")
Back to Journals » Drug Design, Development and Therapy » Volume 16
The Role of Histone H3 Methylation in Acute Kidney Injury
Authors Zhao YB, Wei W, Lin XX, Chai YF, Jin H
Received 30 May 2022
Accepted for publication 27 July 2022
Published 2 August 2022 Volume 2022:16 Pages 2453—2461
DOI https://doi.org/10.2147/DDDT.S376673
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Yi-Bo Zhao, Wei Wei, Xiao-Xi Lin, Yan-Fen Chai, Heng Jin
Department of Emergency Medicine, Tianjin Medical University General Hospital, Tianjin, 300000, People’s Republic of China
Correspondence: Heng Jin; Yan-Fen Chai, Department of Emergency Medicine, Tianjin Medical University General Hospital, Tianjin, 300000, People’s Republic of China, Email [email protected]; [email protected]
Abstract: Acute kidney injury (AKI) is a clinical syndrome in which kidney function declines sharply due to various reasons. Although the morbidity and mortality of AKI are high, the mechanism of occurrence and development of AKI has not been fully elucidated, and precise prevention and treatment measures are lacking. Epigenetics is a branch of genetics that provides a new perspective to explore the pathophysiology of AKI and renal repair. A large amount of literature shows that the methylation mechanism of H3 in histones is closely related to the development of kidney diseases. The sorting out of histone H3 methylation mechanism in AKI and kidney repair can help understand the pathophysiological process of the disease more deeply. It may also provide new ideas for diagnosing and treating of the disease.
Keywords: kidney, Acute kidney injury, histone methylation, epigenetic
Introduction
AKI is a clinical syndrome characterized by a rapid decline in renal function due to several reasons. It is manifested by a quick increase in serum creatinine, a decrease in urine output, or both.1 AKI is a serious threat to human health. Its high treatment costs and many sequelae have caused a heavy economic burden on society: about 1.7 million people die from AKI annually in the world.2,3 The main pathological features of AKI include sublethal and lethal damage of the renal tubular cells,4 plasma membrane is the main damage site.5 The kidney undergoes a repair response involving epithelial cell dedifferentiation, proliferation, and re-differentiation following an injury. However, during severe or persistent injury, the kidney resorts to maladaptive repair characterized by fibrosis, vascular sparing, tubular loss, glomerulosclerosis, and chronic inflammatory infiltration of the kidney.6,7 The kidney undergoes pathological processes such as G2/M cell cycle arrest, cellular senescence, production of pro-fibrotic cytokines, activation of pericytes and interstitial myofibroblasts.6,9 The events ultimately result in chronic kidney disease (CKD) and end-stage renal disease.7,8 Here, epigenetic mechanisms such as histone modifications play essential roles in the pathology of AKI and kidney repair10,11 (Figure 1). The regulatory mechanisms of AKI pathology and renal repair are complex. Studying the methylation of histone H3 amino acid residues can provide new perspectives for the disease.
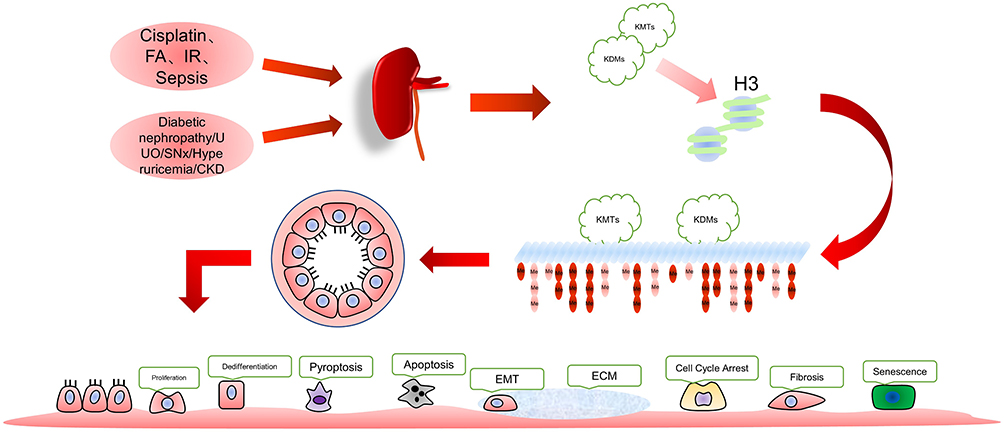 |
Figure 1 Various factors modulate renal cell changes by altering the methylation status of histone H3. |
Acute injury caused by cisplatin, folic acid, ischemia-reperfusion injury (IRI)and sepsis, and chronic kidney injury due to high glucose, Unilateral Ureteral Obstruction (UUO), subtotal nephrectomy (SNx), hyperuricemia, etc., affect the methylation level of histone H3 in the kidney. Intervention of methylation at this site can effectively modulate AKI prognosis.
