")
Back to Archived Journals » Hypoxia » Volume 3
The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy
Authors Muz B, de la Puente P, Azab F, Azab AK
Received 30 July 2015
Accepted for publication 4 November 2015
Published 11 December 2015 Volume 2015:3 Pages 83—92
DOI https://doi.org/10.2147/HP.S93413
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Barbara Muz, Pilar de la Puente, Feda Azab, Abdel Kareem Azab
Department of Radiation Oncology, Cancer Biology Division, Washington University School of Medicine in St Louis, MO, USA
Abstract: Hypoxia is a non-physiological level of oxygen tension, a phenomenon common in a majority of malignant tumors. Tumor-hypoxia leads to advanced but dysfunctional vascularization and acquisition of epithelial-to-mesenchymal transition phenotype resulting in cell mobility and metastasis. Hypoxia alters cancer cell metabolism and contributes to therapy resistance by inducing cell quiescence. Hypoxia stimulates a complex cell signaling network in cancer cells, including the HIF, PI3K, MAPK, and NFκB pathways, which interact with each other causing positive and negative feedback loops and enhancing or diminishing hypoxic effects. This review provides background knowledge on the role of tumor hypoxia and the role of the HIF cell signaling involved in tumor blood vessel formation, metastasis, and development of the resistance to therapy. Better understanding of the role of hypoxia in cancer progression will open new windows for the discovery of new therapeutics targeting hypoxic tumor cells and hypoxic microenvironment.
Keywords: hypoxia, cancer, metastasis, angiogenesis, treatment resistance
Introduction
Twenty-five percent of deaths in the United States are caused by cancer, and the number of incidences increases due to population growth, prolonged life expectancy, and an abundance of risk factors including smoking, a lack of activity, and obesity.1 A common feature of most tumors is a low level of oxygen, called hypoxia, the severity of which varies between tumor types (Table 1). In intensively proliferating and expanding tumor tissue, oxygen demand is surpassed by oxygen supply, and the distance between cells and the existing vasculature increases, hampering oxygen diffusion and creating even more hypoxic milieu.2,3 It is generally accepted that the oxygen level in hypoxic tumor tissues is poorer than the oxygenation of the respective normal tissues and on average it is between 1%–2% O2 and below (Table 1). However, tumor oxygen level depends on the initial oxygenation of the tissue, the size and stage of the tumor, the method of oxygen measurement, and in which part of the heterogenic tumor tissue the measurement was performed (Table 1).4–7 Tissue normoxia, also known as physoxia, is the oxygenation in healthy tissues, which varies widely between the organs due to diversified blood vessel network and metabolic activity. Oxygen concentration in humans ranges between approximately 9.5% O2 in the renal cortex7 to 4.6% O2 in the brain with neurons extremely sensitive to hypoxia.8–10 These oxygen values are far from the experimental in vitro conditions. The oxygen concentration commonly used in the laboratory setting is 20.9% O2, which means that cell culture is performed in hyperoxic rather than physoxic conditions of respective organs. In order to better understand principles of oxygenation in vitro and in vivo, basic knowledge of the physics of gases is required for newcomers in the hypoxia research field which has been neatly described in a recently published review.11
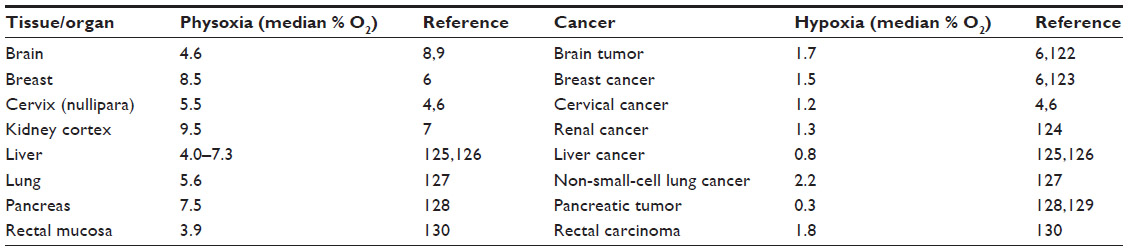 | Table 1 Comparison of the oxygenation in organs and respective tumors |
Cancer cells respond differently to decreased oxygenation leading to cell death or cell survival which partially depends on the time of exposure to hypoxia. The discrepancy and lack of consistency in experimental oncology regarding the definition of acute versus chronic hypoxia often with different biological consequences was thoroughly reviewed.12,13 In general, it is accepted that acute hypoxia is an abrupt and brief exposure to short-term hypoxia which occurs when blood vessel occlusion lasts for at least several minutes.14 It is reversible and often leads to oxygen fluctuations called cycling hypoxia. In acute hypoxia in vitro, cells are usually exposed to continuous hypoxia between a few minutes and up to 72 hours.12 Short-term hypoxia allows cells to survive in these adverse conditions by activating autophagy, an apoptotic and metabolic adaptation of cells. Autophagy is achieved by decreasing oxidative metabolism.15,16 On the contrary, others have shown that cycling hypoxia led to increased reactive oxygen species (ROS) production, what contributed to tumor cell survival and progression.17,18 Moreover, both short- and long-term hypoxia was shown to increase radio-resistance of cancer cells both in vitro17,19 and in vivo.17,20 In addition, acute hypoxia was associated with more aggressive tumor phenotype through induction of spontaneous metastasis.12,21,22
Enduring changes in blood flow and low oxygen availability resulting in chronic hypoxia are especially pronounced in larger tumors and contribute to long-term cellular changes. In experimental settings, chronic conditions are considered when the cells are incubated in hypoxia between a few hours and as long as several weeks.12 Longer exposure to hypoxia is associated with high frequency of DNA breaks, accumulation of DNA replication errors since hypoxia hampers DNA repair systems including homologous recombination and mismatch repair, potentially leading to genetic instability and mutagenesis.23–25 Of note, acute hypoxia also leads to genomic instability due to delayed DNA damage response and rapid p53-dependent apoptosis.26 It was suggested that cells lacking functional p53 are more susceptible to genomic instability and potentially tumorigenesis if they experience reoxygenation after acute exposure to hypoxia.26
Nonetheless, cycling hypoxia represents the situation of oxygenation in tumor tissues. Oxygen fluctuation occurs at irregular intervals in cancer with sporadic reoxygenation periods due to dysfunctional tumor vascularity and heterogenic blood supply.27,28 Undoubtedly, both chronic and acute hypoxic regions in tumors directly affect clinical responses to therapy by influencing tumor growth, ability to metastasize, and resistance to cell death.
