is complex, usually arising as a result of more than one cause. Nondrug causes include prolonged or severe prerenal states (eg, hypovolemia, decreased cardiac output, sepsis), pyelonephritis, and a variety of immunologic mechanisms primarily affecting the glomerulus.9,33
Acute tubular necrosis
Acute tubular necrosis is common in critically ill patients and frequently results from prolonged prerenal insults or nephrotoxins. Histological changes show that when there is necrosis of the renal tubule epithelium, the necrotic cells are shed into the tubule lumen and can result in extensive damage. Regeneration is possible if the tubular basement membrane remains intact. The damage is usually reversible, but may take days to weeks, and short-term renal replacement therapy may be required. If ischemia is severe and prolonged, the damage may be irreversible.34
Drug-induced ATN is most commonly caused by aminoglycosides (AGs), amphotericin B (AmB), radiocontrast agents, cocaine, cisplatin, and several antiretroviral agents (adefovir, cidofovir, tenofovir, foscarnet). AGs’ cationic charge facilitates binding to tubular epithelial cell membrane phospholipids.35 Once transported intracellularly, the AG binds to acidic phospholipids, which may aggregate and inhibit phospholipid activity, and, thus, cellular functions. Synthesis and reabsorption of proteins, mitochondrial function, and the Na–K–ATPase pump may be interrupted.36–38 Further nephrotoxicity may occur as the lysosomal membrane ruptures and releases the drug, toxins, and other lysosomal enzymes into the cytosol.36,39 It is postulated that the number of cationic groups on the AG molecule positively correlates with its nephrotoxic potential secondary to an increased degree of transport across the cell membrane.40,41 This concept has been supported by comparison studies indicating that neomycin is associated with a significant toxicity burden; amikacin, gentamicin, and tobramycin are considered intermediate, with streptomycin being the least toxic.41–44 Despite a continued decline in the use of AGs, due to newer, less-toxic antimicrobials, they still have a role in therapy. Appropriate monitoring, including drug levels, should be incorporated to decrease the incidence of toxicity. Additionally, appropriate evidence-based dosing regimens, including extended interval dosing, may ensure effective therapy while minimizing drug exposure.
Amphotericin B is another antiinfective that is associated with a significant nephrotoxic potential. Beyond its nephrotoxic effects, AmB is frequently associated with adverse effects such as infusion-related reactions (fever, hypotension), metabolic disorders, cardiotoxicity, and death.45 The development of lipid formulations of AmB was driven by the high rate of nephrotoxicity seen with the conventional formulation (30%–80%).46 A reduction in nephrotoxicity with the liposomal formulations of AmB is well documented. Amphotericin is a polyene antiinfective that alters the permeability of the fungal cell membrane by binding to ergosterol. Subsequent cell content leakage leads to cell death. Amphotericin also binds to mammalian cell membranes via cholesterol molecules with a lesser affinity, potentially leading to a similar effect seen with fungal cells. These effects are seen mainly in the distal tubule but also in the proximal tubule.47,48 Consequentially, these pores or ion channels allow for transmembrane ion and metabolite fluxes, with sodium influx being the dominant process. When the Na–K–ATPase pump runs short on available adenosine triphosphate in its attempt to maintain appropriate balances, the downstream result is free radical generation, cellular calcium overload, impaired acidification, and potential cell death.49
Contrast administration accounts for a third of AKIs seen in hospitalized patients.5,50 There are several risk factors associated with contrast-induced nephropathy (CIN), and adequate fluid administration with either 0.9% sodium chloride or sodium bicarbonate is critical in reducing its incidence in high-risk patients.51 Chronic kidney disease has been described as a leading cause. Other risk factors include advanced heart failure, hypovolemia, elevated blood glucose, anemia, nephrotoxic drugs, and multiple myeloma. In addition, the amount and type of contrast agent have been associated with the likelihood of developing CIN. The best approach in reducing the risk of CIN is likely limiting exposure (volume and repeated administration) and the choice of a nonionic, low-osmolality agent for high-risk patients.52,53 In addition, repeated studies requiring contrast administration should be planned several days apart if the clinical circumstance permits.54 A scoring system has been developed to predict the risk of CIN in patients undergoing percutaneous coronary intervention.55 See Figure 1.