Histone Methylation and Demethylation in Renal Injury
H3k27
The protein Enhancer of Zeste Homolog 2(EZH2) is a methyltransferase that catalyzes the trimethylation of the 27th amino acid (lysine) of the H3 histone (H3K27) and 3-deazaneplanocin A(3-DZNep) is an inhibitor of EZH2. Blocking EZH2 with 3-DZNep attenuates the renal tubular cell death in the proximal tubule cells after renal ischemia-reperfusion injury or FA(folic acid)-induced injury in experimental mice.12 Later studies have shown that EZH2 inhibition inactivates p38, decreasing the level of active caspase-3 and pro-inflammatory molecules and reducing cell apoptosis.13 Cisplatin, a chemotherapy drug, is one of the common causes of AKI. Animal studies or cell experiments have confirmed the following: 1. Cisplatin mediates the increased expression of the trimethylated form of histone H3 protein at the 27th amino acid (lysine) (H3K27me3) in the kidney cells. 2. 3-DZNep demonstrates a protective effect on the renal tubular cells in response to the damage induced by cisplatin. 3. The basal concentration of EZH2 has a protective effect on the kidney cells.14 Interestingly, cisplatin did not mediate the up regulation of EZH2 in renal cells in vitro, and 3-DZNep did not change the EZH2 level in the cells after the intervention. However, in animal experiments, cisplatin administration increased EZH2, and 3-DZNep dependently inhibited EZH2 expression. Thus, the protective effect of 3-DZNep on the cisplatin-mediated renal injury was not achieved by EZH2 and/or H3K27 trimethylation.14 At the same time, 3-DZNep treatment did not activate ERK1/2, p38 and JNK1/2. Instead, it inhibited cisplatin-induced renal tubular epithelial cell (RTEC) apoptosis and AKI through an E-cadherin dependent mechanism.14 E-cadherin is the main component of the tubular adhesion protein. It maintains intercellular contact and cell polarity in epithelial tissue, and has a protective effect on cisplatin-induced AKI.15 H3K27me3 and H3K9me3 participate in regulating its expression regulation.16,17 According to evidence, 3-DZNep inhibits H3K27me3 and reduces the levels of H3K9 me3, H3K36 me3 and H3K4 me3.18 Thus, in cisplatin- induced AKI, H3K9 me3 play a crucial role in regulating the mechanism of 3-DZNep on the repair of E-cadherin. Concerning cisplatin-related AKI, new in vitro studies on the NRK-52E cells (rat RTECs) have demonstrated that EZH2 inhibition increases the transcription by reducing H3K27 me3 in the DEPTOR promoter region. This event further inhibits the activities of mTORC1 and mTORC2, down-regulates the expression of the apoptosis inhibitory genes, and finally leads to renal tubular cell apoptosis. Meanwhile, the inhibition of EZH2 aggravates cisplatin-induced renal tubular cell injury by inactivating the mTOR complex.19 The mechanism of H3K27 me3 in cisplatin-related AKI has been investigated further. New studies indicated that EZH2 inhibition reduces NOX4, attenuates ROS-mediated pyrolysis and protects the kidneys by regulating oxidative stress.20 These studies provide new ideas for understanding the mechanism of EZH2 in AKI.