Signaling pathways related to tumor hypoxia
Hypoxia induces a number of complex intracellular signaling pathways such as the major hypoxia-inducible factor (HIF) pathway. Other hypoxia-associated pathways include PI3K/AKT/mTOR,29,30 MAPK also known as ERK pathways,31–33 and the NFκB.34 These pathways are involved in cell proliferation, survival, apoptosis, metabolism, migration, and inflammation.
PI3K/AKT/mTOR, MAPK, and NFκB signaling pathways are also stimulated in a hypoxia-independent manner by a number of factors such as cytokines, chemokines, and growth factors which bind to receptor tyrosine kinases, G protein-coupled receptors, toll-like receptors (TLR), and alarmins receptors on the cell surface, which eventually may also lead to HIF-1α activation (Figure 1). In addition, in cancer cells epigenetic changes and acquired mutations of the pathways’ members and overactivation/overstimulation of receptors cause uncontrollable cancer cell growth.35 Targeting non-HIF pathways provides a promising target for anti-neoplastic therapy and each pathway is a vast topic on its own. More information regarding the role of the non-HIF pathways in cancer can be found elsewhere. This review will mainly concentrate on the HIF pathway and its involvement in tumor progression.
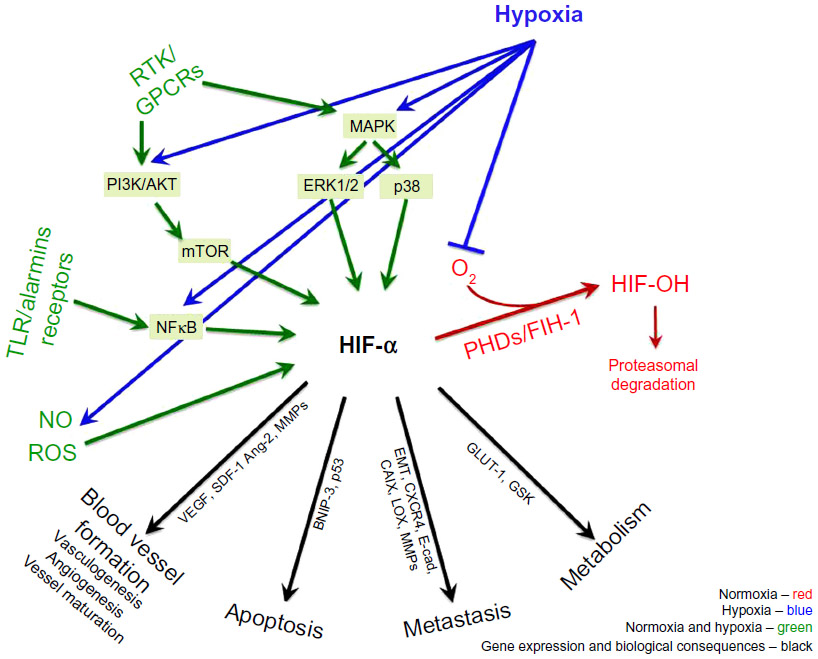 | Figure 1 Regulation of HIF in normoxic and hypoxic conditions. |
HIF pathway
Cellular adaptation to hypoxia is primarily mediated by a family of transcriptional regulators, HIF, which was identified 2 decades ago.36 The hypoxic induction and protein stabilization of HIF-α subunits (HIF-1α, HIF-2α, and HIF-3α) is regulated by oxygen sensors, including PHD and FIH-1 enzymes. PHDs and FIH-1 are upstream of HIF-α and their activity is oxygen-dependent.37 In oxygenated cells, HIF-α subunits are hydroxylated by PHDs and FIH-1, which facilitate the tagging of HIF-α-OH by pVHL, polyubiquitination and protein degradation of HIF-α by the proteasome.38 When the oxygen level drops, the PHD enzymes lose their activity, the hydroxylation of the HIF-α subunit is inhibited and the degradation halted. The non-hydroxylated, stabilized HIF-α subunits translocate to the nucleus where they dimerize with constitutively expressed HIF-β subunit, bind to DNA and initiate gene transcription of the adaptive pathways (Figure 1).37
Apart from hypoxia, the HIF pathway is modulated in a hypoxia-independent manner. HIF-α stabilization and activity is regulated by epigenetic changes and mutations, which lead to a loss of tumor-suppressor functions (ING4, p53, PTEN, VHL) and a gain of oncogene functions (Ras, Raf, Src, mTOR, and Myc).39–41 Hypoxia-independent HIF-α regulation occurs in response to cytokines, lipopolysaccharides, and growth factors, mediated by PI3K/AKT/mTOR,29,30 MAPK,41 and NFκB pathways.42,43 In addition, mitochondrial ROS44,45 and nitric oxide (NO)46 were shown to up- or downregulate HIF-1α accumulation (Figure 1).