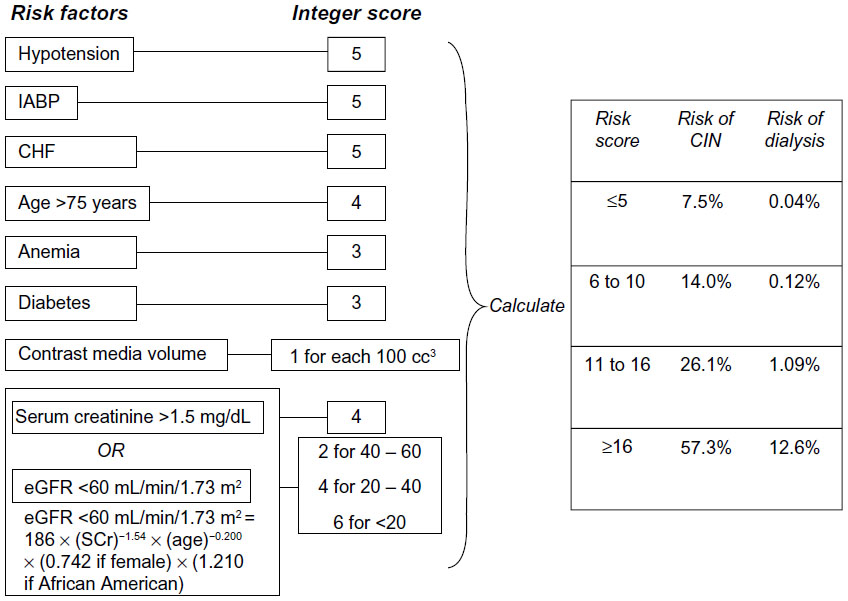 | Figure 1 Scheme to define contrast-induced nephropathy (CIN) risk score. |
Acute interstitial nephritis
Acute interstitial nephritis may affect both the renal tubules and the interstitium. It is characterized by interstitial inflammatory infiltrates and edema. The glomeruli and vessels retain a normal appearance since they are unaffected.56 The triad of fever, rash, and eosinophilia that is associated with AIN is only seen in 10%–30% of patients.57 Defining the incidence is difficult because a definitive diagnosis includes a renal biopsy. Available data suggest that between 0.5% and 2.6% of all renal biopsies are proven to be AIN.58 Biopsies performed at the time of AKI show an increased rate of AIN, 5%–18% seen in retrospective analyses.59–62 Causes of AIN vary, but drugs are implicated in the majority of cases. Other causes include infection, systemic diseases, and idiopathic causes.56 Recovery is usually seen in weeks to months after the offending agent is removed and/or disease treated, if known. One exception, as detailed earlier, is AIN associated with the chronic use of CIs, which is often irreversible.
Drug-induced AIN has a mean delay of onset occurring approximately 10 days after the initiation of the offending agent,57 though the latent period may be as short as a single day for some antibiotics or up to several months with NSAIDs.56 The most common drugs implicated are ²-lactam antibiotics, sulfonamides, and NSAIDs. Their incidences and associations have been thoroughly discussed elsewhere.56,63 With the ever-increasing use of proton pump inhibitors (PPIs) over the last two decades, their association with AIN has become more evident. Following the first published report of PPI-induced AIN in 1992, dozens more have been added to the literature.63,64 In a study performed at the Mayo Clinic, they found that PPI-induced AIN resulted in less severe AKI than that caused by antibiotics; however, the likelihood of recovery by 6 months was significantly lower.65
Vancomycin’s exact mechanism of nephrotoxicity is not fully known, but it is more commonly associated with AIN.66–68 Other mechanisms of injury have been reported, and include ATN, most of which has been reported in the pediatric population.69–71 Rodent models suggest that the injury with vancomycin may be due to oxidative stress. Vancomycin was shown to increase malondialdehyde levels, signifying oxidative stress, while decreasing the levels of superoxide dismutase and catalase, antioxidative enzymes.72
Management begins with removing the causative agent. Although renal recovery is seen, it may be incomplete in many cases. Observational studies and case reports have suggested that corticosteroid treatment can accelerate recovery.56 The lack of prospective, randomized, controlled trials, in addition to several retrospective analyses suggesting a lack of correlation, has led to the use of corticosteroids being controversial.57,73 The lack of benefit seen in some retrospective studies may be tied to the delay in initiation of corticosteroid therapy. Multivariate analysis suggests that an interval greater than 7 days between drug withdrawal and onset of corticosteroid treatment as well as the severity of interstitial fibrosis were associated with an increased risk of incomplete recovery of baseline renal function.74
Acute glomerular injury