H3k4
In sepsis induced AKI, the level of H3K4 me3 decreases in kidneys, and the effects of anti-anxiety, sedation and analgesia are improved. α Dexmedetomidine (DEX), an α2-adrenergic receptor agonist, inhibits the histone demethylase KDM5A, increasing the level of H3K4 me3 and protecting renal function. Thus, DEX inhibits histone deacetylase (histone deacetylase, HDAC) by participating in apparent acetylation. Further, DEX modulates AKI through histone methylation.21 DEX also improves renal IRI (ischemia-reperfusion injury) and its inflammatory responses by inhibiting the activation of T cells and the level of inflammatory factors.13,22 However, the relationship between the level of histone methylation and the levels of immune cells and inflammatory factors is not yet clear. The exploration of this aspect may illuminate the role of epigenetics in kidney disease. In addition, methylation of H3K4 regulates the HMG-CoA reductase activity during AKI and abnormal cholesterol metabolism.23,24 Table 1 shows the changes and roles of histone methylation targets in injury.
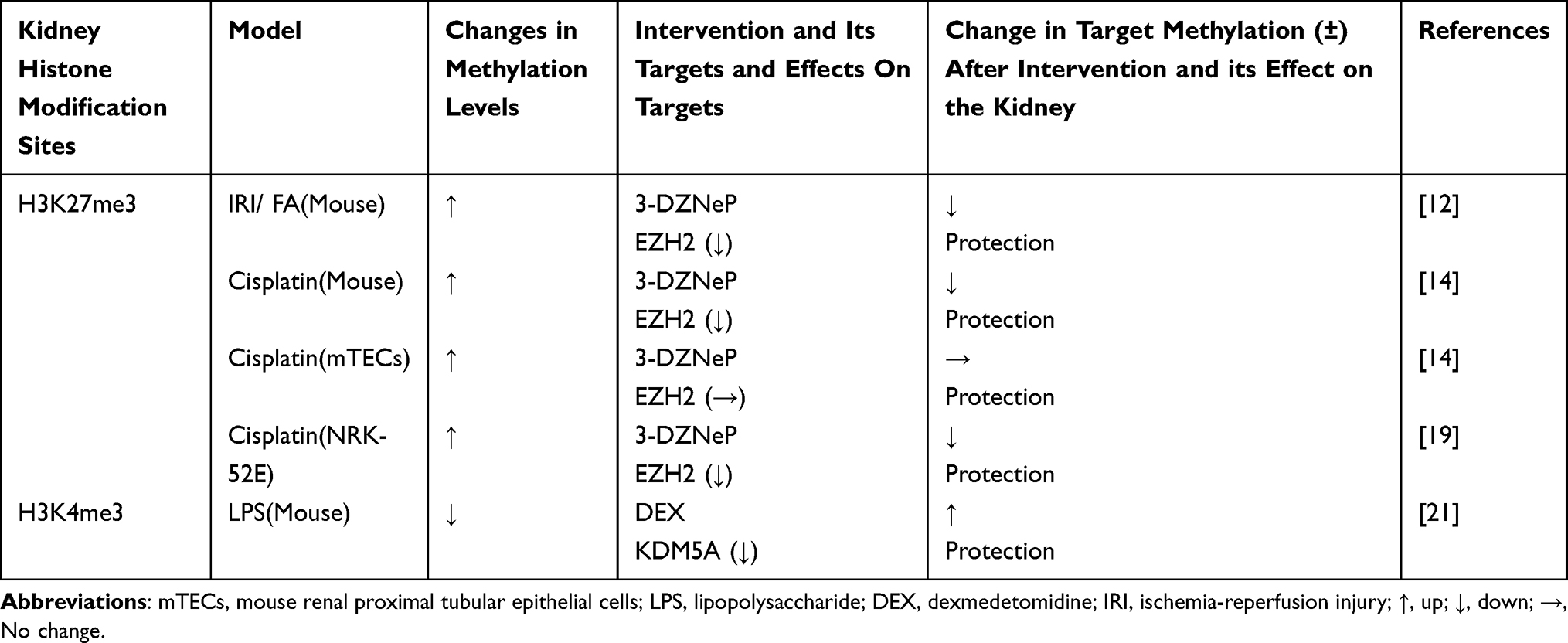 |
Table 1 Changes and Roles of Histone Methylation Targets in Injury |
In addition to regulating the stage of kidney injury, changes in histone methylation levels also participate in post-injury repair processes. The regulation of the methylation level of histone H3 can improve the repair, which provides new thinking for blocking pathological processes such as fibrosis caused by incomplete repair. In addition to H3K27 and H3K4, H3K79, H3K9, and H3K36 are also involved.