Due to the diversified character of tumors including hypoxic and inflammatory phenotype, signaling pathways are activated simultaneously and they frequently share a number of target genes. HIF-1α and NFκB together regulate over 1,000 genes, and thus control malignant and metastatic phenotype of cancer cells since they both: i) enhance cell survival via a number of growth factors and inhibition of pro-apoptotic pathways, ii) contribute to tumor neovascularization via VEGF, VEGF receptors, COX-2, iNOS, iii) regulate cell detachment via downregulation of adhesion molecules such as cadherins, and iv) induce cell migration and invasion through matrix degrading enzymes.43 The HIF and NFKB pathways are controlled by a negative feedback loop mechanism and also intersect via alarmins. Tissue damage and necrosis, which can be also induced by hypoxia, increases the presence of alarmins, the endogenous markers for damage, which are recognized by receptor for advanced glycation endproducts (RAGE) and some of TLRs. In addition, the expression of RAGE receptor is also upregulated by HIF-1a. In turn, alarmin receptors strongly activate NFkB and proinflammatory gene expression. Moreover, the basal HIF-1a mRNA expression is regulated by NFKB in non-hypoxic conditions since HIF-1a promoter was shown to be responsive to certain NFKB subunits.39
The HIF pathway is required during physiological processes and is implicated in cancer biology by regulating hundreds of genes.47–49 This master regulator facilitates tumor growth by promoting angiogenesis via VEGF and SDF-1,50,51 metabolism via regulation of GLUT-1, GLUT-3, and glycolytic enzymes,52–54 and regulating cell apoptosis and cell survival via BNIP-3,55 p53,56,57 TGF-β, and bFGF.3 Moreover, HIF-α contributes to cancer metastasis by altering cancer cell adhesion and motility through regulation of epithelial-to-mesenchymal transition (EMT) and E-cad, ZEB1, -2 and TCF3 expression,58 as well as migration and invasion abilities through CXCR4,59 CAIX,60 LOX,61 MMP-2, and MMP-9.47,62,63
The role of hypoxia in progression and metastasis in cancer
Pathological hypoxia is a common microenvironment factor in tumors that facilitates cell survival and propagation of the tumor. Key cellular responses to hypoxia triggered by overexpression of HIF-1α and HIF-2α subunits and their downstream targets increase blood vessel formation, aggressiveness, metastasis, and resistance to treatment.
Blood vessel formation
Blood vessels create a network of tubes and capillaries which nourish the entire body with oxygen and nutrients. Thus, the way they are formed and function is crucial in embryogenesis and physiology. Blood vessels consists of endothelial cells (ECs) which create a tight barrier between the blood and tissue, and interact with ECM. In embryogenesis, blood vessels are formed de novo by vasculogenesis involving bone marrow-derived endothelial progenitor cells (EPCs)64 followed by angiogenesis, a process where new blood vessels are created from pre-existing vasculature.65 Lastly, the vessels undergo maturation which includes physical interaction with smooth muscle cells and pericytes. Abnormal angiogenesis is a feature of pathological conditions including tumor progression, where hyperproliferating cancer cells surpass their blood supply and become hypoxic. Hypoxia induces the imbalance between pro- and anti-angiogenic factors’ production, which leads to enhanced, rapid and chaotic blood vessel formation. Hypoxia and potent transcription factors HIF-1α and HIF-2α have been shown to be involved in all steps of blood vessel formation.36,64,65 i) Hypoxia and HIF-α subunits contribute to the EPCs’ recruitment from the bone marrow and induction of their differentiation into ECs by regulation of VEGF, a primary regulator of vasculogenesis. This is also mediated through stimulation of pro-angiogenic molecule production such as VEGF-R2 (Flk-1), members of the FGF family and PDGF, important in the primitive vascular network formation.64,66 ii) Hypoxia and HIF-α are also involved in the angiogenesis process by inducing enzymes’ expression (ie, MMPs) in order to sprout and split the pre-existing vessels. In turn, neovessels allows ECs to migrate in response to chemoattractants across ECM. Additionally hypoxia induces ECs’ proliferation by regulation of VEGF-R1 (Flt-1), Ang-1 and Ang-2 expression.67 iii) Finally, hypoxia and HIF-α support vessel maturation via induction of Ang-1, PDGF, and TGF-β to recruit supporting cells such as smooth muscle cells and pericytes creating mature and stable blood vessels.67
However, in tumors, neovessels are often abnormal, immature, and leaky. They are either insufficient or excessive depending on the tumor type.65 Neovasculogenesis maintains blood flow to the growing tumor tissue that expands rapidly, providing nutrients and oxygen for thriving cancer cells; however, more cells means more demand causing even more hypoxia. Again, hypoxia in turn stimulates angiogenesis to ameliorate hypoxic condition, closing the vicious circle. As a consequence the tumor tissue ends up being highly hypoxic with excessive but dysfunctional vasculature.68
Folkman was the first to propose anti-angiogenic therapy to treat cancer in 1971.69 A successful use of monoclonal antibody against VEGF (bevacizumab) approved for treatment of metastatic colorectal cancer70 followed by multiple solid tumors, has stimulated development of other anti-angiogenic therapies. However, long-term exposure to these agents revealed not only reducing tumor growth, but also more malignant and invasive cancer phenotype increasing metastasis.71 Long-term exposure to anti-angiogenic agents reduce tumor; however, at the same time induce more aggresive and metastatic tumor phenotype.71,72
Metastasis
Enhanced angiogenesis is associated with metastasis since permeable and heterogeneous vasculature facilitates the extravasation, circulation, and relocation of tumor cells of tumor cells to new and unaffected tissues escaping the hostile hypoxic environment.68 Tumor oxygenation is a critical factor of cancer progression and the overexpression of HIF-α subunits in tumors and their metastases is associated with the aggressiveness of a majority of human cancers and correlates with poor overall survival.49,73,74