Glomerulonephritis (GN) encompasses several glomerular diseases that are brought about by various immunologic mechanisms. Although uncommon, there are four types of drug-induced glomerulonephritis: nephrotic syndrome, focal segmental glomerulosclerosis (FSGS), membranous nephropathy, and membranoproliferative glomerulonephritis.9 With nephrotic syndrome, the damage to the glomeruli is secondary to inflammation. It is theorized that it is a cell-mediated immune process of inflammation; however, the exact etiology is not clear. Drugs that have been associated with this form of GN include NSAIDs, ampicillin, rifampin, foscarnet, and lithium.75–80 Management includes supportive therapies and often includes immunosuppressive drugs such as corticosteroids to treat the immune process causing the renal injury. Focal segmental glomerulosclerosis has a presentation that is similar to nephrotic syndrome; however, it is characterized by areas of sclerosis in some glomeruli, as well as the renal interstitial inflammation and fibrosis. Lithium and pamidronate are the two drugs most commonly associated with FSGS.80,81 The effective treatment for FSGS remains unclear; however, prednisone, cyclosporine, and mycophenolate mofetil have been used with some success for the nephrotic symptoms of the disease.82–85 Membranous nephropathy is the most common of the drug-induced glomerular diseases occurring as a result of an immune complex deposition along the glomerular capillary loops that causes thickening of part of the basement membrane. Remission typically occurs when the offending agent (NSAID, gold therapy, penicillamine) is discontinued.86 Membranoproliferative GN is rare and typically correlates with hydralazine-induced systemic lupus erythematosis. These patients most commonly present with nephrotic syndrome, and the condition is reversed in most with discontinuation of hydralazine therapy.87,88
Postrenal injury
Tubular obstruction
This form of failure results from precipitation of proteins or crystals in the lumen of the renal tubule, with the obstruction occurring anywhere between the renal pelvis and external urethral meatus. It may be seen with multiple myeloma (monoclonal light chains), tumor lysis syndrome (uric acid), or with several medications. Drugs associated with tubular obstruction include acyclovir, methotrexate (MTX), sulfadiazine, several antiretrovirals (foscarnet, indinavir, tenofovir), and triamterene. The most common risk factors associated with tubular obstruction include chronic kidney disease and volume depletion.
AKI associated with acyclovir administration is typically seen when volume depletion is present. If renal insufficiency is present, the dose should be reduced and additional nephrotoxicants avoided. It is primarily cleared by the kidney through glomerular filtration and tubular secretion as unchanged drug, approximately 62%–91%.89,90 When blood flow through the kidney is reduced, acyclovir may precipitate in the renal tubule with subsequent formation of crystals that result in obstruction. Damage caused by acyclovir is usually reversible following its discontinuation and adequate hydration.
Tubular obstruction seen with MTX is associated with high-dose therapy and preexisting renal disease. During low-flow states the drug precipitates in renal tubules as it is cleared by the kidney at >90%. Its solubility is dependent on urine pH and is most soluble when the urine is alkaline.91 Although leucovorin is commonly administered following high-dose MTX administration, it does not treat or prevent AKI.
Table 3 summarizes the potential causes of AKI and its recovery.