Histone Methylation and Demethylation in Kidney Repair
H3k27
In the pathological process of kidney repair, H3K27me demonstrates similar effects in different kidney cells. In renal fibroblasts from UUO-treated mice and humans with CKD, EZH2 and H3K27 me3 are highly expressed. Moreover, inhibiting the increase in EZH2 and H3K27 me3 reduces the activation of the renal fibroblasts induced by TGFβ-1 and reduces the kidney fibrosis progress after UUO injury.25 When the specific Jumonji domain-containing protein-3(JMJD3) (a lysine-specific demethylase of H3K27 me3) is inhibited, the renal interstitial fibroblasts are activated and renal fibrosis after UUO and SNx is aggravated.26 Thus, the H3K27 me3 level increase in the kidney after UUO injury, promoting fibrosis after incomplete repair. New studies show that H3K27 me3 regulates the dedifferentiation of the glomerular podocytes in kidney injury caused by diabetic nephropathy, adriamycin kidney and SNx, and protects the kidneys. The loss of H3K27 me3 promotes the dedifferentiation of podocytes after mitosis and accelerates glomerular disease. Moreover, the inhibition of lysine-specific demethylase may help improve the progression of glomerular disease.27 Subsequent studies confirmed that the H3K27me demethylase KDM6B regulates in vitro podocyte differentiation. The podocytes in the glomeruli of patients with renal disease exhibited increased KDM6B content and decreased H3K27me3 levels,28 consistent with the previous results. EZH2 inhibition promotes kidney oxidative stress and fibrosis induced by high glucose.29 In the pathology of UUO injury, inhibition of EZH2 minimizes renal fibrosis by reducing renal tubular cell arrest in the G2/M phase and epithelial-mesenchymal transition (EMT).25,30 Moreover, EZH2 is inhibited by TGF-β through miR-101b.31 In the pathology of CKD induced by hyper uric acid, EZH2 blockade reduces the activation and proliferation of renal interstitial fibroblasts, renal tubular cell damage, apoptosis, and renal interstitial fibroblast activation and extracellular matrix (ECM) deposition.32 The upregulation of aging-related H3K27 me3 in elderly mice, is primarily due to reduced JMJD3 expression in aging kidneys. Klotho is a one-way transmembrane protein highly expressed in the kidney, H3K27me3 directly binds to the Klotho promoter and inhibits Klotho gene expression.33 It prevents and alleviates damage, promotes recovery, and inhibits fibrosis to reduce repair after kidney damage through exogenous supplementation or stimulation of endogenous Klotho recovery.34 Briefly, in repair after kidney damage induced by factors such as folic acid, cisplatin, UUO, SNx, high glucose and aging, the presence of H3K27me3 promotes fibrosis and deteriorates the disease; inhibiting H3K27me3 has a protective effect on the kidneys. However, sepsis is one of the critical causes of AKI, and the role of trimethylation at H3K27 in kidney repair after AKI has not been elucidated.
H3k79
Disruptor of telomeric silencing 1-like (DOT1L) protein is the methyltransferase of H3K79.35 It regulates cell cycle and cell senescence,36,37 and increases the expression of the senescence-associated secretory phenotype (SASP).37 Deleting the DOT1L protein by gene editing increases the renal collecting duct intercalated cell/primary cell ratio and urine volume and decreases urinary osmolality, but electrolyte metabolism remains normal in the mice.38 The targeted destruction of DOT1L, the progenitor cell of the nephron, leads to congenital renal dysplasia. The kidney with a DOT1L mutation exhibits the phenotype of congenital nephron defect and cystic dysplasia nephropathy, indicating the role of DOT1L in the maintenance and differentiation of the progenitor cells into epithelial nephrons.39 A study on the repair after kidney injury found that the G2/M phase tubular epithelial cell cycle arrest mediates renal fibrosis after injury.9 Inhibition of DOT1L can blocks renal interstitial fibroblast activation and EMT along with the injury-induced inhibition of the G2/M phase cell cycle arrest. Additionally, blockade of DOT1L reduces the expression of Snail, Twist and Notch1; and inhibits injury to the kidney. Inhibition of DOT1L also inactivates several pro-fibrotic signaling molecules like Smad3, epidermal growth factor receptor, platelet-derived growth factor receptor, signal transducer and activator of transcription 3, protein kinase B, and NF-κB. At the same time, DOT1L inhibition increases the expression of PTEN (phosphatase and tensin homolog) and prevents the reduction in the renoprotective factors Klotho and Smad7.40 In addition to the above methods, inhibition of DOT1L alleviates oxidative stress by blocking the PI3K/AKT pathway, reducing renal ischemia-reperfusion injury and post-injury fibrosis.41 In conclusion, Dot1L is indispensable for kidney growth and development, but its inhibition in adult mice ameliorates fibrosis after kidney injury.