It was demonstrated previously that hypoxic cells are more aggressive and invasive with better ability to metastasize. For instance, multiple myeloma cancer cells cultured in hypoxic conditions in vitro and injected into mice were able to spread to the new bone marrow faster than the cells cultured in normoxic conditions.63,75 Also, exposing an orthotopic mouse model of cervical carcinoma to a dozen cycles of 10 minutes 7% O2, which was followed by 10 minutes of air exposure daily, increased the number of lymph node metastases.76 Similar observations were recorded in mice bearing sarcoma tumors, where exposure to acute hypoxia augmented the lung metastases.77
Mechanistically, hypoxia was shown to influence invasive and migratory behavior of cancer cells via EMT, a trans-differentiation of cells in order to acquire plastic and mobile abilities, a process which alters their gene expression prior to migration.78 EMT is physiologically active during embryogenesis and tissue regeneration, as well as in cancerogenesis in many types of solid tumors79 and hematologic malignancies.63 Hypoxia-induced EMT is characterized by a decrease in epithelial-associated gene expression, such as E-cad, β-catenin80 and an increase in mesenchymal-like gene expression, such as N-cad,81 vimentin, SMA,82 and CXCR4.63,75 EMT is promoted by a master regulator TGF-β, also increased by hypoxia, which activates downstream transcription factors such as Smads, Snail, Slug, and Twist, and inhibits expression of E-cad.63,83 Interestingly, radio- and chemoresistance was also shown to be associated with EMT phenotype; expression of Snail and Slug antagonizes p53-mediated apoptosis and promotes resistance to radiation and chemotherapeutic agents such as paclitaxel and cisplatin in ovarian cancer cells.84
Moreover, HIF-1α was shown to be expressed in 90% of human gastric cancer biopsies at the front edge of the invading tumor compared to HIF-1α negative normal tissues.74 HIF inhibition significantly reduced the metastasis of gastric cancer cells in vivo, and HIF deficient cells were less motile, invasive, and adhesive in vitro.74 High involvement of the main hypoxic regulator, HIF-α, in all steps of metastasis led to many trials of inhibiting this molecule to diminish cancer cell trafficking thus reducing metastasis. Inhibition of HIF-1 activity using antisense oligonucleotide (EZN-2968) gave effective results and a safe toxicity in a Phase I clinical trial in metastatic, advanced solid malignancy.85 Targeting hypoxic cells with a pro-drug, activated only in a hypoxic environment, is one of the newest and highly promising strategies to reduce metastasis, currently undergoing phase I/II clinical trials in multiple myeloma, a model of a process of metastasis.86
Apart from targeting HIF-α molecules, another strategy to inhibit metastasis is to target genes downstream of HIF-α. For instance, CAIX is a hypoxia-inducible enzyme widely present in tumors; it is crucial in regulating intra- and extra-cellular pH, thus CAIX promotes survival and invasion of cancer cells.87 It was demonstrated that, inhibition of CAIX decreased tumor growth and metastasis in pre-clinical breast tumor models.87 LOX is another protein elevated in hypoxic human tumor cells (such as breast, and head and neck cancer) and is HIF-α-dependent.61 LOX secreted by tumor cells accumulates at new sites and mediates bone marrow-derived cell recruitment which forms a “pre-metastatic niche” for future metastasis.88 Inhibition of LOX reduced hypoxia-induced recruitment and metastasis in the breast cancer mouse model.88 Another target candidate induced by hypoxia in cancer cells is CXCR4 involved in cell trafficking.89,90 It was demonstrated that metastatic tumor cells expressing high levels of CXCR4 home to tissues rich in its ligand, SDF-1,63,91 and inhibitors of CXCR4/SDF-1 axis resulted in disruption of the metastatic process (Figure 2).90
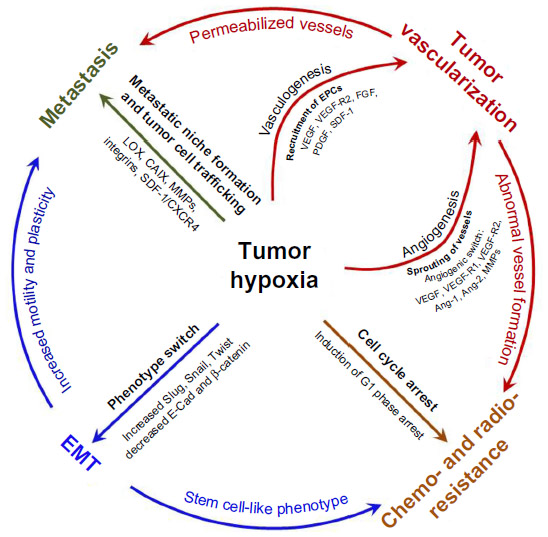 | Figure 2 Hypoxia as a driving force of tumor progression and metastasis. |
Radiation and drug resistance
Resistance of cancer cells to treatment-induced apoptosis is one of the biggest obstacles in cancer therapy.92 A vast number of cancer patients relapse and suffer from recurring tumors as a result of micro-residual disease, the resistant subpopulation of cancer cells, leading to local recurrence and/or metastasis.93 Tumor hypoxia develops due to uncontrollable cell proliferation, altered metabolism, and abnormal tumor blood vessels resulting in reduced transport of oxygen and nutrients.13 Hypoxia is one of the main features of solid tumors and was shown to correlate with poor prognosis of cancer patients.73 While hypoxia is lethal for many cells, a subpopulation of tumor cells is able to not only adapt to hypoxic conditions but also become resistant to chemo- and radiotherapy. The role of hypoxia in the phenomenon of therapy resistance has been acknowledged for at least 60 years.94,95
Hypoxia confers treatment resistance of cancer cells by regulating processes such as i) inducing cell cycle arrest (quiescence), a state of reduced cell proliferation which protects the cells from external stress,96,97 ii) inhibiting apoptosis and senescence of cells, iii) controlling autophagy, p53, and mitochondrial activity.94,97 Apart from cellular adaptations influenced by hypoxia, lowered oxygenation of the tumor tissue confers chemoresistance by affecting iv) drug delivery and cellular uptake through associated acidity and drug efflux pump expression such as P-gp,98 as well as by the v) lack of oxygen required for the cytotoxicity of a number of chemotherapeutics.99
