")
Back to Journals » Drug Design, Development and Therapy » Volume 12
The role of IL-1 inhibition in systemic juvenile idiopathic arthritis: current status and future perspectives
Authors Toplak N, Blazina Š , Avčin T
Received 11 February 2018
Accepted for publication 12 April 2018
Published 8 June 2018 Volume 2018:12 Pages 1633—1643
DOI https://doi.org/10.2147/DDDT.S114532
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Nataša Toplak,1,2 Štefan Blazina,1 Tadej Avčin1,2
1Department of Allergology, Rheumatology and Clinical Immunology, University Children’s Hospital, University Medical Centre Ljubljana, Ljubljana, Slovenia; 2Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia
Abstract: The pathogenesis, clinical course, and response to treatment in systemic juvenile idiopathic arthritis (SJIA) differ from other types of juvenile idiopathic arthritis and are similar to other interleukin-1 (IL-1)-mediated diseases. The main cytokine involved in the pathogenesis of SJIA is IL-1β, which can be neutralized by targeted anti-IL-1 therapy. In SJIA, no antibodies have been found and there is growing evidence that it is mainly an autoinflammatory and not an autoimmune disease. Before the era of biologic therapy, treatment of SJIA was primarily based on long-term treatment with high doses of glucocorticosteroids (GCS). The side effects of GCS could have a significant impact on the outcome of the disease and could cause long-term damage. Treatment with anti-IL-1 agents early in the disease course has revolutionized the management principles of SJIA. However, not all SJIA patients respond equally well to anti-IL-1 therapy, and it has been shown that age at the onset of disease, duration of the disease, number of affected joints, neutrophil count, and ferritin level can predict the response to anti-IL-1 therapy. In particular, an elevated ferritin level should prompt testing for macrophage activation syndrome (MAS), the most severe complication of SJIA. Anti-IL-1 therapy has been shown to be effective also in patients with MAS. Although anti-IL-1 agents are currently not recommended as first-line treatment, there is growing evidence that anti-IL-1 agents introduced at the beginning of SJIA could enable lower doses and a shorter duration of GCS therapy, change the long-term disease outcome, and even influence molecular disease patterns. There are currently three anti-IL-1 agents available: anakinra, canakinumab, and rilonacept. In this review, we present the current knowledge on the pathogenesis of SJIA, the rational for anti-IL-1 treatment, and future perspectives on the treatment of SJIA.
Keywords: anti-IL-1 therapy, anakinra, canakinumab, rilonacept, systemic juvenile idiopathic arthritis
Introduction
Inflammation in systemic juvenile idiopathic arthritis (SJIA) and adult-onset Still’s disease (AOSD) is mediated by the interleukin-1 (IL-1) family of cytokines, which is closely related to the innate inflammatory immune response. The IL-1 family of cytokines includes 11 cytokines; of these, IL-1β has a major role as a therapeutic target in several autoinflammatory diseases, including SJIA. Neutralization of IL-1β causes a rapid and sustained blockade of inflammation.1,2 Other IL-1 family cytokines involved in the pathogenesis of SJIA are IL-1α, IL-18, and IL-1 receptor antagonist (IL-1Ra). IL-1Ra is a naturally occurring antagonist of IL-1α and IL-1β, whereas IL-18 is a proinflammatory cytokine. Several recent reports revealed that IL-18 could be used as a diagnostic biomarker distinguishing SJIA from other febrile illnesses.3 Furthermore, it has been shown that substantially increased levels of IL-18 may be associated with the risk of macrophage activation syndrome (MAS), a potentially lethal complication of SJIA.4
SJIA is a pediatric counterpart of AOSD, and it seems that both diseases represent variants of the same clinical entity with similar pathogenesis beginning at different ages.5,6 Currently, SJIA is still classified within the group of childhood chronic arthritides collected under the umbrella term juvenile idiopathic arthritis (JIA).7–10 The International League Against Rheumatism’s (ILAR) classification of JIA includes seven subtypes, one of which is SJIA, representing 10%–20% of all JIA cases.7 The ILAR classification criteria for SJIA are presented in Table 1. In SJIA, no autoantibodies have been found and uveitis is very rare.9
 | Table 1 ILAR classification criteria for SJIA |
SJIA has a distinct clinical spectrum compared to other types of JIA, with a different pathogenesis and also a different genetic background.11 Treatment of SJIA could be challenging. Before the era of biologic therapy, only nonsteroidal anti-inflammatory drugs (NSAIDs), disease modifying antirheumatic drugs (DMARDs), and glucocorticosteroids (GCS) were available.12 The development of targeted treatments against inflammatory cytokines has significantly modified the treatment approach to SJIA. In addition to anti-IL-1 therapy, anti-IL-6 therapy has been used with excellent clinical outcomes, especially in cases resistant to anti-IL-1 therapy.13–17 Recent advances in the pathogenesis of SJIA have revealed new possible biologic targets for the treatment of resistant cases, including IL-18, IL-17, and signaling molecules such as enzymes of the Janus kinase (JAK) family.18