H3k4
The absence of H3K4 methylation has no negative impact on the kidney development and function in terms of growth and regeneration in young mice. However, the lack of H3K4 methylation results in the appearance of pathophysiological changes similar to chronic kidney disease in the kidney of old mice; these include podocyte death, reduction, interstitial fibrosis, and proteinuria.42 The methylation of histone H3K4 is also related to the regeneration of RTECs: the renal function of the mice without H3K4 methylation is normal. However, after injury, the renal tubular repair and regeneration ability is severely damaged, mainly causing fibrosis and scars repair.43 In fibrosis, TGF-β1 increases the methylation of H3K4 at the p21 promoter by up-regulating the H3K4 methyltransferase SET domain-containing lysine methyltransferase7/9 (SET7/9). TGF-β1 also promotes its gene expression and increases ECM gene expression.44,45 H3k4me1 was up-regulated in UUO injury, which has been observed in rat RTECS and renal interstitial fibroblasts as well as in vivo experiments in mice. Inhibiting SET7/9 and reducing H3K4me1 alleviates renal fibrosis. The SET7/9 expression is affected by TGF β1 -Smad3 pathway regulation; correspondingly, SET7/9 expression positively correlates with the degree of interstitial fibrosis in human kidney biopsy specimens from patients with IgA nephropathy and membranous nephropathy.46 The histone H3K4 me3 is highly expressed in the nuclei of podocytes, mesangial cells, and endothelial cells of patients with membranous nephropathy.47 To investigate the role of this target methylation in podocytes, mice were treated with lipopolysaccharide. The histone H3K4 me3 increased in the podocytes; there was an increase in podocyte swelling, serum creatinine, and urine albumin. The inhibition of H3K4 methyltransferase reduces H3K4-me3 restores the up-regulation of cathepsin L, decreases the synaptopod in protein and the podocyte swelling induced by lipopolysaccharide.47 Briefly, the presence of a certain amount of H3K4 methylation positively affects kidney repair after injury, but increased H3K4 methylation after injury participates in promoting kidney fibrosis.
H3k9
The role of methylation of H3K9 in kidney repair completely various among different research groups. Initial studies reported a TGF-β1-induced reduction in the H3K9me2/3 level of ECM-related gene promoters in RMC.44 However, subsequent studies identified TGF-β1-induced upregulation of H3K9 methyltransferase G9a expression via Smad3 in the kidney of mice after UUO. Also, TGF-β1 inhibits H3K9 methyltransferase G9a reduces H3K9 me1, and inhibits renal fibrosis. Other reports indicated that the G9a immunostaining region positively correlates with H3K9me1 levels and fibrosis markers in human kidney biopsies.48 The study also showed that H3K9me1, which reduces the Klotho promoter region, retains klotho expression.48 This observation, provides a new perspective on the apparent regulation of Klotho protein expression. New research shows that G9a inhibits the transcription of the histone deacetylase SIRT1 through H3K9 me2.49 Moreover, SIRT1 is regulated by Klotho expression.50,51 Few studies have dealt with H3K9 methylation, and the contradictory of these studies are not well explanation. More in-depth studies may prompt additional information for the analysis of kidney repair after injury.