One of the most important parameters of radio-resistance is oxygenation, the state of cell cycle, and the nature of radiation, whether it is gamma, X-ray, neutron radiation, or linear energy transfer including alpha and beta particles.94,96 When oxygen is abundant, the normoxic cells are sensitive to radiation, due to “oxygen fixation” which happens when available molecules of oxygen react with free radicals in DNA generated by ionizing radiation leading to irreversible DNA damage. Whereas, cells irradiated in hypoxic conditions are resistant to death, due to decreased production of DNA radicals (which can be restored “chemical restitution”) caused by reduced generation of ROS and decreased DNA damage.95 The cell cycle phase determines the radiosensitivity of the tumors. It was shown that cells exposed to ionizing radiation are the least sensitive at the end of S phase, less sensitive in the G1 phase, and the most radiosensitive in the G2/M phase where DNA repair mechanisms are the most prone to malfunction.100 Hence, anticancer treatments, both radio- and chemotherapy, preferentially target the bulk of rapidly proliferating tumor cells. On the other hand, the cells which are most resistant to treatment are quiescent, low-proliferating, stem-cell-like cell fractions residing in the most hypoxic region.82,84,97,101 For instance, it was demonstrated that glioblastoma cancer stem cells (CD133+) showed higher DNA repair and decreased apoptosis in response to irradiation, compared to non-stem-cell-like (CD133−) population.94,96,102
Hypoxia causes slow-proliferating stem-cell-like phenotype of cells, decreases senescence, creates chaotic and malfunctioning blood vessels, and augments metastasis, which all together further induces therapy resistance.96 Currently, assessment of tumor oxygenation and HIF expression pattern helps determine tumor chemo- and radio-sensitivity.94,103 It was reported that head-and-neck cancer samples with high expression of HIF-1α and HIF-2α were more resistant to chemotherapy (carboplatin) compared to biopsies with low HIF-α expression which were chemo-sensitive.104 Patients with oropharyngeal cancer demonstrating high expression of HIF-1α had a lower chance to achieve complete remission after irradiation.103 In addition, irradiation was shown to induce HIF-1 activity, leading to production of angiogenic molecules such as VEGF which protects ECs from irradiation-induced apoptosis.105 Therefore HIF-1 represents a valid predictive marker and therapeutic target for manipulation, in combination with chemotherapeutics and radiotherapy, in order to sensitize the cells to treatments.73
As demonstrated by others, inactivation of HIF-1α in mouse embryonic fibroblasts increased their susceptibility to carboplatin and etoposide compared to wild-type, both in vitro and in vivo.106 Similarly, inhibition of HIF-2α with short hairpin RNA reversed the resistance to doxorubicin and etoposide of human clear cell renal cell cancer cells (one of the most resistant tumors) by restoring p53.57 It was reported that treating tumor-bearing mice with HIF-1 inhibitor (YC-1) induced radiation-induced vessel damage.105 Similarly, treatment of glioma, squamous and pancreatic cancer cells with the HIF-1α inhibitor (PX-478) radio-sensitized hypoxic cells.107 Silencing of HIF-1α with siRNA in mouse embryonic fibroblasts increased susceptibility to irradiation;106 and also, HIF-2α inhibition was shown to enhance radiation-induced apoptosis due to HIF-2-mediated increase of p53 activity and accumulation of ROS, thus DNA damage (Figure 2).108
Tumor microenvironment
The tumor would not thrive without the interaction, cross-talk, and support with the tumor microenvironment including cellular components such as stromal cells, immune cells, ECs as well as non-cellular components including ECM, cytokines, and other mediators.109 Thus, in hypoxic tumor tissue, not only cancer cells but also the tumor microenvironment is affected by hypoxia-inducible changes.110
Hypoxia was shown to induce metabolic and molecular changes in ECs, increasing expression of pro-angiogenic molecules, blood vessel formation, and thus providing more oxygen and nutrients for tumor cells. Hypoxia also regulates inflammatory mediators and growth factors, which then stimulate platelet, leukocyte, and smooth muscle cell activity. One of the most significant changes is increase in adhesiveness of ECs to neutrophils facilitating NK cell trafficking and local inflammatory reaction.111 Depending on the duration of oxygen depletion, hypoxia regulates expression of NO synthase expression contributing to vasoconstriction.112 Since blood vessels nourish tumors, targeting ECs will prevent or reverse tumor growth.110,113–115
Stromal cells, on the other hand, facilitate tumor growth and tumor dissemination mostly by regulating cancer cell adhesion and contributing to cell proliferation and survival. It was shown that hypoxia induces stromal cells to produce a number of factors including Ang-2, ANGPTL-4, PDGF, VEGF, SDF-1, LOX, and SCF (KIT-ligand), influencing ECs and EPCs thus promoting new blood vessel formation and lymphangiogenesis. Also, stromal-derived SDF-1 attracts cancer cells and thus facilitates metastasis.115,116
It was demonstrated that hypoxia leads to immune-resistance and immune-suppression, which help tumor cells to escape from immune surveillance.117,118 Some of the immune-suppressive effects include: 1) shedding of immune-recognition molecules by tumor hypoxia, which results in decreased sensitivity to T- and NK-mediated killing;119 2) inhibition of T cells’ and dendritic cells’ maturation and cytokine production;120 3) and promotion of suppressive cells such as regulatory T cells and tumor-associated macrophages, which block immune effector cells.121
Therefore, there is an increasing importance of the hypoxic phenotype of stromal and immune cells in the tumor microenvironment providing non-cancer cells as potential novel targets in the fight against the tumor.110
Conclusion
Pathological hypoxia affects both cancer cells and the tumor microenvironment, and plays a pivotal role in the process of cancer progression and dissemination. Hypoxia regulates tumor neovascularization, metabolism, cell survival, and cell death. In addition, hypoxia contributes to EMT-like cancer cell migration and cancer stem-cell-like properties including resistance to treatment, one of the nightmares in the medical field. Each step of the cancer adaptive processes is controlled by hypoxia-activating transcriptional programs involving HIF, NFκB, PI3K, and MAPK pathways.
Since hypoxia signifies increased tumor progression and aggressiveness hampering patients’ survival, direct and indirect methods of measuring hypoxia combined with clinical observations may help to predict patients’ outcome as well as identify patients who could benefit from hypoxia/HIF-targeted treatments. Better understanding of hypoxic phenomenon and dissecting out the hypoxia-inducible responses and signaling pathways will grant numerous novel targets in the near future.