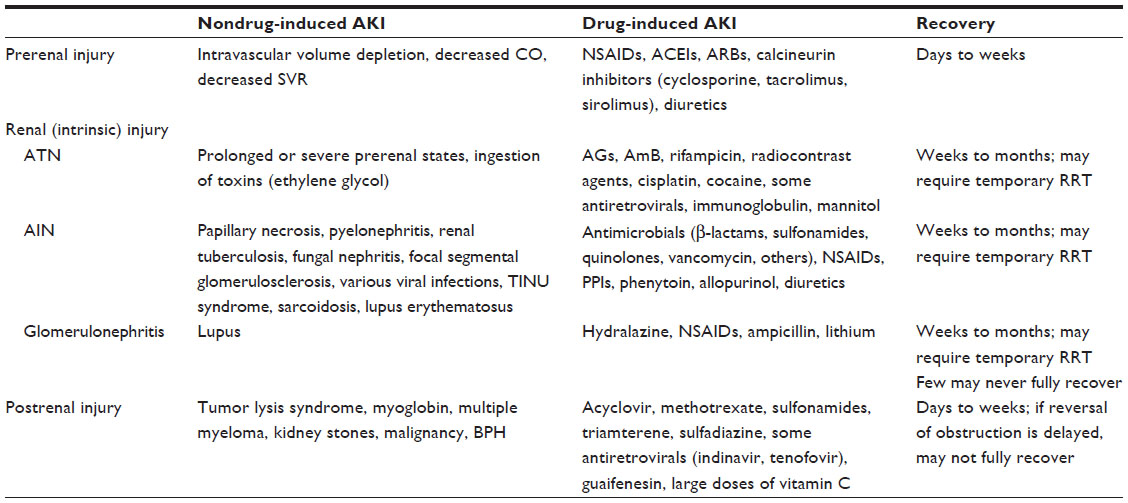 | Table 3 Potential causes of AKI and recovery expectations |
Pharmacists’ role
Numerous investigators have documented the pharmacists’ role in identifying drug-related problems and interventions to avoid these unwanted complications, both retrospectively and prospectively. The beneficial impact has been seen in both acute and chronic forms of renal failure, as well as in the setting of inpatient92–97 and outpatient care.98–100 Recommendation for renal drug adjustment is one of the more common interventions provided by pharmacists,92–95,99,100 along with educational interventions.95–97,98–100 Cost-avoidance, secondary to these interventions, has also been well described.94,101 Pharmacists are an important member of the health care team and are appropriately trained to make rational drug-dose adjustments in patients with acute and chronic kidney disease.
Conclusion
The kidneys play a pivotal role in metabolism byproduct waste removal. They help regulate hemodynamics and maintain electrolytes and acid–base balance. AKI is common, and clinicians should be aware of the many risk factors associated with its development. Determining the likely location of injury (prerenal, renal, and/or postrenal) can assist in identifying the cause of failure. Drugs are often implicated or contribute to the development of AKI, whether by affecting hemodynamics or by direct and indirect damage. Identifying patients at greatest risk and identifying drugs most commonly associated with AKI can minimize damage to the kidney if appropriate actions are taken. Clinicians should weigh the benefits and risks of each drug before choosing to start or continue its therapy. Drugs cleared by the kidney should be assessed for dose adjustments and levels monitored when clinically indicated. Pharmacists often perform these tasks while the patient is hospitalized, and plans should be developed for continued monitoring as transitions occur to other facilities or the patient is discharged home if the risk or injury still persists.
Disclosure
The authors report no conflict of interest related to this work.
References
Bellomo R, Ronco C, Kellum JA, et al; Acute Dialysis Quality Initiative Workgroup. Acute renal failure – definition, outcome measures, animal models, fluid therapy and information technology needs: the Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care. 2004;8(4):R204–R212. | |
Lassnigg A, Schmidlin D, Mouhieddine M, et al. Minimal changes of serum creatinine predict prognosis in patients after cardiothoracic surgery: a prospective cohort study. J Am Soc Nephrol. 2004;15(6):1597–1605. | |
Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16(11):3365–3370. | |
Coca SG, Peixoto AJ, Garg AX, Krumholz HM, Parikh CR. The prognostic importance of a small acute decrement in kidney function in hospitalized patients: a systematic review and meta-analysis. Am J Kidney Dis. 2007;50(5):712–720. | |
Nash K, Hafeez A, Hou S. Hospital-acquired renal insufficiency. Am J Med. 2002;39(5):930–936. | |
Clermont G, Acker CG, Angus DC, Sirio CA, Pinsky MR, Johnson JP. Renal failure in the ICU: comparison of the impact of acute renal failure and end-stage renal disease on ICU outcomes. Kidney Int. 2002;62(3):986–996. | |
Ricci Z, Ronco C. Today’s approach to the critically ill patient with acute kidney injury. Blood Purif. 2009;27(1):127–134. | |
Mehta RL, Kellum JA, Shah SV, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31. | |
Taber SS, Pasko DA. The epidemiology of drug-induced disorders: the kidney. Expert Opin Drug Saf. 2008;7(6):679–690. | |