H3k36
Few studies have investigated the role of H3K36 in kidney repair. The lysine methyltransferase SET and MYND domain protein 2 (SMYD2) mediates the methyltransferase H3K36 me3. The inhibition of this protein prevents renal fibrosis and inhibits the activation and proliferation of renal interstitial fibroblasts. SMYD2 inhibition also hinders the transformation of epithelial cells to the fibrogenic phenotype, and prevents the down-regulation of the renal protective factor Smad7 under in vivo and in vitro conditions.52 The role of H3K36 methylation in kidney repair needs further exploration.
Among the drugs used in the clinical treatment of kidney diseases, traditional Chinese medicine prescription QianyangYuyin granules are found to regulate the level of H3K4me3, inhibit the proliferation of kidney epithelial cells, and prevent kidney damage caused by hypertension.53 Table 2 shows the changes and roles of histone methylation targets in post-injury repair.
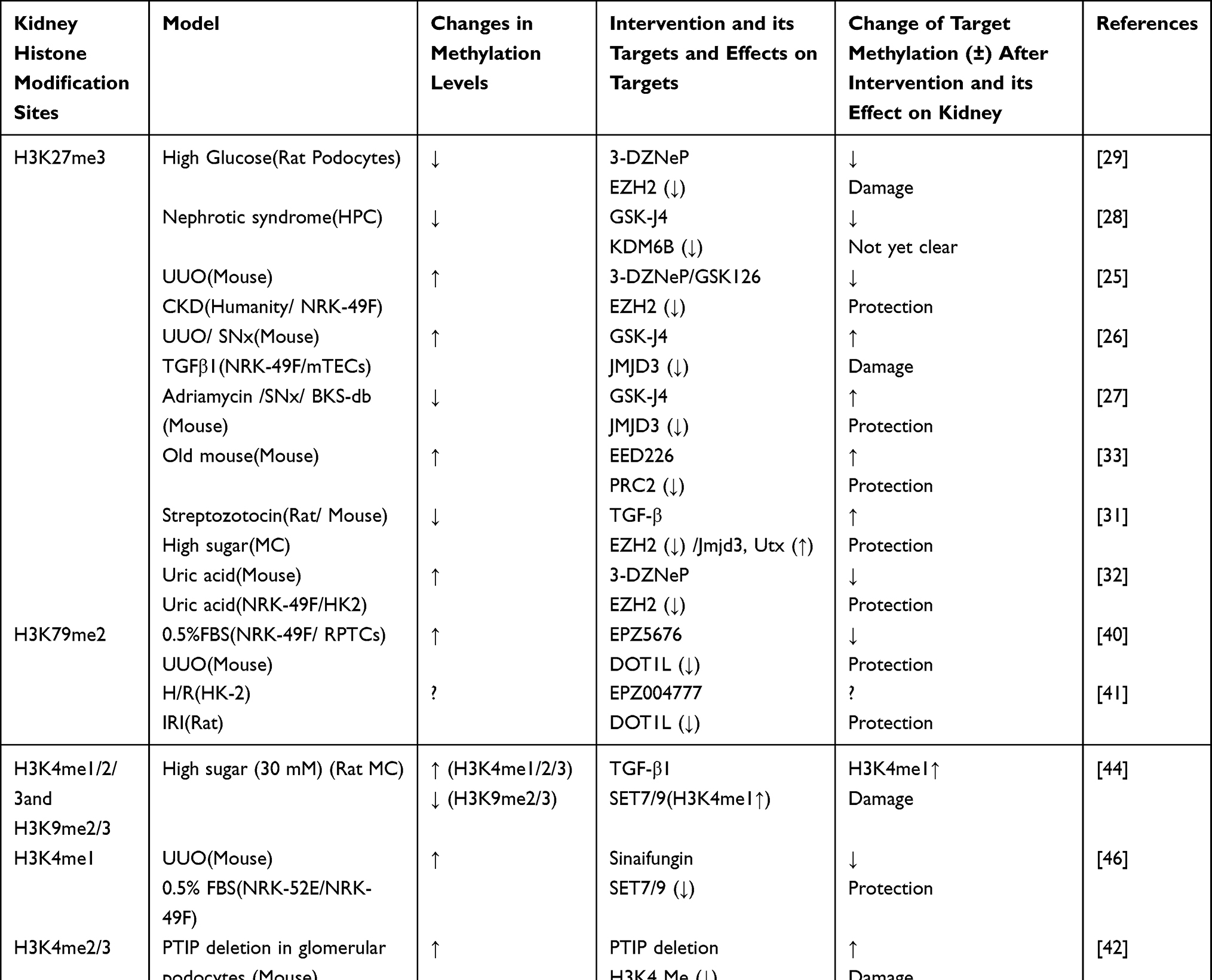 |
Table 2 Changes and Roles of Histone Methylation Targets in Post-Injury Repair |
Conclusion
Epigenetics is an emerging discipline that provides new ideas for understanding AKI and kidney repair. Histone H3 methylation is an essential regulatory point, and its research has produced many new results. A certain amount of methylation of amino acid residues on histone H3 is critical for healthy kidney development. However, methylation levels are generally elevated in sepsis-induced-kidney injury and injury due to other reasons. Studies support the observation that inhibition of methylation attenuates AKI and impedes renal fibrosis resulting from incomplete repair after injury. In addition to affecting the repair after kidney injury through the traditional fibrosis pathway, H3 methylation also participates in the process of kidney repair by regulating the expression of Klotho. Thus, histone methylation controls the cellular aging pathological process. Some studies support that histone methylation regulates inflammatory factor levels, and sepsis is one of the common causes of AKI in clinical practice. However, few studies discuss sepsis-induced AKI and repair after renal injury. The lack of such studies could be due to the complex injury mechanism of the animal model of sepsis kidney injury, the relative difficulty of modeling and the high mortality of animals after modeling.
