")
Back to Journals » Journal of Asthma and Allergy » Volume 13
The Role of Mast Cells in Aspirin-Exacerbated Respiratory Disease (AERD) Pathogenesis: Implications for Future Therapeutics
Authors Kuruvilla ME , Vanijcharoenkarn K, Levy JM
Received 20 August 2020
Accepted for publication 23 September 2020
Published 12 October 2020 Volume 2020:13 Pages 463—470
DOI https://doi.org/10.2147/JAA.S237463
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Luis Garcia-Marcos
Merin E Kuruvilla,1 Kristine Vanijcharoenkarn,1 Joshua M Levy2
1Division of Pulmonary, Allergy, Critical Care & Sleep Medicine, Emory University School of Medicine, Atlanta, GA, USA; 2Department of Otolaryngology – Head & Neck Surgery, Emory University School of Medicine, Atlanta, GA, USA
Correspondence: Joshua M Levy
Emory University School of Medicine, 550 Peachtree St NE, 11th Floor, Atlanta, GA 30308, USA
Email [email protected]
Abstract: Mast cells (MC) have recently been demonstrated to play an integral role in the pathogenesis of aspirin-exacerbated respiratory disease (AERD). When activated, MCs release pre-formed granules of many pro-inflammatory mediators, including histamine, serotonin, and various chemokines and cytokines including tumor necrosis factor (TNF)-α, interferon ɣ (IFN ɣ), macrophage inhibitory factor, transforming growth factor, interleukin (IL) 1, 3– 6, 9, 10, 13 and 16. These mediators promote inflammation in AERD by recruiting or activating a network of cells involved in acute and chronic inflammatory pathways, such as endothelial, epithelial, stromal, and other immune cells. Several studies have implicated multifactorial pathways for MC activation in AERD beyond classical IgE mediated mechanisms. The elucidation of these complex networks therefore represents important targets for innovative patient therapeutics. This review summarizes classic and alternative pathways of MC activation in AERD with a special focus in relation to new and emerging treatment strategies.
Keywords: mast cells, aspirin-exacerbated respiratory disease, asthma, drug sensitivity, humanized monoclonal antibody
Introduction
Dysregulated mast cell (MC) activity directly contributes to the pathophysiology and symptoms associated with many respiratory diseases, including Aspirin-exacerbated respiratory disease (AERD). AERD is characterized by adult-onset asthma, chronic rhinosinusitis with nasal polyposis, and non-IgE-mediated anaphylactic reactions to cyclooxygenase-1 (COX-1) inhibitors, including nonsteroidal anti-inflammatory drugs (NSAIDS). The prevalence of aspirin intolerance is estimated to be 7.2% among adult asthma patients and increases to 14.9% in those with severe, persistent disease.1 AERD adversely affects quality of life, with the most common symptoms cited as chronic nasal symptoms followed by loss of sense of smell. Additionally, many patients report respiratory reactions to several common foods, including alcohol, tea, and certain spices, which may persist even after aspirin desensitization. Although the exact mechanism is unclear, it is postulated that polyphenols found in alcoholic drinks may inhibit COX-1, and foods such as spices and mint act through thermoreceptor transient receptor potential ion channels (eg, TRPV2, TRPV4, TRPM8) on the surface of MCs, activating them and leading to these symptoms.2
MCs originate from CD34+ pluripotent hematopoietic cells in the bone marrow. IL-3 and IL-6 increase the number of these CD34+ progenitor cells and begin the process of mast cell differentiation.3 However, other than in the setting of mast cell leukemia, mast cells do not fully mature until they arrive at their terminal tissue. The binding of stem cell factor (SCF) produced by stromal cells to the c-Kit (CD117) receptor is the main driver of the growth and final differentiation of mast cells at their tissue of destination (such as the skin, gastrointestinal tract, and respiratory mucosa).3,4