Guo X, Nzerue C. How to prevent, recognize, and treat drug-induced nephrotoxicity. Cleve Clin J Med. 2002;69(4):289–290. | |
Schetz M, Dasta J, Goldstein S, Golper T. Drug-induced acute kidney injury. Curr Opin Crit Care. 2005;11(6):555–565. | |
Farrugia E, Larson T. Drug-induced renal toxicity. Postgrad Med. 1991; 90(2):241–248. | |
Mehta RL, Pascual MT, Soroko S, et al. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int. 2004;66: 1613–1621. | |
Guerin C, Girard R, Selli JM, Perdrix JP, Ayzac L. Initial versus delayed acute renal failure in the intensive care unit. A multicenter prospective epidemiological study. Rhone-Alpes Area Study Group on Acute Renal Failure. Am J Respir Crit Care Med. 2000;161(3):872–879. | |
Uchino S, Bellomo R, Goldsmith D, Bates S, Ronco C. An assessment of the RIFLE criteria for acute renal failure in hospitalized patients. Crit Care Med. 2006;34(7):1913–1917. | |
Watnick S, Dirkx T. Kidney Disease. In: Papadakis MA, McPhee SJ, Rabow MW, editors. CURRENT Medical Diagnosis and Treatment 2014. New York, NY: McGraw-Hill; 2014. http://accessmedicine.mhmedical.com/content.aspx?bookid=330&Sectionid=44291024. Accessed August 19, 2014. | |
Hilton R. Acute renal failure. BMJ. 2006;333(7572):786–790. | |
Clive DM, Stoff JS. Renal syndromes associated with nonsteroidal antiinflammatory drugs. N Engl J Med. 1984;310(9):563–572. | |
Oates JA, FitzGerald GA, Branch RA, et al. Clinical implications of prostaglandin and thromboxane A2 formation. N Engl J Med. 1988; 319(11):689–698. | |
Patrono C, Dunn MJ. The clinical significance of inhibition of renal prostaglandin synthesis. Kidney Int. 1987;32(1):1–12. | |
Cecka JM, Terasaki PI. The UNOS scientific renal transplant registry – 1991. In: Terasaki PI, Cecka JM, editors. Clinical Transplants 1991. Los Angeles, CA: UCLA Tissue Typing Laboratory; 1991:1–11. | |
Deray G, Benhmida M, Le Hoang P, et al. Renal function and blood pressure in patients receiving long-term, low-dose cyclosporine therapy for idiopathic autoimmune uveitis. Ann Int Med. 1992;117(7):578–583. | |
Lebwohl M, Ellis C, Gottlieb A, et al. Cyclosporine consensus conference: with emphasis on the treatment of psoriasis. J Am Acad Dermatol. 1998;39(3):464–475. | |
Bunchman TE, Brookshire CA. Cyclosporine-induced synthesis of endothelin by cultured human endothelial cells. J Clin Invest. 1991; 88(1):310–314. | |
Auch-SchwelkW, Duske E, Hink U, Betz M, Unkelbach M, Fleck E. Vasomotor responses in cyclosporin A-treated rats after chronic angiotensin blockade. Hypertension. 1994;23(6 Pt 2):832–837. | |
Lanese DM, Conger JD. Effects of endothelin receptor antagonist on cyclosporine-induced vasoconstriction in isolated rat renal arterioles. J Clin Invest. 1993;91(5):2144–2149. | |
Bennett WM, Burdmann EA, Andoh TF, Houghton DC, Lindsley J, Elzinga LW. Nephrotoxicity of immunosuppressive drugs. Nephrol Dial Transplant. 1994;9(Suppl 4):S141–S145. | |
Mel E, Davidovits M, Dagan O. Long-term follow-up evaluation of renal function in patients treated with peritoneal dialysis after cardiac surgery for correction of congenital anomalies. J Thorac Cardiovasc Surg. 2014;147(1):451–455. | |
Thind GS. Renal insufficiency during angiotensin converting enzyme inhibitor therapy in hypertensive patients with no renal artery stenosis. J Clin Hypertens. 1985;1(4):337–343. | |
Bakris GL, Weir MR. Angiotensin-converting enzyme inhibitor-associated elevations in serum creatinine: is this a cause for concern? Arch Intern Med. 2000;160(5):685–693. | |
Kaufman J, Dhakal M, Patel B, Hamburger R. Community-acquired acute renal failure. Am J Kidney Dis. 1991;17(2):191–198. | |
Liano F, Pascual J. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Kidney Int. 1996;50(3):811–818. | |