This article presents the use of anti-IL-1 treatment in SJIA and MAS. Currently, three anti-IL-1 drugs (canakinumab, anakinra, and rilonacept) are available in the market, but only canakinumab is licensed for the treatment of SJIA. Canakinumab neutralizes only IL-1β, whereas anakinra and rilonacept neutralize both IL-1β and IL-1α.13,14
IL-1 cytokine family and signaling pathway
The IL-1 family of cytokines comprises major mediators of the innate immune system, with IL-1 being the first cytokine identified as a strong inducer of fever and inflammation.19 Major sources of IL-1β include tissue macrophages, blood monocytes, and dendritic cells.20 IL-1 has the ability to induce the synthesis of potent inflammatory mediators such as cyclooxygenase type 2 (COX-2), type 2 phospholipase A, and inducible nitric oxide synthase, which accounts for prostaglandin-E2, platelet activating factor, and NO production. In addition, IL-1 has angiogenic properties.21
Initially, the IL-1 family included only IL-1α and IL-1β but later expanded to 11 members. Seven members act as proinflammatory cytokines (IL-1α, IL-1β, IL-18, IL-36α, IL-36β, IL-36γ, and IL-33), whereas three have an anti-inflammatory function (IL-1Ra – blocks IL-1α and β, IL-36Ra – blocks IL-36α, β, and γ, and IL-37). The function of IL-38 is not known. Except for IL-1Ra, all members of the IL-1 family are first synthesized as precursors without a signal peptide for processing and secretion. The precursor forms of IL-18 and IL-1β require cleavage by either intracellular caspase-1 or extracellular neutrophilic proteases for activation and receptor binding.1,22
In the IL-1 family, two cytokines have a dual function: IL-1α and IL-33. In addition to binding to their respective cell surface receptors, the intracellular precursor forms translocate to the nucleus and influence the transcription of proinflammatory genes.23
The IL-1 family of cytokines mediate the effect by binding to the IL-1 family receptor subunit, which subsequently recruits an accessory receptor to activate downstream signaling. The IL-1 family of receptors is unique due to the presence of the Toll-IL-1 receptor (TIR) domain in the cytoplasmatic segment of each member. Because members of Toll-like receptors (TLRs) and IL-1 receptors retained their TIR intracellular signaling domain, they were grouped in the IL-1 receptor/TLR superfamily.24
The potent proinflammatory effect of IL-1α and IL-1β follows three major steps: synthesis and release, membrane receptor binding, and intracellular signal transduction. The activation of both cytokines can be blocked by IL-1Ra. After ligand binding to the receptor, a complex sequence of events is triggered, including phosphorylation and ubiquitination, which results in the activation of nuclear factor kappa B and AP-1-dependent expression of pro-inflammatory cytokines, chemokines, and secondary mediators of inflammatory response. IL-1 targeted genes include IL-6, IL-8, MCP-1, COX-2, IL-1α, and IL-1β. Most intracellular components that participate in the cellular response to IL-1 also mediate responses to other cytokines (IL-18, IL-33), TLRs, and many other forms of cytotoxic stresses.1,25
A decade ago, new types of intracellular receptors were discovered: nucleotide-binding oligomerization domain-like receptors. They function as recognition receptors for endogenous danger signals and can form multimolecular complexes termed inflammasomes. In addition to recognizing danger signals, these complexes can also activate proteolytic caspases that cleave and activate cytokines important in early immune response.26 Activation and secretion of IL-1β and IL-18 require inflammasome/caspase-1-dependent processing. Other members of the IL-1 family are not dependent on this mechanism.
SJIA
SJIA is the third most common type of chronic idiopathic arthritis in children and represents 10%–20% of JIA patients. There is no gender predominance in SJIA; boys are affected as often as girls.8 In Asia, the percentage of SJIA patients is much higher compared to Europe or North America and could represent over 40% of all JIA cases.27
Recently, results of an international genome-wide association study found no evidence of shared genetic risk factors between SJIA and other JIA categories.11 Single-nucleotide polymorphisms were tested for association with SJIA in 770 children and the genetic architecture of SJIA patients was compared to other JIA subtypes.