Disclosure
Dr AK Azab receives research support from Verastem, Selexys, Karyopharm and Cell Works, and is the founder and owner of Targeted Therapeutics LLC and Cellatrix LLC. No potential conflicts of interest were disclosed by B Muz, P de la Puente and F Azab.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | |
Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. 1955;9(4):539–549. | |
Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. 2000;35(2):71–103. | |
Hockel M, Schlenger K, Knoop C, Vaupel P. Oxygenation of carcinomas of the uterine cervix: evaluation by computerized O2 tension measurements. Cancer Res. 1991;51(22):6098–6102. | |
Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 2011;15(6):1239–1253. | |
Vaupel P, Hockel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antiox Redox Signal. 2007; 9(8):1221–1235. | |
Muller M, Padberg W, Schindler E, et al. Renocortical tissue oxygen pressure measurements in patients undergoing living donor kidney transplantation. Anesth Analg. 1998;87(2):474–476. | |
Dings J, Meixensberger J, Jager A, Roosen K. Clinical experience with 118 brain tissue oxygen partial pressure catheter probes. Neurosurgery. 1998;43(5):1082–1095. | |
Hoffman WE, Charbel FT, Edelman G. Brain tissue oxygen, carbon dioxide, and pH in neurosurgical patients at risk for ischemia. Anesth Analg. 1996;82(3):582–586. | |
Cervos-Navarro J, Diemer NH. Selective vulnerability in brain hypoxia. Crit Rev Neurobiol. 1991;6(3):149–182. | |
Wenger RH, Kurtcuoglu V, Scholz CC, Marti HH, Hoogewijs D. Frequently asked questions in hypoxia research. Hypoxia. 2015(3):35–43. | |
Bayer C, Vaupel P. Acute versus chronic hypoxia in tumors: Controversial data concerning time frames and biological consequences. Strahlenther Onkol. 2012;188(7):616–627. | |
Vaupel P, Harrison L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist. 2004;9 Suppl 5:4–9. | |
Chaplin DJ, Olive PL, Durand RE. Intermittent blood flow in a murine tumor: radiobiological effects. Cancer Res. 1987;47(2):597–601. | |
Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22(2):177–180. | |
Rouschop KM, Ramaekers CH, Schaaf MB, et al. Autophagy is required during cycling hypoxia to lower production of reactive oxygen species. Radiother Oncol. 2009;92(3):411–416. | |
Hsieh CH, Lee CH, Liang JA, Yu CY, Shyu WC. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signal transduction activity. Oncol Rep. 2010; 24(6):1629–1636. | |
Hsieh CH, Shyu WC, Chiang CY, Kuo JW, Shen WC, Liu RS. NADPH oxidase subunit 4-mediated reactive oxygen species contribute to cycling hypoxia-promoted tumor progression in glioblastoma multiforme. PloS One. 2011;6(9):e23945. | |
Kato Y, Yashiro M, Fuyuhiro Y, et al. Effects of acute and chronic hypoxia on the radiosensitivity of gastric and esophageal cancer cells. Anticancer Res. 2011;31(10):3369–3375. | |
Chaplin DJ, Durand RE, Olive PL. Acute hypoxia in tumors: implications for modifiers of radiation effects. Int J Radiat Oncol Biol Phys. 1986;12(8):1279–1282. | |
Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009; 29(10):2570–2581. | |
Rofstad EK, Gaustad JV, Egeland TA, Mathiesen B, Galappathi K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int J Cancer. 2010;127(7):1535–1546. | |
Chan N, Koritzinsky M, Zhao H, et al. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res. 2008;68(2):605–614. | |
Luoto KR, Kumareswaran R, Bristow RG. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013;4(1):5. | |
Kondo A, Safaei R, Mishima M, Niedner H, Lin X, Howell SB. Hypoxia-induced enrichment and mutagenesis of cells that have lost DNA mismatch repair. Cancer Res. 2001;61(20):7603–7607. | |
Pires IM, Bencokova Z, Milani M, et al. Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 2010;70(3):925–935. | |
Brurberg KG, Graff BA, Olsen DR, Rofstad EK. Tumor-line specific pO(2) fluctuations in human melanoma xenografts. Int J Radiat Oncol Biol Phys. 2004;58(2):403–409. | |
Kimura H, Braun RD, Ong ET, et al. Fluctuations in red cell flux in tumor microvessels can lead to transient hypoxia and reoxygenation in tumor parenchyma. Cancer Res. 1996;56(23):5522–5528. | |
Agani F, Jiang BH. Oxygen-independent regulation of HIF-1: novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr Cancer Drug Targets. 2013;13(3):245–251. | |
Courtnay R, Ngo DC, Malik N, Ververis K, Tortorella SM, Karagiannis TC. Cancer metabolism and the Warburg effect: the role of HIF-1 and PI3K. Mol Biol Rep. 2015;42(4):841–851. | |
Seta KA, Spicer Z, Yuan Y, Lu G, Millhorn DE. Responding to hypoxia: lessons from a model cell line. Sci STKE. 2002;2002(146):re11. | |
Sanchez A, Tripathy D, Yin X, et al. p38 MAPK: a mediator of hypoxia-induced cerebrovascular inflammation. J Alzheimers Dis. 2012;32(3):587–597. | |
Minet E, Arnould T, Michel G, et al. ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett. 2000;468(1):53–58. | |
Koong AC, Chen EY, Giaccia AJ. Hypoxia causes the activation of nuclear factor kappa B through the phosphorylation of I kappa B alpha on tyrosine residues. Cancer Res. 1994;54(6):1425–1430. | |
Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133(3):403–414. | |
Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992; 12(12):5447–5454. | |
Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–1340. | |
Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. | |
van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J. 2008;412(3):477–484. | |
Kilic-Eren M, Boylu T, Tabor V. Targeting PI3K/Akt represses Hypoxia inducible factor-1alpha activation and sensitizes Rhabdomyosarcoma and Ewing’s sarcoma cells for apoptosis. Cancer Cell Int. 2013;13:36. | |
Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999;274(46):32631–32637. | |
Royds JA, Dower SK, Qwarnstrom EE, Lewis CE. Response of tumour cells to hypoxia: role of p53 and NFkB. Mol Pathol. 1998;51(2):55–61. | |
Tafani M, Pucci B, Russo A, et al. Modulators of HIF1alpha and NFkB in Cancer Treatment: Is it a Rational Approach for Controlling Malignant Progression? Front Pharmacol. 2013;4:13. | |
Chandel NS, McClintock DS, Feliciano CE, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275(33):25130–25138. | |