MCs are found in almost all tissues, including the brain. They are found in particularly high numbers in tissues that interact with the outside environment, such as skin and mucosal tissues. Furthermore, they are located near blood vessels, neurons, and lymphatic vessels, which allow them to regulate inflammation in both local tissue and systemic sites.5 When activated, MCs release pre-formed granules of many pro-inflammatory mediators, including histamine, serotonin, cytokines and chemokines including tumor necrosis factor (TNF)-α, interferon ɣ (IFN ɣ), macrophage inhibitory factor, transforming growth factor, IL 1, 3–6, 9, 10, 13, 16, numerous proteases such as tryptase, chymase, carboxypeptidase, as well as leukotrienes (such as LTC4, LTB4) and prostaglandins (such as PGD2, PGE2). These mediators further promote inflammation by recruiting or activating a network of cells involved in acute and chronic inflammatory pathways, such as endothelial, epithelial, stromal, and other immune cells.5
MCs are critical to the pathophysiology of AERD, as evidenced by their ability to elaborate large quantities of key mediators including cysteinyl leukotrienes (CysLTs) as well as prostaglandin D2 (PGD2). CysLTs are generated through the 5-lipoxygenase (5-LO)/Leukotriene C4 (LTC4) synthase pathway, and MC metabolizes arachidonic acid through cyclooxygenase (COX) to synthesize PGD2. These mediators act synergistically to activate various type 2 inflammatory pathways in AERD. Stable urinary metabolites of both CysLTs and PGD2 are measured at significantly higher levels among AERD patients, reflective of MC activation.6,7 MC activation in the respiratory tract is also a feature of aspirin-induced respiratory reactions associated with elevated plasma tryptase during aspirin challenges.8 Finally, increased numbers of MCs have been demonstrated in bronchial biopsies of AERD subjects as compared with aspirin tolerant controls.9 However, the mechanisms underlying this sustained activation of MC are to be fully elucidated. In addition to traditional signaling through the high-affinity IgE receptor (FcεR1), MCs in AERD may also be activated by non-classical mechanisms, as reviewed below, and outlined in Figure 1.
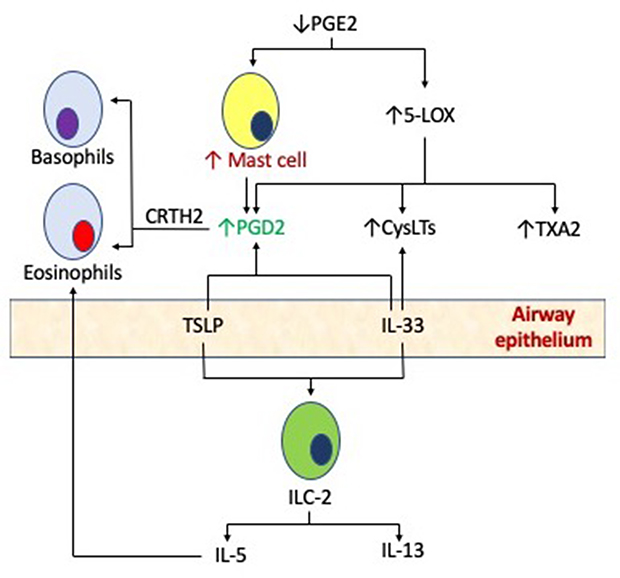 |
Figure 1 Mast cells in the pathogenesis of AERD. Pro-inflammatory mast cells may be activated in AERD by both classic, high-affinity IgE receptor binding, or alternative mechanisms such as PGE2, cysteinyl leukotrienes (CysLTs) and the production of innate cytokines such as thymic stromal lymphopoietin (TSLP) and interleukin 33 (IL-33) via type 2 innate lymphoid cells (ILC-2). |
Pathogenesis of MC Activation in AERD
Classical Activation
Local Antibody Production
Classical MC activation through FcεRI signaling may play a role in AERD pathogenesis, although the nature of the cytokine milieu driving IgE production is not fully understood. While many AERD patients lack classic atopic sensitization, they tend to have overall elevated serum and local tissue IgE.10 Tissue levels of IgE and IgG4 are higher in AERD subjects as compared with aspirin tolerant nasal polyps, chronic sinusitis, and healthy controls, with evidence of local B cell activation and antibody secretion.11 The driving factors and the consequences of antibody production within polyp tissue is unknown. However, higher IgE levels are associated with aggressive nasal polyp growth and accelerated recurrence following sinus surgery.
Elevated local IgE concentrations in AERD may be related to Staphylococcus aureus enterotoxins that act as superantigens and induce polyclonal IgE. As many as 87% of patients with AERD are colonized with Staph. aureus, and a subset of these produce IgE directed against staphylococcal superantigens.12 Levels of Staph. aureus superantigen IgE are significantly increased in AERD as compared with other nasal polyps.13 Staph. aureus has also been implicated in defective mucosal barrier function and innate immune system activation, as will be discussed later.