Clinical presentation of SJIA differs from other JIA subtypes in several aspects: it includes typical quotidian fever, arthritis, and “salmon pink” rash that appears with the fever and fades away when the fever subsides (Figure 1). Lymphadenopathy, hepatosplenomegaly, and serositis, such as pericardial effusion, are also hallmarks of the disease and are not seen in other forms of JIA.7 SIJA patients are more prone to develop MAS. Some SJIA patients have a monophasic course of the disease with only one episode of fever, others have a polycyclic course, and in some patients the disease persists after the initial attack of fever and follows the severe polyarticular course.28 Systemic signs of the disease can sometimes be more amenable to treatment than chronic arthritis, which can be unresponsive to various treatment approaches.29 Early predictors of joint damage and poor outcome are young age at diagnosis (<18 months of age), longer disease duration, persistent systemic use of GCS, thrombocytosis, and high inflammatory parameters.30–32 With a long-standing disease that is refractory to treatment, the impact on growth can be significant (Figure 2). Growth retardation is thought to be multifactorial, including altered nutritional status, prolonged immobilization, endocrinologic abnormalities, and GCS treatment. A growing body of evidence suggests that inflammation itself has a detrimental effect on the growing skeleton.33 Observations from the pre-steroid era indicate that stunted growth is seen during periods of disease activity with catch-up growth during remission.
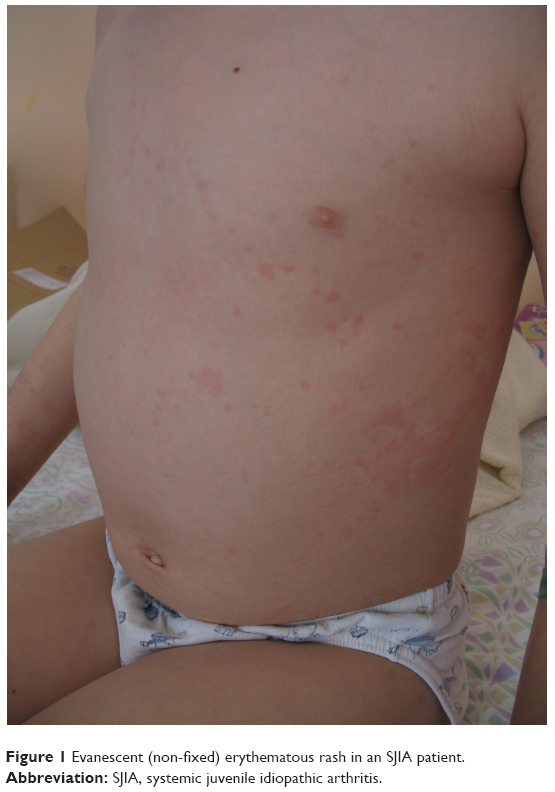 | Figure 1 Evanescent (non-fixed) erythematous rash in an SJIA patient. |
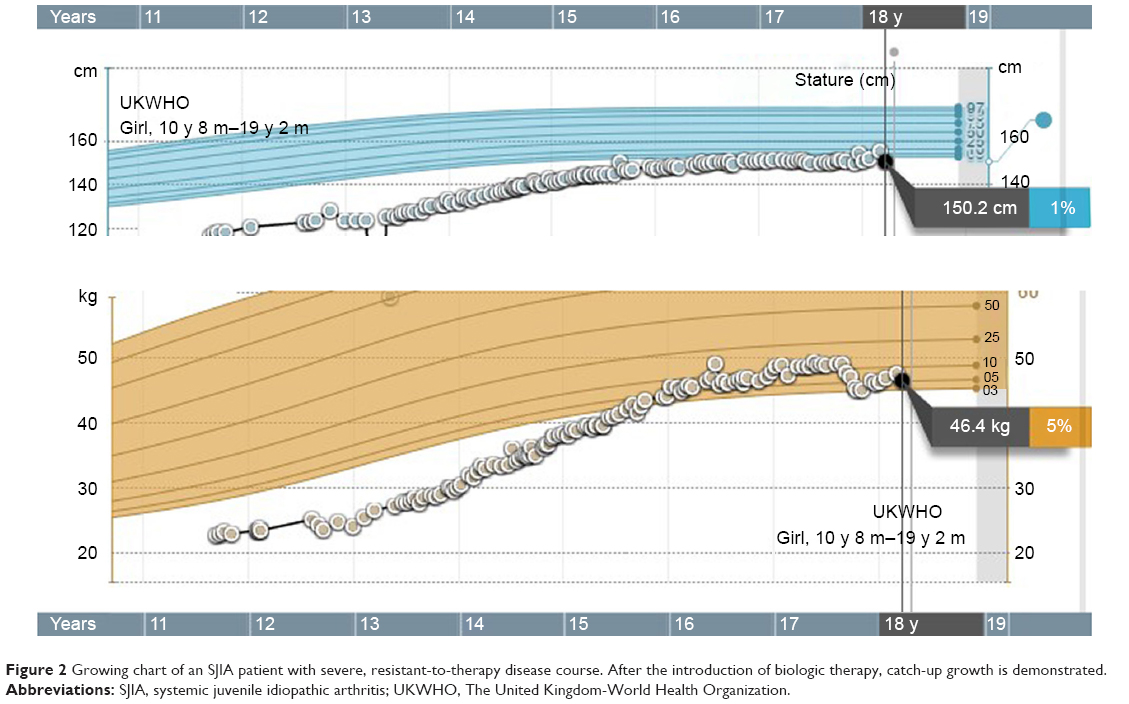 | Figure 2 Growing chart of an SJIA patient with severe, resistant-to-therapy disease course. After the introduction of biologic therapy, catch-up growth is demonstrated. |
Systemic signs of the disease can precede development of arthritis by weeks, months, or even years.34 The clinical presentation may mimic infectious or malignant diseases; therefore, a multidisciplinary approach is mandatory.
Markedly elevated C-reactive protein (CRP) is typical of SJIA, which is not the case in other JIA subtypes. Further laboratory changes include leukocytosis, thrombocytosis, elevation of the erythrocyte sedimentation rate, a low hemoglobin level, and a low albumin level.10 Inflammatory parameters influenced by cytokines such as IL-1 are monitored to evaluate disease activity and response to therapy. Markers for active SJIA also include IL-18, myeloid-related protein (MRP) 8, MRP 14, and S100A12; these could be useful as biomarkers for monitoring treatment response in daily clinical practice.35 An elevated ferritin level should prompt testing for MAS, a potentially life-threatening complication of SJIA.
Characteristic clinical features of MAS include high, continuous fever, generalized lymphadenopathy, hepatosplenomegaly, central nervous system dysfunction, and hemorrhagic diathesis. The signs of severe inflammatory reaction are caused by massive hypersecretion of proinflammatory cytokines, the so-called “cytokine storm,” with uncontrolled and dysfunctional immune response causing continuous activation and expansion of T lymphocytes and macrophages.36 Although the prevalence of MAS in SJIA patients is about 10%, some reports suggest that subclinical MAS may occur in as many as 30%–40% of SJIA patients.37 Recently, the classification criteria for MAS in SJIA were developed by a collaborative initiative of the European League Against Rheumatism, American College of Rheumatology (ACR), and Paediatric Rheumatology International Trials Organization.38
Regular measurement of disease activity in SJIA patients is important for monitoring the disease course and assessing therapy response. At present, clinical measurement tools specifically validated for use in SJIA patients are lacking.39 In randomized controlled trials evaluating drug efficacy in SJIA patients, the adapted pediatric ACR 30/50 was used for measuring therapeutic response. Resolution of fever (≥38°C) during the week preceding the evaluation or the absence of fever in the previous 2 weeks and the reduction of the systemic GCS dose by at least 10% from the baseline dose were added to six core set variables. Over the last decade, the Juvenile Arthritis Disease Activity Score (JADAS) has been recognized as an important tool for measuring disease activity in children with JIA. However, JADAS has not been validated for children with SJIA and active systemic features of the disease.40
Therapeutic strategies in the treatment of SJIA
NSAIDs are the first choice of treatment for SJIA, as they are for other JIA subtypes.12,41,42 Systemic GCS should be withheld until diagnostic procedures are completed. Infections, malignant diseases, and other rheumatic diseases, such as rheumatic fever and Kawasaki disease, should be excluded. In patients who fail to respond to NSAIDs, GCS are used for the treatment of persistent systemic signs. As soon as the inflammation is under control – usually within 2–4 weeks – GCS should be tapered and stopped in about 6–8 weeks, depending on the severity of the disease. In cases with persistent arthritis as the leading clinical feature of the disease, methotrexate (MTX) is the drug of choice after systemic signs have subsided. In patients with a polycyclic course with relapses of systemic features during tapering of GCS, biologic therapy is recommended (Figure 3).42
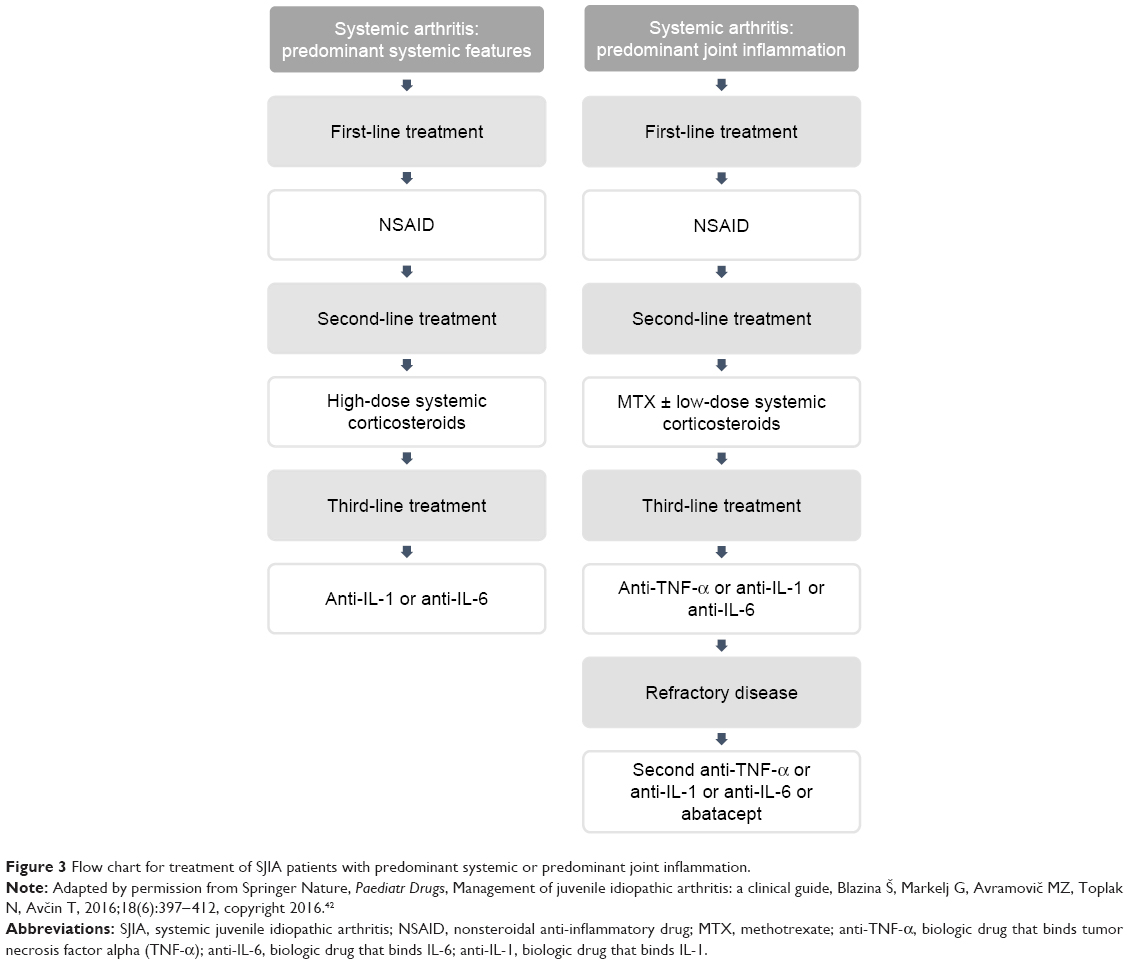 | Figure 3 Flow chart for treatment of SJIA patients with predominant systemic or predominant joint inflammation. |