Bentley ML. Acute kidney insufficiency in the critically ill. J Pharm Pract. 2011;24(1):61–69. | |
Moore EM, Bellomo R, Nichol AD. The meaning of acute kidney injury and its relevance to intensive care and anaesthesia. Anaesth Intensive Care. 2012;40(6):929–948. | |
Humes HD, Weinberg JM, Krauss TC. Clinical and pathophysiologic aspects of aminoglycoside nephrotoxicity. Am J Kidney Dis. 1982;2(1):5–29. | |
Mingeot Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. 1999;43(5):1003–1012. | |
Swan SK. Aminoglycoside nephrotoxicity. Semin Nephrol. 1997;17(1):27–33. | |
Cronin R, Nix K, Ferguson E, Southern PM, Henrich WL. Renal cortex ion composition and Na-K-ATPase activity in gentamicin nephrotoxicity. Am J Physiol. 1982;242(5):F477–F483. | |
Fanos V, Cataldi L. Renal transport of antibiotics and nephrotoxicity: a review. J Chemother. 2001;13(5):461–472. | |
Humes HD. Aminoglycoside nephrotoxicity. Kidney Int. 1988;33(4):900–911. | |
Bennett WM, Wood CA, Houghton DC, Gilbert DN. Modification of experimental aminoglycoside nephrotoxicity. Am J Kidney Dis. 1986; 8(5):292–296. | |
Matzke GR, Lucarotti RL, Shapiro HS. Controlled comparison of gentamicin and tobramycin nephrotoxicity. Am J Nephrol. 1983;3(1):11–17. | |
Smith CR, Lipsky JJ, Laskin OL, et al. Double-blind comparison of the nephrotoxicity and auditory toxicity of gentamicin and tobramycin. N Engl J Med. 1980;302(20):1106–1109. | |
Meyer RD. Risk factors and comparisons of clinical nephrotoxicity of aminoglycosides. Am J Med. 1986;80(6B):119–125. | |
Johansen HK, Gotzsche PC. Amphotericin B lipid soluble formulations vs amphotericin B in cancer patients with neutropenia. Cochrane Database Syst Rev. 2000;(3):CD000969. | |
Kleinberg M. What is the current and future status of conventional amphotericin B? Int J Antimicrob Agents. 2006;27(Suppl 1):12–16. | |
Zager RA. Polyene antibiotics: relative degrees of in vitro cytotoxicity and potential effects on tubule phospholipid and ceramide content. Am J Kidney Dis. 2000;36(2):238–249. | |
Zager RA, Bredl CR, Schimpf BA. Direct amphotericin B-mediated tubular toxicity assessments of selected cytoprotective agents. Kidney Int. 1992;41(6):1588–1594. | |
Bagnis CI, Deray G. Amphotericin B nephrotoxicity. Saudi J Kidney Dis Transpl. 2002;13(4):481–491. | |
Bellomo R. Defining, quantifying, and classifying acute renal failure. Crit Care Clin. 2005;21(2):223–237. | |
Massicotte A. Contrast medium-induced nephropathy: strategies for prevention. Pharmacotherapy. 2008;28(9):1140–1150. | |
Manske CL, Sprafka JM, Strony JT, Wang Y. Contrast nephropathy in azotemic diabetic patients undergoing coronary angiography. Am J Med. 1990;89(5):615–620. | |
Schwab SJ, Hlatky MA, Pieper KS, et al. Contrast nephrotoxicity: a randomized controlled trial of a nonionic and an ionic radiographic contrast agent. N Engl J Med. 1989;320(3):149–153. | |
Lautin EM, Freeman NJ, Schoenfeld AH, et al. Radiocontrast-associated renal dysfunction: a comparison of lower-osmolality and conventional high-osmolality contrast media. AJR Am J Roentgenol. 1991;157(1):59–65. | |
Mehran R, Aymong ED, Nikolsky E, et al. A simple risk score for prediction of contrast-induced nephropathy after percutaneous coronary intervention: development and initial validation. J Am Coll Cardiol. 2004;44(7):1393–1399. | |
Praga M, González E. Acute interstitial nephritis. Kidney Int. 2010; 77(11):956–961. | |
Rossert J. Drug-induced acute interstitial nephritis. Kidney Int. 2001;60(2):804–817. | |