Nasal polyp IgE and its association with aggressive recurrence may be instrumental to local MC activation in AERD. Locally produced IgE may allow FcεRI-expressing effector cells including MCs and basophils to respond to microbial or other antigens. Polyp IgE has been demonstrated to be functional and induces basophil activation with histamine release and upregulation of the surface marker CD63.14 The effects of nasal polyp IgE specifically on FcεRI responses in MCs are yet to be elucidated.
Non-Classical Activation
Eicosanoids and MCs
PGE2 is a homeostatic and ubiquitous product of the COX pathway that is constitutively generated and induced at extremely high levels during inflammatory reactions. This PGE2 response to inflammation is dependent on inducible COX2 that acts in conjunction with microsomal prostaglandin E2 synthase 1 (mPGES1) to generate PGE2. As compared with the constitutive expression of COX-1, COX-2 is less sensitive to blockade by aspirin in AERD.15 Selective COX2 inhibitors appear to be well tolerated in most patients with NSAID hypersensitivity, especially those with AERD.16
A deficiency of homeostatic PGE2 appears to be the central defect in AERD, through baseline reductions in both PGE2 production and activity. PGE2 is critical in inhibiting MC activation through its target receptor EP2.17 Impaired respiratory tract PGE2 production or signaling leads to 5-lipoxygenase activation and MC stimulation. PGE2 inhibits MC activation in response to FcεRI through the E-prostanoid 2 (EP2) and EP4 receptors, specifically by attenuating the PLC-γ and ERK1/2 signaling pathways.18 Lower levels of PGE2 have been linked with MC hyperresponsiveness.19 Furthermore, transgenic mPGES1(-/-) knockout mice are used as an animal model to study AERD, further highlighting the importance of PGE2 in the pathogenesis of this disease.20 However, the reasons underlying the dysregulation of PGE2 in AERD are yet to be fully described.
Cysteinyl leukotriene receptors CysLT1R and CysLT2R are broadly expressed by structural and hematopoietic cells, including MCs.21 The overproduction of CysLTs is pathognomonic of AERD, and also plays a role in MC activation. A substantial body of evidence supports the effect of CysLTs on MC activation and mediator release. The stimulation of cord-blood MCs with CysLTs elicits strong calcium influx accompanied by mediator release22 as well as chemotaxis of MC progenitors.23 More recently, Lazarinis et al directly demonstrated prominent increases in urinary PGD2 excretion and other MC metabolites in response to LTE4-triggered bronchoconstriction in mild asthmatics.24 This response was abrogated by montelukast, a CysLT1R antagonist. This confirms a role for LTE4 in MC activation through CysLT1R, with the consequent generation of MC mediators. A similar effect on mast cell PGD2 production was also demonstrated by Banakova et al through indirect binding of LTE4 to epithelial GPR99 receptors (CysLTR3) with consequent MC activation.25 Taken together, these findings imply that CysLTs induce MC activation in AERD, and elicit MC release of contractile COX-derived mediators.
Further, the gene encoding 15-lipoxygenase, ALOX15, has selective and significantly greater expression in AERD. A novel mediator, 15-oxo-ETE was also recently identified in AERD that is a product of the 15-lipoxygenase pathway.26 MCs appear to be critical in this process using MC-derived hematopoietic PGD2 synthase (HPGDS) to generate 15-oxo-ETE.
There is an accumulating body of evidence that supports the role of innate cytokines in AERD through type 2 innate lymphoid cell (ILC2) activation. Various models of type 2 innate immunity initiated by IL-33, IL-25 and TSLP also strongly implicate MC activation.27 TSLP and IL-33 appear to play unique roles in AERD including COX-1-inhibitor-induced reactions. More recently, both TSLP and IL-33-mediated MC activation has been found to elicit eicosanoid generation in cultures.6,28
The airway epithelium in AERD shows impaired barrier integrity when compared with normal epithelium following exposure to Staph. aureus. While the precise role of Staph. aureus in AERD is unclear, epithelial repair following this exposure to Staph exoproducts is inferior in nasal polyp subjects when compared with healthy controls.29 This exposure increases the expression of epithelium-derived innate type 2 cytokines including IL-33 and TSLP in polyp tissue.30 MCs are localized just beneath the epithelium and may even be intraepithelial, and thus poised to respond to epithelium-derived TSLP and IL-33.31
In contrast to classical activation mechanisms, innate type 2 cytokines do not induce MC degranulation and exocytosis. Rather, TSLP and IL-33 prominently induce Th2-cytokine production by MCs as well as lipid mediator synthesis.27,32,33 In particular, TSLP drives MC-derived PGD2 production, and when combined with IL33 results in a synergistic rise in PGD2 generation. Dysregulation of the innate inflammatory system is a significant contributor to AERD pathophysiology.6
IL-33 acts through ST2 receptors on the MC surface to activate MAP kinase and NF-κB pathways, with consequent activation and leukotriene production. In a murine model of AERD, MC activation with synthesis of PGD2 and LTs hinged upon IL-33 release34 and LTE4 induced MC activation through an IL-33 dependent mechanism. Interestingly, IL-33 induced eicosanoid synthesis in MCs in this model was critically dependent upon constitutive COX-1 activity. PGE2 synthase deficient mice upregulated IL33 and MC activation in the airway epithelium.35 Deletion of LTC4 synthase in this model abrogated IL33 production as well as MC activation. In addition, IL-33 promotes the synthesis of preformed granule mediators, thus augmenting IgE-mediated MC activation.36
In another model, IL-33-induced release of arachidonic acid and all its downstream products (including PGD2 and LTC4) was sharply abrogated by the treatment of MCs with a COX-1 inhibitor.28 These COX-1 inhibitor-treated mice showed absence of changes in airway resistance and eicosanoid levels following aspirin challenge suggesting desensitization.