Inflammatory cytokines (tumor necrosis factor [TNF]-α, IL-1, IL-6, IL-18) have a central role in the pathogenesis of SJIA; cytokine blockers such as anti-IL-1 and anti-IL-6 biologics have excellent efficacy for systemic signs of SJIA.13–16,43–54
Drugs blocking IL-1 cytokine
Three anti-IL-1 drugs are available on the market, but only one has been licensed for treatment of SJIA (Table 2). The European Medicines Agency and the US Food and Drug Administration approved canakinumab for treatment of SJIA and AOSD in 2013 and 2016, respectively.
 | Table 2 Dose, route of administration, and interval of administration of the IL-1 blocking agents in the treatment of SJIA |
The first reports and landmark studies which revealed effective treatment with anti-IL-1 agent in SJIA and AOSD were performed with anakinra, the first discovered anti-IL-1 drug.
Anakinra
Anakinra is a recombinant form of the naturally occurring IL-1Ra. It competitively inhibits the IL-1 type I receptor and neutralizes the biological activity of IL-1α and IL-1β. It is approved for treatment of rheumatoid arthritis (RA) in adults and can be used in combination with MTX in cases with an inadequate response to MTX. A recently published study in patients with RA showed that anakinra, in combination with MTX, was not effective in early, active RA.55 However, substantial evidence exists to demonstrate that anti-IL-1 agents are successful in treating AOSD. A recent comprehensive review of the literature revealed a strong steroid sparing effect with a reduction of the side effects of GCS. The overall response rate to anti-IL-1 drugs is high, up to 90%, especially in resistant AOSD patients.44
Although anakinra was not licensed for the treatment of SJIA, several reports underline the efficacy of anakinra in this disease.45–49 The use of anakinra is currently recommended in SJIA patients with persistent systemic signs of the disease who are refractory to GCS treatment.41,42
After the first two reports of successful therapy with anakinra in a child with SJIA and in a patient with SJIA and MAS, the first study was published by Pascual et al in 2005.45,56 In 2008, a retrospective study of 21 SJIA patients demonstrated an excellent response to treatment with anakinra in 10 out of 21 patients.46 In this study, two groups of patients were identified. The first group demonstrated a dramatic response with normalization of CRP in the first week of treatment and a rapid improvement of arthritis. The second group experienced only a transient improvement or no improvement in the CRP level and arthritis. The differences between the two groups included an increased absolute neutrophil count and milder arthritis in the responder group. It was suggested that a better response to anakinra can be expected in patients with arthritis in only a few joints compared to those with polyarthritis.
A larger retrospective study including 46 patients revealed that anakinra was able to achieve complete clinical response in about 60% of patients. This study included 10 children who received anakinra as initial monotherapy without GCS; of these, 80% had complete response.48
To date, only one small randomized control trial (RCT) of anakinra has been published (ANAJIS trial), including 12 patients in both the placebo and anakinra groups.49 ACR pediatric 30 response was achieved in eight out of 12 patients in the anakinra group and only in one patient in the placebo group at month 1 (p=0.003). Ten patients from the placebo group switched to anakinra and nine of them were responders at month 2. Twelve months after the beginning of the study, only 3/12 patients who started anakinra at day 0, and 4/10 who started in the placebo group and were later switched to anakinra at month 1, were still considered responders. The explanation for the loss of response over time was that in a majority of the included patients, severe polyarthritis without fever was present as the main clinical feature. The other reason could also have been that lower doses of anakinra were used in this study (2 mg/kg subcutaneously daily, maximum dose 100 mg), which could be insufficient in younger patients. Moreover, the study design allowed faster tapering of GCS and precluded concomitant use of DMARDs. Blood gene expression profiling at enrollment and at 6 months showed one set of dysregulated genes that reverted to normal values in responders. Interestingly, another set of genes, including interferon-inducible genes, were found in a majority of the patients treated with anakinra, regardless of the clinical response to treatment.49
The first prospective study in which anakinra was used as first-line therapy in 20 consecutive SJIA patients, before the use of GCS, showed excellent response in nearly all patients within 3 months.50 After 1 year, 17/20 patients had a clinically inactive disease. Thirteen of these patients met the criteria for inactive disease while receiving monotherapy with anakinra, but seven patients required additional therapy along with anakinra due to persistent disease activity.