Praga M, Sevillano A, Aunon P, Gonzalez E. Changes in the aetiology, clinical presentation and management of acute interstitial nephritis, an increasingly common cause of acute kidney injury. Nephrol Dial Transplant. Epub October 16, 2014. | |
Prakash J, Sen D, Kumar NS, Kumar H, Tripathi LK, Saxena RK. Acute renal failure due to intrinsic renal diseases: review of 1122 cases. Ren Fail. 2003;25(2):225–233. | |
Kazi JI, Mubarak M, Akhter F, et al. Spectrum of pathological lesions in acute renal failure. J Coll Physicians Surg Pak. 2003;13(1):22–24. | |
Goicoechea M, Rivera F, López-Gómez JM, et al. Increased prevalence of acute tubulointerstitial nephritis. Nephrol Dial Transplant. 2013;28: 112–115. | |
López-Gómez JM, Rivera F; Spanish Registry of Glomerulonephritis. Renal biopsy findings in acute renal failure in the cohort of patients in the Spanish Registry of Glomerulonephritis. Clin J Am Soc Nephrol. 2008;3(3):674–681. | |
Perazella MA, Markowitz GS. Drug-induced acute interstitial nephritis. Nat Rev Nephrol. 2010;6(8):461–470. | |
Brewster UC, Perazella MA. Proton pump inhibitors and the kidney: critical review. Clin Nephrol. 2007;68(2):65–72. | |
Muriithi A, Leung N, Valeri A, et al. Clinical characteristics, causes and outcomes of acute interstitial nephritis in the elderly. Kidney Int. 2015;87:458–464. | |
Salazar MN, Matthews M, Posadas A, Ehsan M, Graeber C. Biopsy proven interstitial nephritis following treatment with vancomycin: a case report. Conn Med. 2010;74(3):139–141. | |
Hong S, Valderrama E, Mattana J, et al. Vancomycin-induced acute granulomatous interstitial nephritis: therapeutic options. Am J Med Sci. 2007;334(4):296–300. | |
Wai AO, Lo AM, Abdo A, Marra F. Vancomycin-induced acute interstitial nephritis. Ann Pharmacother. 1998;32(11):1160–1164. | |
Wu CY, Wang JS, Chiou YH, Chen CY, Su YT. Biopsy proven acute tubular necrosis associated with vancomycin in a child: case report and literature review. Ren Fail. 2007;29(8):1059–1061. | |
Shah-Khan F, Scheetz MH, Ghossein C. Biopsy-proven acute tubular necrosis due to vancomycin toxicity. Int J Nephrol. 2011;2011: 436856. | |
Belen C, Budhiraja P, Bracamonte E, Popovtzer M. Biopsy-proven acute tubular necrosis associated with vancomycin in an adult patient. Ren Fail. 2012;34(4):502–505. | |
Oktem F, Arslan MK, Ozguner F, et al. In vivo evidence suggesting the role of oxidative stress in pathogenesis of vancomycin-induced nephrotoxicity: protection by erdosteine. Toxicology. 2005;215(3):227–233. | |
Clarkson MR, Giblin L, O’Connell FP, et al. Acute interstitial nephritis: clinical features and response to corticosteroid therapy. Nephrol Dial Transplant. 2004;19(11):2778–2783. | |
González E, Gutiérrez E, Galeano C, et al. Early steroid treatment improves renal function recovery in patients with drug-induced acute interstitial nephritis. Kidney Int. 2008;73(8):940–946. | |
Wright NG, Nash AS. Experimental ampicillin glomeruloneprhopathy. J Comp Pathol. 1984;94(3):357–361. | |
Baum M, Peil CF, Goodman JR. Antibiotic-associated interstitial nephritis and nephrotic syndrome. Am J Nephrol. 1986;6(2):149–151. | |
Kistler A, Lappin DW, Coward RA. Therapeutic dilemma: crescentic mesangiocapillary glomerulonephritis type 1 in a patient on antituberculosis therapy with rifampicin. Nephrol Dial Transplant. 1999;14: 243–244. | |
Kohler LJ, Gohara AF, Hamilton RW, Reeves RS. Crescentic fibrillary glomerulonephritis associated with intermittent rifampin therapy for pulmonary tuberculosis. Clin Nephrol. 1994;42(4);263–265. | |
Yong JL, Killingsworth M. Diffuse glomerulonephritis associated with rifampicin treatment for tuberculosis. Pathology. 2002;34(3):295–297. | |