Mast Cell Derived Mediators in AERD
PGD2 is the major COX product of MCs and induces bronchoconstriction as well as chemotaxis of type 2 inflammatory cells. Prostaglandin DP1 and CRTH2 receptors are overexpressed in the sinonasal tissue of AERD patients and MC-derived PGD2 is a major effector of type 2 immune responses in AERD.37 AERD has even been labeled a syndrome of MC-mediated overproduction of PGD2,38 with significantly increased tissue expression in AERD as compared with other forms of eosinophilic sinusitis and controls.39 PGD2 triggers bronchospasm through the thromboxane receptor.6 PGD2 also drives chemotaxis of Th2 cells, eosinophils and basophils, and also promotes type 2 cytokine release from Th2 cells and ILC2s. Of note, while PGD2 was exclusively attributed to MC production until recently, eosinophils were also discovered to be a dominant producer in AERD through high expression of hematopoietic PGD2 synthase (HPGDS).6
MCs also express both 5-lipoxygenase and leukotriene C4 synthase and are hence able to catalyze the generation of LTC4 from arachidonic acid. While they may contribute to the CysLT burden in AERD, no significant abnormalities in CysLT production were found in AERD subjects.40
Clinical Implications
MC activation in the respiratory tract is a well-recognized feature of aspirin-induced respiratory reactions and plays a role in desensitization. A subset of patients manifests severe reactions during aspirin challenges associated with elevated plasma tryptase that develop despite prophylaxis with a leukotriene receptor antagonist.8 It has been hypothesized that MC activation is responsible for PGD2-mediated bronchospasm and potential systemic symptoms in these cases and the tryptase level was inversely correlated with the decrease in FEV1 in this patient subset.
MC activation in aspirin challenges has also been associated with an acute increase in local and systemic production of eicosanoids – specifically PGD2 metabolites and LTE4.41 Peak levels of PGD2 synthesized during reactions correlate with symptom severity,6 and patients unable to tolerate desensitization generate significantly more PGD2 during reactions. Increased PG metabolites, particularly PGD2, were documented in a subgroup of patients resistant to desensitization.42 Differences in eicosanoid synthesis may account for the variance in clinical response to desensitization. This aligns with reports of PGD2 dominance in an AERD sub-phenotype associated with more serious cutaneous and gastrointestinal reactions to aspirin.42
MC-derived PGD2 promotes the rapid recruitment of CRTH2+ ILC2s, eosinophils and basophils into the respiratory tissue during aspirin desensitization,43 with concurrent decrease in blood numbers of these effector cells.44 Additional MC and cys-LT independent mechanisms and mediators are also possible contributors to AERD in general and in particular the reactions to COX-1 inhibitors.
Therapeutic Implications
MC Response During Long-Term Aspirin Therapy
With longer-term high-dose aspirin therapy, one proposed mechanism of clinical benefit is a reduction in PGD2 activity.45,46 However, high-dose aspirin therapy does not appear to rectify the overall dysregulation in eicosanoid synthesis and MC homeostasis characteristic of AERD. The clinical benefit of daily high-dose aspirin in AERD appears to be independent of a reduction in MC activation and CysLT production. In fact, a paradoxical increase in serum tryptase and urinary LTE4 has been demonstrated following desensitization.46 Thus, the clinical benefit of high-dose aspirin is disconnected from a global correction of the arachidonic acid pathway.