Recently, another retrospective study with anakinra in 25 SJIA patients from a single center has been published.51 Patients who were treated with anakinra for at least 6 months were included. The primary end point was the number of patients who achieved a clinically inactive disease at 6 months; of the 25 included patients, 14 had inactive disease at that time. In this study, the only variable significantly associated with response to treatment was the time from the onset of disease to the beginning of anakinra therapy, with earlier treatment initiation being associated with a better outcome.
Canakinumab
Canakinumab is a human monoclonal antibody that neutralizes the biological activity of IL-1β, but not IL-1α. It binds selectively and with high affinity to human IL-1β, preventing IL-1β-induced gene activation and inflammatory mediator production.57 It is the only anti-IL-1 agent licensed for the treatment of SJIA. It was approved for use as monotherapy or in combination with MTX for SJIA patients who are at least 2 years old, have active disease, and have responded inadequately to previous therapy with NSAIDs or systemic GCS. In a Phase II multicenter open-label study, 23 children with active SJIA were included. ACR pediatric 50 response was achieved at day 15 after the first canakinumab dose in 60% of children. It was demonstrated that the dose of 4 mg/kg was associated with rapid and sustained clinical improvement. GCS could be tapered to 50% by month 5.13 Responders had fewer active joints and a higher white blood cell count at baseline compared to nonresponders. These findings were in line with a previous study published by Gattorno et al, in which anakinra was the anti-IL-1 agent under investigation.46
The results of the Phase II multicenter study have provided the grounds for further studies. Two randomized trials showed the efficacy of canakinumab in SJIA with active systemic features.14 In the first trial, patients were randomized to receive a single subcutaneous dose of canakinumab or placebo. At day 15, significantly more patients in the canakinumab group had ACR 30 response compared to the placebo group (84% vs 10%). In the second trial, patients who had a clinical response were randomly assigned to continued treatment with canakinumab or placebo. The risk of flare was lower among patients who continued to receive canakinumab than among those who were switched to placebo: 74% of patients in the canakinumab group had no flare, compared to only 25% of patients in the placebo group. The average dose of GCS was significantly reduced in canakinumab-treated patients and discontinued in 33% of patients. Infections were more common in the canakinumab group compared to the placebo group. MAS occurred in seven patients, including two in the placebo group.
Rilonacept
Rilonacept is a soluble decoy receptor that neutralizes mainly IL-1β and to a lesser extent also IL-1α. So far, two studies in children with rilonacept have been conducted.58,59 In the first study, efficacy and safety were evaluated during 23 months of open-label treatment after a 4-week, double-blind, placebo-controlled phase. Twenty-four patients were included in the first 4 weeks of the double-blind, placebo-controlled part, and 23 entered during the open-label period. The median age of the included patients was 14 years (4–20 years). In the double-blind phase, no significant differences between the rilonacept and placebo groups were observed. However, during the open-label treatment period, fever and rash completely resolved by month 3 in all patients. Adapted ACR pediatric 30, 50, and 70 response rates at 3 months were 78%, 60%, and 34%, respectively, and were maintained over the study duration. The prednisone dose was decreased or prednisone therapy discontinued in 22 of the 23 patients. Treatment with rilonacept had a substantial steroid sparing effect. No serious adverse events were observed.58
In the second randomized, double-blind, placebo-controlled study, 71 patients with active arthritis in at least two joints were included. They were randomized in the rilonacept arm and in the placebo arm. An initial 4-week, double-blind, placebo phase was incorporated into a 24-week randomized multicenter design, followed by the open-label phase. Patients in the placebo arm received placebo for 4 weeks, followed by a loading dose of rilonacept 4.4 mg/kg at week 4, and then followed by weekly maintenance doses of 2.2 mg/kg subcutaneously. Patients in the rilonacept arm received a rilonacept loading dose, followed by a weekly maintenance dose. The primary end point of the study was time to response. Adapted ACR pediatric 30 score was used, together with the absence of fever and tapering of the dose of GCS. The time to response was shorter in the rilonacept arm compared to the placebo arm (p=0.007). The number of patients fulfilling the response criteria was significantly higher at week 4 in the rilonacept arm compared to the placebo arm (p=0.016). Exacerbation of SJIA was the most common severe adverse event. Rilonacept was generally well tolerated. Efficacy of the drug was confirmed in active SJIA.59
In comparison with the other two anti-IL-1 agents, rilonacept is not commonly used for the treatment of SJIA and it is not approved for this indication. Recently, a meta-analysis of RCTs of biologic agents in SJIA was published. Five RCTs were eligible for inclusion; one study each for anakinra, canakinumab, and tocilizumab, and two for rilonacept. All studies were placebo-controlled and showed the efficacy of the tested drug; however, the meta-analysis demonstrated that canakinumab and also the anti-IL-6 agent tocilizumab were more effective than rilonacept. Biologic agents seem to be safe and comparable for the risk of severe adverse events in short-term observations.54
Other cytokine blockers effective in the treatment of SJIA
Anti-TNF-α agents are used successfully in the treatment of several subtypes of JIA, but they do not demonstrate the same efficacy in SJIA patients with predominantly systemic clinical features. In a subset of SJIA patients with well-controlled systemic features and persistent arthritis, anti-TNF-α drugs are more effective and often used if the main clinical feature is arthritis. Similarly to MTX, anti-TNF-α drugs have a very limited effect, or no effect at all, on systemic signs of the disease. Among 45 SJIA patients treated with anti-TNF-α agents, only 24% experienced remission and only 13% experienced a sustained benefit.60 Furthermore, the results of a recent French study showed that introduction of anti-IL-1 or anti-IL-6 inhibitor, instead of anti-TNF-α agents as the first-line biologic therapy in SJIA patients, significantly increases the chance of these patients achieving remission.61