Movafagh S, Crook S, Vo K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: new developments in an old debate. J Cell Biochem. 2015;116(5):696–703. | |
Mateo J, Garcia-Lecea M, Cadenas S, Hernandez C, Moncada S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem J. 2003;376(Pt 2):537–544. | |
Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33(4):207–214. | |
Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8(12):967–975. | |
Muz B, de la Puente P, Azab F, Luderer M, Azab AK. The role of hypoxia and exploitation of the hypoxic environment in hematologic malignancies. Mol Cancer Res. 2014;12(10):1347–1354. | |
Siemeister G, Weindel K, Mohrs K, Barleon B, Martiny-Baron G, Marme D. Reversion of deregulated expression of vascular endothelial growth factor in human renal carcinoma cells by von Hippel-Lindau tumor suppressor protein. Cancer Res. 1996;56(10):2299–2301. | |
Zagzag D, Krishnamachary B, Yee H, et al. Stromal cell-derived factor-1alpha and CXCR4 expression in hemangioblastoma and clear cell-renal cell carcinoma: von Hippel-Lindau loss-of-function induces expression of a ligand and its receptor. Cancer Res. 2005;65(14):6178–6188. | |
Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12(2):149–162. | |
Iliopoulos O, Levy AP, Jiang C, Kaelin WG Jr, Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A. 1996;93(20):10595–10599. | |
Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123(9):3664–3671. | |
Guo K, Searfoss G, Krolikowski D, et al. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001;8(4):367–376. | |
An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1 alpha. Nature. 1998;392(6674):405–408. | |
Roberts AM, Watson IR, Evans AJ, Foster DA, Irwin MS, Ohh M. Suppression of hypoxia-inducible factor 2alpha restores p53 activity via Hdm2 and reverses chemoresistance of renal carcinoma cells. Cancer Res. 2009;69(23):9056–9064. | |
Krishnamachary B, Zagzag D, Nagasawa H, et al. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006;66(5):2725–2731. | |
Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425(6955):307–311. | |
Grabmaier K, MC AdW, Verhaegh GW, Schalken JA, Oosterwijk E. Strict regulation of CAIX(G250/MN) by HIF-1alpha in clear cell renal cell carcinoma. Oncogene. 2004;23(33):5624–5631. | |
Erler JT, Bennewith KL, Nicolau M, et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440(7088):1222–1226. | |
Krishnamachary B, Berg-Dixon S, Kelly B, et al. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003;63(5):1138–1143. | |
Azab AK, Hu J, Quang P, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119(24):5782–5794. | |
Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res. 2001;49(3):507–521. | |
Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438(7070):932–936. | |
de la Puente P, Muz B, Azab F, Azab AK. Cell Trafficking of Endothelial Progenitor Cells in Tumor Progression. Clin Cancer Res. 2013; 19(3):3360–3368. | |
Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. 2011;2(12):1117–1133. | |
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. | |
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–1186. | |
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–2342. | |
Mountzios G, Pentheroudakis G, Carmeliet P. Bevacizumab and micrometastases: revisiting the preclinical and clinical rollercoaster. Pharmacol Ther. 2014;141(2):117–124. | |
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. | |
Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29(5):625–634. | |
Rohwer N, Lobitz S, Daskalow K, et al. HIF-1alpha determines the metastatic potential of gastric cancer cells. Br J Cancer. 2009; 100(5):772–781. | |
Muz B, de la Puente P, Azab F, Ghobrial IM, Azab AK. Hypoxia promotes dissemination and colonization in new bone marrow niches in Waldenstrom macroglobulinemia. Mol Cancer Res. 2015;13(2):263–272. | |
Cairns RA, Hill RP. Acute hypoxia enhances spontaneous lymph node metastasis in an orthotopic murine model of human cervical carcinoma. Cancer Res. 2004;64(6):2054–2061. | |
Cairns RA, Kalliomaki T, Hill RP. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHT murine tumors. Cancer Res. 2001;61(24):8903–8908. | |
Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–142. | |
Mulholland DJ, Kobayashi N, Ruscetti M, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012;72(7):1878–1889. | |
Kim K, Lu Z, Hay ED. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol Int. 2002; 26(5):463–476. | |
Hsu M, Andl T, Li G, Meinkoth JL, Herlyn M. Cadherin repertoire determines partner-specific gap junctional communication during melanoma progression. J Cell Sci. 2000;113(Pt 9):1535–1542. | |
Manotham K, Tanaka T, Matsumoto M, et al. Transdifferentiation of cultured tubular cells induced by hypoxia. Kidney Int. 2004;65(3):871–880. | |
Tian M, Neil JR, Schiemann WP. Transforming growth factor-beta and the hallmarks of cancer. Cell Signal. 2011;23(6):951–962. | |
Tamara Marie-Egyptienne D, Lohse I, Hill RP. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: Potential role of hypoxia. Cancer Lett. 2013;341(1):63–72. | |
Greenberger LM, Horak ID, Filpula D, et al. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol Cancer Ther. 2008;7(11):3598–3608. | |
Hu J, Handisides DR, Van Valckenborgh E, et al. Targeting the multiple myeloma hypoxic niche with TH-302, a hypoxia-activated prodrug. Blood. 2010;116(9):1524–1527. | |
Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011;71(9):3364–3376. | |
Erler JT, Bennewith KL, Cox TR, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15(1):35–44. | |
Azab AK, Azab F, Blotta S, et al. RhoA and Rac1 GTPases play major and differential roles in stromal cell-derived factor-1-induced cell adhesion and chemotaxis in multiple myeloma. Blood. 2009; 114(3):619–629. | |
Azab AK, Runnels JM, Pitsillides C, et al. CXCR4 inhibitor AMD3100 disrupts the interaction of multiple myeloma cells with the bone marrow microenvironment and enhances their sensitivity to therapy. Blood. 2009;113(18):4341–4351. | |
Azab AK, Weisberg E, Sahin E, et al. The influence of hypoxia on CML trafficking through modulation of CXCR4 and E-cadherin expression. Leukemia. 2013;27(4):961–964. | |
Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11(6):393–410. | |
Munshi NC, Anderson KC. Minimal residual disease in multiple myeloma. J Clin Oncol. 2013;31(20):2523–2526. | |
Rohwer N, Cramer T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Updat. 2011;14(3):191–201. | |
Gray LH, Conger AD, Ebert M, Hornsey S, Scott OC. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol. 1953;26(312):638–648. | |
Vaupel P, Kelleher DK, Hockel M. Oxygen status of malignant tumors: pathogenesis of hypoxia and significance for tumor therapy. Semin Oncol. 2001;28(2 Suppl 8):29–35. | |
Das B, Tsuchida R, Malkin D, Koren G, Baruchel S, Yeger H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells. 2008;26(7):1818–1830. | |
Abraham J, Salama NN, Azab AK. The role of P-glycoprotein in drug resistance in multiple myeloma. Leuk Lymphoma. 2015;56(1):26–33. | |
Cosse JP, Michiels C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med Chem. 2008;8(7):790–797. | |
Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59(4):928–942. | |
Muz B, de la Puente P, Azab F, Luderer M, Azab AK. Hypoxia promotes stem cell-like phenotype in multiple myeloma cells. Blood Cancer J. 2014;4:e262. | |
Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. | |
Aebersold DM, Burri P, Beer KT, et al. Expression of hypoxia-inducible factor-1alpha: a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001; 61(7):2911–2916. | |
Koukourakis MI, Giatromanolaki A, Sivridis E, et al. Hypoxia-inducible factor (HIF1A and HIF2A), angiogenesis, and chemoradiotherapy outcome of squamous cell head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2002;53(5):1192–1202. | |
Moeller BJ, Cao Y, Vujaskovic Z, Li CY, Haroon ZA, Dewhirst MW. The relationship between hypoxia and angiogenesis. Semin Radiat Oncol. 2004;14(3):215–221. | |
Unruh A, Ressel A, Mohamed HG, et al. The hypoxia-inducible factor-1 alpha is a negative factor for tumor therapy. Oncogene. 2003; 22(21):3213–3220. | |
Schwartz DL, Powis G, Thitai-Kumar A, et al. The selective hypoxia inducible factor-1 inhibitor PX-478 provides in vivo radiosensitization through tumor stromal effects. Mol Cancer Ther. 2009; 8(4):947–958. | |
Bertout JA, Majmundar AJ, Gordan JD, et al. HIF2alpha inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc Natl Acad Sci U S A. 2009;106(34):14391–14396. | |
Sounni NE, Noel A. Targeting the tumor microenvironment for cancer therapy. Clin Chem. 2013;59(1):85–93. | |
Casazza A, Di Conza G, Wenes M, Finisguerra V, Deschoemaeker S, Mazzone M. Tumor stroma: a complexity dictated by the hypoxic tumor microenvironment. Oncogene. 2014;33(14):1743–1754. | |
Baudry N, Danialou G, Boczkowski J, Vicaut E. In vivo study of the effect of systemic hypoxia on leukocyte-endothelium interactions. Am J Respir Crit Care Med. 1998;158(2):477–483. | |
Kourembanas S, Bernfield M. Hypoxia and endothelial-smooth muscle cell interactions in the lung. Am J Respir Crit Care Med. 1994; 11(4):373–374. | |
Michiels C, Arnould T, Remacle J. Endothelial cell responses to hypoxia: initiation of a cascade of cellular interactions. Biochim Biophys Acta. 2000;1497(1):1–10. | |
Verdegem D, Moens S, Stapor P, Carmeliet P. Endothelial cell metabolism: parallels and divergences with cancer cell metabolism. Cancer Metab. 2014;2:19. | |
Azab AK, Sahin I, Moschetta M, et al. CXCR7-dependent angiogenic mononuclear cell trafficking regulates tumor progression in multiple myeloma. Blood. 2014;124(12):1905–1914. | |
Semenza GL. Cancer-stromal cell interactions mediated by hypoxia-inducible factors promote angiogenesis, lymphangiogenesis, and metastasis. Oncogene. 2013;32(35):4057–4063. | |
Lee CT, Mace T, Repasky EA. Hypoxia-driven immunosuppression: a new reason to use thermal therapy in the treatment of cancer? Int J Hyperthermia. 2010;26(3):232–246. | |
Noman MZ, Hasmim M, Messai Y, et al. Hypoxia: a key player in anti-tumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol Cell Physiol. 2015;309(9):C569–C579. | |
Siemens DR, Hu N, Sheikhi AK, et al. Hypoxia increases tumor cell shedding of MHC class I chain-related molecule: role of nitric oxide. Cancer Res. 2008;68(12):4746–4753. | |
Yang M, Ma C, Liu S, et al. Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology. 2009;128 (1 Suppl):e237–e249. | |
Sitkovsky MV. T regulatory cells: hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol. 2009; 30(3):102–108. | |
Rampling R, Cruickshank G, Lewis AD, Fitzsimmons SA, Workman P. Direct measurement of pO2 distribution and bioreductive enzymes in human malignant brain tumors. Int J Radiat Oncol Biol Phys. 1994;29(3):427–431. | |
Vaupel P, Schlenger K, Knoop C, Hockel M. Oxygenation of human tumors: evaluation of tissue oxygen distribution in breast cancers by computerized O2 tension measurements. Cancer Res. 1991; 51(12):3316–3322. | |
Lawrentschuk N, Poon AM, Foo SS, et al. Assessing regional hypoxia in human renal tumours using 18F-fluoromisonidazole positron emission tomography. BJU Int. 2005;96(4):540–546. | |
Leary TS, Klinck JR, Hayman G, Friend P, Jamieson NV, Gupta AK. Measurement of liver tissue oxygenation after orthotopic liver transplantation using a multiparameter sensor. A pilot study. Anaesthesia. 2002;57(11):1128–1133. | |
Brooks AJ, Eastwood J, Beckingham IJ, Girling KJ. Liver tissue partial pressure of oxygen and carbon dioxide during partial hepatectomy. Br J Anaesth. 2004;92(5):735–737. | |
Le QT, Chen E, Salim A, et al. An evaluation of tumor oxygenation and gene expression in patients with early stage non-small cell lung cancers. Clin Cancer Res. 2006;12(5):1507–1514. | |
Koong AC, Mehta VK, Le QT, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys. 2000;48(4):919–922. | |
Graffman S, Bjork P, Ederoth P, Ihse I. Polarographic pO2 measurements of intra-abdominal adenocarcinoma in connection with intraoperative radiotherapy before and after change of oxygen concentration of anaesthetic gases. Acta Oncol. 2001;40(1):105–107. | |
Kallinowski F, Eble MJ, Buhr HJ, Wannenmacher M, Herfarth C. Intraoperative radiotherapy for primary and recurrent rectal cancer. Eur J Surg Oncol. 1995;21(2):191–194. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.