Despite this paradoxical increase in MC activation following aspirin desensitization, COX products (PGD2 and thromboxane metabolites) appear to be suppressed.46 Markedly decreased levels of urinary eicosanoids (including PGE2, PGD2 and thromboxane A2 metabolites) implies that decreased synthesis of these PGs may facilitate clinical improvement on high-dose aspirin therapy. This is accompanied by prominent increases in blood eosinophils and basophils, a presumed reflection of diminished PGD2-mediated chemotaxis of these effector cells into the respiratory tissue.42 Further, the rise in plasma tryptase correlates with increases in peripheral blood eosinophils and basophils.46
Thus, a predominant mechanism by which aspirin therapy is hypothesized to work is the loss of PGD2-mediated chemotactic gradient and prevention of cells homing into respiratory tissues. MC-intrinsic COX-1 amplifies IL-33-induced activation, potentially mediating the effects of aspirin desensitization. The suppression of PGD2 may be secondary to MC stabilization through inhibition of constitutive COX-1 activity and impaired MC responses to endogenous IL-33.28 Therefore, while aspirin might increase MC activation as it exerts its therapeutic benefit, the dominant effects of PGD2 and thromboxane A2 suppression may overcome this increased mast cell activity.
Notably, systemic PGE2 levels have not been shown to change significantly and even decreased in some reports.46 This has been attributed to high renal PGE2 production that masks fluctuations in respiratory tissue provoked by aspirin.47 It is hypothesized that this PGE2 depletion allows for increasingly activated MCs during long-term high-dose aspirin therapy.
Pharmacotherapeutic Implications
MC directed therapy may translate into clinical improvement for AERD subjects. A recent study reported that anti-IgE treatment with omalizumab improved sinonasal symptoms in patients with AERD and also decreased urinary PGD2 and LTE4 levels, with ~90% coming from MCs.48 This decrease in urinary PGD2 metabolites was observed as early as within 24 hours of administration. Omalizumab also suppressed the overproduction of PGD2 characteristic of aspirin challenges in these patients.
The predominant role of eosinophils versus mast cells in AERD has long been debated. Anti-eosinophil agents that are clinically available are helpful in some scenarios. In addition to a direct anti-eosinophil effect, the IL-5 receptor is expressed on local antibody secreting cells that elaborate local IgE.11 The decrease in MC activation following IL-5 blockade may contribute to the therapeutic benefit of these agents. The efficacy of dupilumab in nasal polyps, including AERD, may similarly derive in part from decreased IgE generation secondary to IL4/IL13 blockade, with consequent decrease in MC activation.
In the future, treatment mechanisms that inhibit PGD2 pathways may thus afford similar clinical improvement. Randomized controlled trials of CRTH2 antagonists are underway. It remains to be seen whether these agents accomplish clinical benefits comparable with high-dose aspirin, while circumventing the risks of desensitization and long-term high-dose therapy. PGD2-induced bronchoconstriction is responsive to blockade of the thromboxane receptor49 and PGD2 signaling may be modified by the selective thromboxane receptor antagonist ifetroban.
Several other emerging therapies may also prove beneficial at decreasing MC activity in AERD (Table 1). For instance, IL-33 appears to be the bridge between CysLT overproduction and MC activation in AERD and this may also be a pharmacotherapeutic target. Similarly, a PGE2 analogue that selectively targets the EP2 receptor may restore the central dysregulation of this disorder and inhibit MC activity, although decreased EP2 expression may limit its utility in some cases. Hopefully, the results of clinical trials using these medications will provide more definitive evidence for the contribution of MCs in AERD.
 |
Table 1 Pharmacologic Treatments for AERD |
Another future avenue for treatment may target Staph. aureus induced barrier dysfunction and type 2 inflammation in AERD. While antibiotic treatment directed at Staph. aureus does not alleviate nasal congestion or anosmia in nasal polyps, targeting its inflammatory pathways may afford a more suitable solution.