Anti-IL-6 agent (tocilizumab) is the first humanized anti-IL-6 receptor antibody. A recently published meta-analysis of RCTs of biologic agents in SJIA revealed that tocilizumab seems to be more appropriate in SJIA patients with extensive joint involvement.54 The efficacy and safety of tocilizumab have been proven in several studies.15,16,52,53 It can be used alone or with MTX in children older than two.29,62 In 2005, a Japanese study showed prompt response in 90% (10/11) of children with SJIA who were treated with tocilizumab.52 Later, tocilizumab’s efficacy was confirmed in the RCT followed by a long-term open-label extension phase.15,53 Similar results were reported in studies conducted in Europe and in North and South America. Within 3 months of the beginning of tocilizumab therapy, 70% of children treated with tocilizumab improved clinically by at least 70%, compared to only 8% of those who were receiving placebo.16
Treatment of MAS
There is currently no evidence-based treatment for MAS.63 The highest level of evidence for treatment comes from case series. High-dose GCS with or without cyclosporine have been frequently reported as the first-line therapy. Promising responses were reported also with anakinra treatment.36 In a recent systematic literature review assessing the performance of 2016 MAS classification criteria for patients with SJIA who developed MAS while being treated with biologic therapy, research demonstrated that substantial alteration in MAS features may limit the utility of defined criteria for diagnoses in SJIA patients treated with biologics.64
Biologic rationale for IL-1 cytokine as a therapeutic target in SJIA
SJIA is different from other types of JIA, so it is unsurprising that the therapeutic approach employed in other forms of JIA is not applicable to it. Over the last decade, several studies have shown that SJIA is primarily an autoinflammatory and not an autoimmune disease. The clinical course and laboratory results, including a very high CRP level, more closely resemble autoinflammation or severe infection than an autoimmune disease. Due to the pronounced activation of a patient’s innate immune system, it has been postulated that SJIA is likely a polygenic autoinflammatory syndrome.65 The disease course can follow a periodic relapsing course similar to diseases belonging to the group of periodic fever syndromes, such as cryopyrin-associated periodic fever syndromes. The cause of these diseases is gain-of-function NLRP3 mutation, which is associated with the activation of the inflammasome, leading to overexpression of IL-1β. The NLRP3 gene encodes the multimeric protein complex cryopyrin, a crucial protein of the inflammasome. Cryopyrin controls the activation of caspase-1, which catalyzes the cleavage of pro-IL-1β into IL-1β.
It has been shown that, also in SJIA, IL-1β is a crucial cytokine, but the source of excess of this cytokine remains unknown and its role in chronic arthritis is not entirely clear.66 In the landmark study published in 2005, Pascual et al showed that serum from SJIA patients induced the transcription of innate immunity genes, including IL-1 in healthy peripheral blood mononuclear cells.45 It is, however, interesting that IL-1β serum levels in patients were as low as those of healthy controls. It was assumed that serum cytokine levels may not reflect a cytokine role in the pathogenesis of the disease. Researchers administered recombinant IL-1R antagonist to nine SJIA patients refractory to other therapies; complete remission was obtained in 7/9 patients, showing that blockade of IL-1 cytokine could be an effective target therapy in SJIA.45
Recent studies have revealed that SJIA pathogenesis likely follows a biphasic course.67 In the initial phase, the systemic disease is driven by an innate immune response with IL-1β as a key cytokine. In the second phase, which is probably dominated by adaptive immunity and cytokine IL-17A, chronic arthritis becomes the leading clinical feature. Based on these facts, anti-IL-1 therapy appears to be a rational therapeutic approach in the first phase of SJIA, but in the case of a persistent disease with chronic arthritis, other biologic drugs might have a better effect.
Future perspectives – the role of early IL-1 blockade and possible new biological targets for the treatment of SJIA
Despite the current evidence showing success of early treatment with anti-IL-1 therapy, at the present moment this therapy is still not standard practice in the early treatment of SJIA. However, in the case of a severe disease course and complications such as MAS, early treatment with anti-IL-1 therapy is advised. On the grounds of a prospective study published recently, in which anakinra was used as the first-line therapy and showed an excellent response in nearly all patients, and based on recent reports that SJIA pathogenesis follows a biphasic course with innate immune response in the initial phase and adaptive immune response in the second phase, there appears to be a window of opportunity for treatment with anti-IL-1 therapy early in the disease course.50,67 If the window of opportunity is missed, the late course of the disease is dominated by adaptive immunity and cytokine IL-17A with chronic arthritis as the main clinical feature; in this case, other therapies are more suitable to control the disease. It has been shown that in SJIA patients in whom severe arthritis is the predominant feature anti-IL-1 therapy is usually not successful.46
It has recently been shown that canakinumab treatment in SJIA patients results in a rapid reduction of the expression of genes related to inflammation and in the reduction of the inflammatory cytokine level.68 The level of IL-6 declined by day 3 and remained suppressed, and IL-18 declined on day 57. Samples used in this study were collected from the two pivotal trials on SJIA.13,68 The strongest clinical response at day 15 (≥50 adapted ACR JIA response criteria) was observed in patients with higher baseline expression of dysregulated genes and a strong transcriptional response on day 3. It was suggested that canakinumab can, at least to some extent, reverse the molecular disease pattern.
Modern treatment of JIA, including the systemic form, should follow the treat-to-target treatment approach with the achievement of complete clinical and laboratory disease remission, or in cases where this is not possible, with minimal disease activity.69 Because long-term disability closely correlates with the duration of the active disease, early aggressive treatment is important to achieve the goal of the treat-to-target approach.
The other aspects of treatment are the side effects of the therapy. While GCS still hold the main position among the most potent anti-inflammatory drugs with prompt resolution of inflammation, the long-term side effects of GCS treatment limit their prolonged use. Except for a higher risk of infection in the treatment with anti-IL-1 drugs, no such long-term side effects that can be devastating in prolonged use of GCS are seen in the prolonged use of IL-1 inhibition. If it is still not possible to completely avoid GCS in some patients, early treatment with IL-1 blockade could at least significantly lower the cumulative dose of GCS in the majority of patients. This is even more important in children than in patients with AOSD because of the impact of GCS therapy on the growth and development of a child.
It is anticipated that in the near future, data from multicenter studies and patient registries will provide additional, adequately strong evidence for changing the recommended treatment approach in SJIA patients to early introduction of IL-1 blockade.