Markowitz GS, Radhakrishnan J, Kambham N, Valeri AM, Hines WH, D’Agati VD. Lithium nephrotoxicity: a progressive combined glomerular and tubulointerstitial nephropathy. J Am Soc Nephrol. 2000;11(8):1439–1448. | |
Markowitz GS, Appel GB, Fine PL, et al. Collapsing focal segmental glomerulosclerosis following treatment with high-dose pamidronate. J Am Soc Nephrol. 2001;12(6):1164–1172. | |
Choi MJ, Eustace JA, Gimenez LF, et al. Mycophenolate mofetil treatment for primary glomerular disease. Kidney Int. 2002;61: 1098–1114. | |
Burgess E. Management of focal segmental glomerulosclerosis: evidence-based recommendations. Kidney Int. 1999;55(Suppl 70):S26–S32. | |
Korbet SM. Primary focal segmental glomerulosclerosis. J Am Soc Nephrol. 1998;9:1333–1340. | |
Korbet SM, Schwartz MM, Lewis EJ. Primary focal segmental glomerulosclerosis: clinical course and response to therapy. Am J Kidney Dis. 1994;23(6):773–783. | |
Wasserstein AG. Membranous glomerulonephritis. J Am Soc Nephrol. 1997;8:664–674. | |
Almroth G, Enestrom S, Hed J, Samuelsson I, Sjostrom P. Autoantibodies to leucocyte antigens in hydralazine associated nephritis. J Intern Med. 1992;23(1)1:37–42. | |
Ihle BU, Whitworth JA, Dowling JP, Kincaid-Smith P. Hydralazine and lupus nephritis. Clin Nephrol. 1984;22(5):230–238. | |
Sawyer MH, Webb DE, Balow JE, Straus SE. Acyclovir-induced renal failure. Clinical course and histology. Am J Med. 1988;84(6):1067–1071. | |
Keeney RE, Kirk LE, Bridgen D. Acyclovir tolerance in humans. Am J Med. 1982;73(1A):176–181. | |
Treon SP, Chabner BA. Concepts in use of high-dose methotrexate therapy. Clin Chem. 1996;42(8 Pt 2):1322–1329. | |
Such Díaz A, Saez de la Fuente J, Alañón Pardo AM, et al. Drug prescribing in patients with renal impairment optimized by a computer-based, semi-automated system. Int J Clin Pharm. 2013;35(6):1170–1177. | |
Bourne RS, Choo CL. Pharmacist proactive medication recommendations using electronic documentation in a UK general critical care unit. Int J Clin Pharm. 2012;34(2):351–357. | |
Gharekhani A, Kanani N, Khalili H, Dashti-Khavidaki S. Frequency, types, and direct related costs of medication errors in an academic nephrology unit in Iran. Ren Fail. 2014;36(8):1268–1272. | |
Baum S, Harder S. Appropriate dosing in patients with impaired renal function on medical wards before and after an educational intervention. Int J Clin Pharmacol Ther. 2010;48(1):29–35. | |
Falconnier AD, Haefeli WE, Schoenenberger RA, Surber C, Martin-Facklam M. Drug dosage in patients in renal failure optimized by immediate concurrent feedback. J Gen Intern Med. 2001;16(6):369–377. | |
Bertsche T, Fleischer M, Pfaff J, Encke J, Czock D, Haefeli WE. Pro-active provision of drug information as a technique to address overdosing in intensive-care patients with renal insufficiency. Eur J Clin Pharmacol. 2009;65(8):823–829. | |
Pai AB. Keeping kidneys safe: the pharmacist’s role in NSAID avoidance in high-risk patients. J Am Pharm Assoc. 2015;55(1):e15–e25. | |
Gheewala PA, Peterson GM, Curtain CM, Nishtala PS, Hannan PG, Castelino RL. Impact of the pharmacist medication review services on drug-related problems and potentially inappropriate prescribing of renally cleared medications in residents of aged care facilities. Drugs Aging. 2014;31:825–835. | |
Balaiche S, Romanet T, Allenet B, Calop J, Zaoui P. Identification of drug-related problems in ambulatory chronic kidney disease patients: a 6-month prospective study. J Nephrol. 2012;25(5):782–788. | |
Hassan Y, Al-Ramahi RJ, Aziz NA, Ghazali R. Impact of a renal drug dosing service on dose adjustment in hospitalized patients with chronic kidney disease. Ann Pharmacother. 2009;43(10):1598–1605. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.