The study of mast cells in other disease processes may also be helpful in elucidating new targets for AERD patients. Mast cell-deficient mouse strains, usually involving a mutation in KIT, have been developed and studied. These models have established the important role of mast cells in allergic diseases, though findings for the underlying pathophysiology have not always been consistent. No humans lacking mast cells have yet been reported, making it more challenging to study this in human patients. However, in diseases such as mastocytosis where reducing mast cell numbers offers clinical relief, therapies involving apoptosis, reducing mast cell migration, differentiation and survival, have been explored. Tyrosine kinase inhibitors are used in aggressive systemic mastocytosis, and siglec-8, a receptor expressed by human mast cells (and eosinophils and basophils) may also be targeted. A siglec-8-specific antibody is currently under investigation for the treatment of mastocytosis (41). Although these therapies are not being specifically studied for AERD, as our knowledge of the role of mast cells in disease increases, variations of these therapies may be applicable for AERD patients in the future.
Acknowledgments
Dr. Levy is supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Numbers UL1TR002378 and KL2TR002381. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rajan JP, Wineinger NE, Stevenson DD, et al. Prevalence of aspirin-exacerbated respiratory disease among asthmatic patients: a meta-analysis of the literature. J Allergy Clin Immunol. 2015;135(3):676–81 e1. doi:10.1016/j.jaci.2014.08.020
2. Ta V, White AA. Survey-defined patient experiences with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol Pract. 2015;3(5):711–718. doi:10.1016/j.jaip.2015.03.001
3. Steinke JW, Payne SC, Borish L. Eosinophils and mast cells in aspirin-exacerbated respiratory disease. Immunol Allergy Clin North Am. 2016;36(4):719–734. doi:10.1016/j.iac.2016.06.008
4. Kirshenbaum AS, Goff JP, Kessler SW, et al. Effect of IL-3 and stem cell factor on the appearance of human basophils and mast cells from CD34+ pluripotent progenitor cells. J Immunol. 1992;148(3):772–777.
5. Siebenhaar F, Redegeld FA, Bischoff SC, et al. Mast cells as drivers of disease and therapeutic targets. Trends Immunol. 2018;39(2):151–162. doi:10.1016/j.it.2017.10.005
6. Buchheit KM, Cahill KN, Katz HR, et al. Thymic stromal lymphopoietin controls prostaglandin D2 generation in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2016;137(5):1566–1576 e5. doi:10.1016/j.jaci.2015.10.020
7. Bochenek G, Stachura T, Szafraniec K, et al. Diagnostic accuracy of urinary LTE4 measurement to predict aspirin-exacerbated respiratory disease in patients with asthma. J Allergy Clin Immunol Pract. 2018;6(2):528–535.
8. Cahill KN, Murphy K, Singer J, et al. Plasma tryptase elevation during aspirin-induced reactions in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2019;143(2):799–803 e2. doi:10.1016/j.jaci.2018.10.007
9. Nasser SM, Pfister R, Christie PE, et al. Inflammatory cell populations in bronchial biopsies from aspirin-sensitive asthmatic subjects. Am J Respir Crit Care Med. 1996;153(1):90–96. doi:10.1164/ajrccm.153.1.8542168
10. Johns CB, Laidlaw TM. Elevated total serum IgE in nonatopic patients with aspirin-exacerbated respiratory disease. Am J Rhinol Allergy. 2014;28(4):287–289. doi:10.2500/ajra.2014.28.4054
11. Buchheit KM, Dwyer DF, Ordovas-Montanes J, et al. IL-5Rα marks nasal polyp IgG4- and IgE-expressing cells in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2020;145(6):1574–1584. doi:10.1016/j.jaci.2020.02.035
12. Van Zele T, Gevaert P, Watelet JB, et al. Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J Allergy Clin Immunol. 2004;114(4):981–3. doi:10.1016/j.jaci.2004.07.013
13. Perez-Novo CA, Kowalski ML, Kuna P, et al. Aspirin sensitivity and IgE antibodies to Staphylococcus aureus enterotoxins in nasal polyposis: studies on the relationship. Int Arch Allergy Immunol. 2004;133(3):255–60. doi:10.1159/000076832