At present, we lack data on treatment with a combination of biologic drugs in severe, therapy-resistant cases of SJIA patients. The possibility of severe infections as a side effect of such a treatment approach probably increases significantly. However, isolated reports of the successful use of combinations in clinical practice do exist. Anakinra in combination with abatacept, which selectively modulates the costimulatory signal (CD80/CD86:CD28) needed for T-cell activity, was successfully used in four treatment-resistant, steroid-dependent SJIA patients, without severe side effects.70
Recent advances in the pathology of SJIA draw attention to other possible therapeutic biologic targets such as IL-18, IL-17, signaling molecules such as enzymes in the JAK family, and interferon-γ in MAS in resistant, difficult-to-treat patients.18,71
Conclusion
Over the last decade, the treatment of SJIA, which belongs to the family of autoinflammatory diseases, has significantly improved with the advent of novel targeted drugs: cytokine blocking agents, with the prime position being held by anti-IL-1 agents. Long-term disease outcome in SJIA patients improved and treatment damage reduced with the introduction of these drugs in the treatment plan. The treat-to-target approach has become an achievable goal. Early introduction of anti-IL-1 therapy, during the first phase of the disease when systemic signs predominate, in the so-called window of opportunity, can induce rapid and sustained blockade of inflammation and significantly change the disease course and its long-term outcome.
Acknowledgment
This review was partially supported by research grants from the University Medical Centre Ljubljana (TP 20160048) and Slovenian Research Agency (L7-8274).
Disclosure
The authors report no conflicts of interest in this work.
References
Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117(14):3720–3732. | ||
Palomo J, Dietrich D, Martin P, Palmer G, Gabay C. The interleukin (IL)-1 cytokine family – balance between agonists and antagonists in inflammatory diseases. Cytokine. 2015;76(1):25–37. | ||
Xia Y, Cui P, Li Q, Liang F, Li C, Yang J. Extremely elevated IL-18 levels may help distinguish systemic-onset juvenile idiopathic arthritis from other febrile diseases. Braz J Med Biol Res. 2017;50(2):e5958. | ||
Shimizu M, Nakagishi Y, Inoue N, et al. Interleukin 18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clin Immunol. 2015;160(2):277–281. | ||
Martini A. It is time to rethink juvenile idiopathic arthritis classification and nomenclature. Ann Rheum Dis. 2012;91(9):1437–1439. | ||
Nigrovich PA, Raychaudhuri S, Thompson SD. Review: genetics and the classification of arthritis in adults and children. Arthritis Rheumatol. 2018;70(1):7–17. | ||
Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31(2):390–392. | ||
Weiss JE, Ilowite NT. Juvenile idiopathic arthritis. Rheum Dis Clin North Am. 2007;33(3):441–470. | ||
Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377(9783):2138–2149. | ||
Cimaz R. Systemic onset juvenile idiopathic arthritis. Autoimmun Rev. 2016;15(9):931–934. | ||
Ombrello MJ, Arthur VL, Remmers EF, et al. Genetic architecture distinguishes systemic juvenile idiopathic arthritis from other forms of juvenile idiopathic arthritis: clinical and therapeutic implications. Ann Rheum Dis. 2017;76(5):906–913. | ||
Malattia C, Martini A. Glucocorticoids in juvenile idiopathic arthritis. Ann N Y Acad Sci. 2014;1318:65–70. | ||
Ruperto N, Quartier P, Wulffraat N, et al. A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 2012;64(2):557–567. | ||
Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2396–2406. | ||
Yokota S, Imagawa T, Mori M, et al. Long term treatment of systemic juvenile idiopathic arthritis with tocilizumab: result of an open label extension study in Japan. Ann Rheum Dis. 2013;72(4):627–628. | ||
De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2385–2395. | ||
Shimizu M, Nakagishi Y, Yachie A. Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine. 2013;61(2):345–348. | ||
Canny S, Mellins E. New frontiers in the treatment of systemic juvenile idiopathic arthritis. F1000Res. 2017;6:971. | ||
Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal. 2010;3(105):cm1. | ||
Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. | ||
Voronov E, Shouval DS, Krelin Y, et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A. 2003;100(5):2645–2650. | ||
Lopez-Castejon G, Brough D. Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev. 2011;22(4):189–195. | ||
Carriere V, Roussel L, Ortega N, et al. IL-33, the IL-1 cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A. 2007;104(1):282–287. | ||
O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008;226:10–18. | ||
Henderson C, Goldbach-Mansky R. Monogenic IL-1 mediated autoinflammatory and immunodeficiency syndrome: finding the right balance in response to danger signals. Clin Immunol. 2010;135(2):210–222. | ||
Jha S, Ting P. Inflammasome associated nucleotide-binding domain, leucine-rich repeat proteins and inflammatory diseases. J Immunol. 2009;183(12):7623–7629. | ||
Huang H, Qian X, Yu H, Li J, Zhang Y. Clinical analysis in 202 children with juvenile idiopathic arthritis. Clin Rheumatol. 2013;32(7):1021–1027. | ||
Singh-Grewal D, Schneider R, Bayer N, Feldman BM. Predictors of disease course and remission in systemic juvenile idiopathic arthritis: significance of early clinical and laboratory features. Arthritis Rheum. 2006;54(5):1595–1601. | ||
Beukelman T. Treatment advances in systemic juvenile idiopathic arthritis. F1000Prime Rep. 2014;6:21. | ||
Spiegel LR, Schneider R, Lang BA, et al. Early predictors of poor functional outcome in systemic onset juvenile rheumatoid arthritis. A multicentre cohort study. Arthritis Rheum. 2000;43(11):2402–2409. | ||
Sandborg C, Holmes TH, Lee T, et al. Candidate early predictors for progression to joint damage in systemic juvenile idiopathic arthritis. J Rheumatol. 2006;33(11):2322–2329. | ||
Russo RA, Katsicas MM. Patients with very early onset systemic juvenile idiopathic arthritis exhibit more inflammatory features and a worse outcome. J Rheumatol. 2013;40(3):329–334. | ||
De Benedetti F, Brunner H, Ruperto N, et al. Catch-up growth during tocilizumab therapy for systemic juvenile idiopathic arthritis: results from a phase III trial. Arthritis Rheumatol. 2015;67(3):840–848. | ||
Giancane G, Minoia F, Davi S, Bracciolini G, Consolaro A, Ravelli A. IL-1 inhibition in systemic juvenile idiopathic arthritis. Front Pharmacol. 2016;7:467. | ||