14. Shamji MH, Thomsen I, Layhadi JA, et al. Broad IgG repertoire in patients with chronic rhinosinusitis with nasal polyps regulates proinflammatory IgE responses. J Allergy Clin Immunol. 2019;143(6):2086–2094 e2. doi:10.1016/j.jaci.2019.02.001
15. Harrington LS, Lucas R, McMaster SK, et al. COX-1, and not COX-2 activity, regulates airway function: relevance to aspirin-sensitive asthma. FASEB J. 2008;22(11):4005–10. doi:10.1096/fj.08-107979
16. Li L, Laidlaw T. Cross-reactivity and tolerability of celecoxib in adult patients with NSAID hypersensitivity. J Allergy Clin Immunol Pract. 2019;7(8):2891–2893 e4. doi:10.1016/j.jaip.2019.04.042
17. Perez-Novo CA, Watelet JB, Claeys C, et al. Prostaglandin, leukotriene, and lipoxin balance in chronic rhinosinusitis with and without nasal polyposis. J Allergy Clin Immunol. 2005;115(6):1189–96. doi:10.1016/j.jaci.2005.02.029
18. Kay LJ, Yeo WW, Peachell PT. Prostaglandin E2 activates EP2 receptors to inhibit human lung mast cell degranulation. Br J Pharmacol. 2006;147(7):707–13. doi:10.1038/sj.bjp.0706664
19. Rastogi S, Willmes DM, Nassiri M, et al. PGE2 deficiency predisposes to anaphylaxis by causing mast cell hyper-responsiveness. J Allergy Clin Immunol. 2020. doi:10.1016/j.jaci.2020.03.046
20. Liu T, Laidlaw TM, Katz HR, et al. Prostaglandin E2 deficiency causes a phenotype of aspirin sensitivity that depends on platelets and cysteinyl leukotrienes. Proc Natl Acad Sci U S A. 2013;110(42):16987–92. doi:10.1073/pnas.1313185110
21. Austen KF, Maekawa A, Kanaoka Y, et al. The leukotriene E4 puzzle: finding the missing pieces and revealing the pathobiologic implications. J Allergy Clin Immunol. 2009;124(3):
22. Paruchuri S, Jiang Y, Feng C, et al. Leukotriene E4 activates peroxisome proliferator-activated receptor γ and induces prostaglandin D2 generation by human mast cells. J Biol Chem. 2008;283(24):16477–87. doi:10.1074/jbc.M705822200
23. Weller CL, Collington SJ, Brown JK, et al. Leukotriene B4, an activation product of mast cells, is a chemoattractant for their progenitors. J Exp Med. 2005;201(12):1961–71. doi:10.1084/jem.20042407
24. Lazarinis N, Bood J, Gomez C, et al. Leukotriene E4 induces airflow obstruction and mast cell activation through the cysteinyl leukotriene type 1 receptor. J Allergy Clin Immunol. 2018;142(4):1080–1089. doi:10.1016/j.jaci.2018.02.024
25. Bankova LG, Lai J, Yoshimoto E, et al. Leukotriene E4 elicits respiratory epithelial cell mucin release through the G-protein–coupled receptor, GPR99. Proc Natl Acad Sci U S A. 2016;113(22):6242–7. doi:10.1073/pnas.1605957113
26. Stevens WW, Staudacher AG, Hulse KE, et al. Activation of the 15-lipoxygenase pathway in aspirin exacerbated respiratory disease. J Allergy Clin Immunol. 2020. doi:10.1016/j.jaci.2020.04.031
27. Allakhverdi Z, Smith DE, Comeau MR, et al. Cutting edge: the ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J Immunol. 2007;179(4):2051–4. doi:10.4049/jimmunol.179.4.2051
28. Pan D, Buchheit KM, Samuchiwal SK, et al. COX-1 mediates IL-33-induced extracellular signal-regulated kinase activation in mast cells: implications for aspirin sensitivity. J Allergy Clin Immunol. 2019;143(3):1047–1057 e8. doi:10.1016/j.jaci.2018.06.033
29. Valera FCP, Ruffin M, Adam D, et al. Staphylococcus aureus impairs sinonasal epithelial repair: effects in patients with chronic rhinosinusitis with nasal polyps and control subjects. J Allergy Clin Immunol. 2019;143(2):591–603 e3. doi:10.1016/j.jaci.2018.05.035
30. Lan F, Zhang N, Holtappels G, et al. Staphylococcus aureus induces a mucosal type 2 immune response via epithelial Cell–derived cytokines. Am J Respir Crit Care Med. 2018;198(4):452–463. doi:10.1164/rccm.201710-2112OC
31. Erjefalt JS. Mast cells in human airways: the culprit? Eur Respir Rev. 2014;23(133):299–307. doi:10.1183/09059180.00005014
32. Allakhverdi Z, Comeau MR, Jessup HK, et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;204(2):253–8. doi:10.1084/jem.20062211
33. Iikura M, Suto H, Kajiwara N, et al. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab Invest. 2007;87(10):971–8. doi:10.1038/labinvest.3700663
34. Liu T, Kanaoka Y, Barrett NA, et al. Aspirin-exacerbated respiratory disease involves a cysteinyl leukotriene-driven IL-33-mediated mast cell activation pathway. J Immunol. 2015;195(8):3537–45. doi:10.4049/jimmunol.1500905
35. Samuchiwal SK, Balestrieri B, Raff H, et al. Endogenous prostaglandin E2 amplifies IL-33 production by macrophages through an E prostanoid (EP)2/EP4-cAMP-EPAC-dependent pathway. J Biol Chem. 2017;292(20):8195–8206. doi:10.1074/jbc.M116.769422
36. Kaieda S, Shin K, Nigrovic PA, et al. Synovial fibroblasts promote the expression and granule accumulation of tryptase via interleukin-33 and its receptor ST-2 (IL1RL1). J Biol Chem. 2010;285(28):21478–86. doi:10.1074/jbc.M110.114991
37. Yamamoto M, Okano M, Fujiwara T, et al. Expression and characterization of PGD2 receptors in chronic rhinosinusitis: modulation of DP and CRTH2 by PGD2. Int Arch Allergy Immunol. 2009;148(2):127–36. doi:10.1159/000155743
38. Borish L. Aspirin-exacerbated respiratory disease: a syndrome of mast cell-mediated PgD2 overproduction. Am J Respir Crit Care Med. 2019;200(6):651–652. doi:10.1164/rccm.201904-0716ED
39. Feng X, Ramsden MK, Negri J, et al. Eosinophil production of prostaglandin D2 in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2016;138(4):1089–1097 e3. doi:10.1016/j.jaci.2016.04.042
40. Adamjee J, Suh Y-J, Park H-S, et al. Expression of 5-lipoxygenase and cyclooxygenase pathway enzymes in nasal polyps of patients with aspirin-intolerant asthma. J Pathol. 2006;209(3):392–9. doi:10.1002/path.1979
41. Szczeklik A, Sladek K, Dworski R, et al. Bronchial aspirin challenge causes specific eicosanoid response in aspirin-sensitive asthmatics. Am J Respir Crit Care Med. 1996;154(6 Pt 1):1608–14.
42. Cahill KN, Bensko JC, Boyce JA, et al. Prostaglandin D(2): a dominant mediator of aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2015;135(1):245–52. doi:10.1016/j.jaci.2014.07.031
43. Sladek K, Szczeklik A. Cysteinyl leukotrienes overproduction and mast cell activation in aspirin-provoked bronchospasm in asthma. Eur Respir J. 1993;6(3):391–9.
44. Eastman JJ, Cavagnero KJ, Deconde AS, et al. Group 2 innate lymphoid cells are recruited to the nasal mucosa in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2017;140(1):101–108 e3. doi:10.1016/j.jaci.2016.11.023
45. Tyrak KE, Kupryś-Lipińska I, Czarnobilska E, et al. Sputum biomarkers during aspirin desensitization in nonsteroidal anti-inflammatory drugs exacerbated respiratory disease. Respir Med. 2019;152:51–59. doi:10.1016/j.rmed.2019.04.021
46. Cahill KN, Cui J, Kothari P, et al. Unique effect of aspirin therapy on biomarkers in aspirin-exacerbated respiratory disease: a prospective trial. Am J Respir Crit Care Med. 2019;200(6):704–711. doi:10.1164/rccm.201809-1755OC
47. Yoshimura T, Yoshikawa M, Otori N, et al. Correlation between the prostaglandin D(2)/E(2) ratio in nasal polyps and the recalcitrant pathophysiology of chronic rhinosinusitis associated with bronchial asthma. Allergol Int. 2008;57(4):429–36. doi:10.2332/allergolint.O-08-545
48. Hayashi H, Fukutomi Y, Mitsui C, et al. Omalizumab for aspirin-hypersensitivity and leukotriene overproduction in aspirin-exacerbated respiratory disease: a randomized trial. Am J Respir Crit Care Med. 2020;201(12):1488–1498. doi:10.1164/rccm.201906-1215OC
49. Larsson AK, Hagfjärd A, Dahlén SE, et al. Prostaglandin D(2) induces contractions through activation of TP receptors in peripheral lung tissue from the guinea pig. Eur J Pharmacol. 2011;669(1–3):136–42. doi:10.1016/j.ejphar.2011.07.046
50. Li KL, Lee AY, Abuzeid WM. Aspirin exacerbated respiratory disease: epidemiology, pathophysiology, and management. Med Sci. 2019;7(3):45.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.