Swart JF, deRoock S, Prakken BJ. Understanding inflammation in juvenile idiopathic arthritis: how immune biomarkers guide clinical strategies in the systemic onset subtype. Eur J Immunol. 2016;46(9):2068–2077. | ||
Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as a part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13(4):289–298. | ||
Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34(5):1133–1138. | ||
Ravelli A, Minoia F, Davi S, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organization collaborative initiative. Ann Rheum Dis. 2016;75(3):481–489. | ||
Minoia F, Consolaro A, Ravelli A. Filling the gap: toward a disease activity tool for systemic juvenile idiopathic arthritis. J Rheumatol. 2018;45(1):3–5. | ||
Consolaro A, Ravelli A. Defining criteria for disease activity states in juvenile idiopathic arthritis. Rheumatology. 2016;55(4):595–596. | ||
Beukelman T, Patkar NM, Saag KG, et al. 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: initiation and safety monitoring of therapeutic agents for the treatment of arthritis and systemic features. Arthritis Care Res (Hoboken). 2011;63(4):465–482. | ||
Blazina Š, Markelj G, Avramovič MZ, Toplak N, Avčin T. Management of juvenile idiopathic arthritis: a clinical guide. Paediatr Drugs. 2016;18(6):397–412. | ||
Grevich S, Shenoi S. Update on the management of systemic juvenile idiopathic arthritis and role of IL-1 and IL-6 inhibition. Adolesc Health Med Ther. 2017;8:125–135. | ||
Junge G, Mason J, Feist E. Adult onset Still’s disease – the evidence that anti-interleukin-1 treatment is effective and well tolerated (a comprehensive literature review). Semin Arthritis Rheum. 2017;47(2):295–302. | ||
Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201(9):1479–1486. | ||
Gattorno M, Piccini A, Lasigle D, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58(5):1505–1515. | ||
Zeft A, Hollister R, LaFleur B, et al. Anakinra for systemic juvenile arthritis: the Rocky Mountain experience. J Clin Rheumatol. 2009;15(4):161–164. | ||
Nigrovich PA, Mannion M, Prince Femke HM, et al. Anakinra as first line disease modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicentre series. Arthritis Rheum. 2011;63(2):545–555. | ||
Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind placebo controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70(5):747–754. | ||
Vastert SJ, de Jager W, Noordman BJ, et al. Effectiveness of first line treatment with recombinant interleukin-1 receptor antagonist in steroid naïve patients with new onset systemic juvenile idiopathic arthritis. Results of a prospective cohort study. Arthritis Rheum. 2014;66(4):1034–1043. | ||
Pardeo M, Pires Marafon D, Insalco A, et al. Anakinra in systemic juvenile idiopathic arthritis: a single-center experience. J Rheumatol. 2015;42(8):1523–1527. | ||
Yokota S, Miyamae T, Imagawa T, et al. Therapeutic efficacy of humanized recombinant anti-interleukin 6 receptor antibody in children with systemic onset juvenile idiopathic arthritis. Arthritis Rheum. 2005;52(3):818–825. | ||
Yokota S, Imagawa T, Mori M, et al. Efficacy and safety of tocilizumab in patients with systemic onset juvenile idiopathic arthritis: a randomised, double blind, placebo controlled, withdrawal phase III trial. Lancet. 2008;371(9617):998–1006. | ||
Tarp S, Amarily G, Foeldvari I, et al. Efficacy and safety of biological agents for systemic juvenile idiopathic arthritis: a systematic review and meta-analysis of randomized trials. Rheumatology (Oxford). 2016;55(4):669–679. | ||
Scott IC, Ibrahim F, Simpson G, et al. A randomised trial evaluating anakinra in early active rheumatoid arthritis. Clin Exp Rheumatol. 2016;34(1):88–93. | ||
Verbsky JW, White AJ. Effective use of the recombinant interleukin 1 receptor antagonist anakinra in therapy resistant systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2004;31(10):2071–2075. | ||
Hoy SM. Canakinumab: a review of its use in the management of systemic juvenile idiopathic arthritis. BioDrugs. 2015;29(2):133–142. | ||
Lovell DJ, Giannini EH, Reiff AO, et al. Long term safety and efficacy of rilonacept in patients with systemic juvenile idiopathic arthritis. Arthritis Rheum. 2013;65(9):2486–2496. | ||
Ilowite NT, Prather K, Lokhnygina Y, et al. Randomized, double blind, placebo controlled trial of the efficacy and safety of rilonacept in the treatment of systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2014;66(9):2570–2579. | ||
Russo RA, Katsicas MM. Clinical remission in patients with systemic juvenile idiopathic arthritis treated with anti-tumor necrosis factor agents. J Rheumatol. 2009;36(5):1078–1082. | ||
Woerner A, Uettwiller F, Melki I, et al. Biological treatment in systemic juvenile idiopathic arthritis: achievement of inactive disease or clinical remission on a first, second or third biological agent. RMD Open. 2015;1(1):e000036. | ||
Barone P, Pignatato R, Garozzo MT, Leonardi S. IL-6 blockers in systemic onset juvenile idiopathic arthritis. Immunotherapy. 2016;8(1):79–87. | ||
Boom V, Anton J, Lahdenne P, et al. Evidence-based diagnosis and treatment of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2015;13:55. | ||
Schulert GS, Minoia F, Bohnsack J, et al. Effect of biologic therapy on clinical and laboratory features of macrophage activation syndrome associated with systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken). 2018;70(3):409–419. | ||
Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Ann Rev Immunol. 2009;27:621–668. | ||
Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol. 2011;7(7):416–426. | ||
Kessel C, Lippitz K, Weinhage T, et al. Proinflammatory cytokine environments can drive interleukin 17 overexpression by gamma/delta T cells in systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 2017;69(7):1480–1494. | ||
Brachat AH, Grom AA, Wulffraat N, et al. Early changes in gene expression and inflammatory proteins in systemic juvenile idiopathic arthritis patients on canakinumab therapy. Arthritis Res Ther. 2017;19(1):13. | ||
Consolaro A, Negro G, Lanni S, Solari N, Martini A, Ravelli A. Toward a treat-to-target approach in the management of juvenile idiopathic arthritis. Clin Exp Rheumatol. 2012;30(4 Suppl 73):S157–S162. | ||
Record JL, Beukelman T, Cron RQ. Combination therapy of abatacept and anakinra in children with refractory systemic juvenile idiopathic arthritis: a retrospective case series. J Rheumatol. 2011;38(1):180–181. | ||
Canna SW, Girard C, Malle L, et al. Life threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol. 2017;139(5):1698–1701. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.