Currently, effective treatments do not exist for AKI; only supportive treatment is available. In addition, efficient prevention and treatment options for various problems in renal repair are not available. With an increasing number of studies on AKI, the injury process of AKI and the aspect of post-injury fibrosis are explained using the epigenetic theory. The experiments have proved that AKI pathology can be improved. Histone methylation plays a vital role in the normal development of the kidney, and the intervention drugs or resources in these experiments have not reached the stage of clinical application. At the same time, drugs presently used in the clinic are used to treat kidney diseases by regulating the methylation of histones21,53. These observations enhance our belief that the research on histone methylation in AKI and kidney repair will help provide therapy for kidney diseases. Numerous histone methylation targets are available, and the mechanism is complex, which provides new ideas for an in-depth understanding of disease progression and the development of new therapeutic strategies. Further, many mechanisms are unclear, and several aspects need exploration.
Funding
The work received financial supports from the National Natural Science Foundation of China, grant: 82072222 (HJ); the Science and Technology Fund of Tianjin Municipal Health Bureau, grant: ZC20180 (HJ).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Ronco C, Bellomo R, Kellum JA. Acute kidney injury. Lancet. 2019;394(10212):1949–1964. doi:10.1016/S0140-6736(19)32563-2
2. Mehta RL, Cerdá J, Burdmann EA, et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet. 2015;385(9987):2616–2643. doi:10.1016/S0140-6736(15)60126-X
3. Negi S, Koreeda D, Kobayashi S, et al. Acute kidney injury: epidemiology, outcomes, complications, and therapeutic strategies. Semin Dial. 2018;31(5):519–527. doi:10.1111/sdi.12705
4. Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z. Regulated cell death in AKI. J Am Soc Nephrol. 2014;25(12):2689–2701. doi:10.1681/ASN.2014030262
5. Agarwal A, Dong Z, Harris R, et al. Cellular and molecular mechanisms of AKI. J Am Soc Nephrol. 2016;27(5):1288–1299. doi:10.1681/ASN.2015070740
6. Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015;11(5):264–276. doi:10.1038/nrneph.2015.3
7. Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol. 2015;26(8):1765–1776. doi:10.1681/ASN.2015010006
8. Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet. 2017;389(10075):1238–1252. doi:10.1016/S0140-6736(16)32064-5
9. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(5):535–543, 531p following 143. doi:10.1038/nm.2144
10. Bomsztyk K, Denisenko O. Epigenetic alterations in acute kidney injury. Semin Nephrol. 2013;33(4):327–340. doi:10.1016/j.semnephrol.2013.05.005
11. Wing MR, Ramezani A, Gill HS, Devaney JM, Raj DS. Epigenetics of progression of chronic kidney disease: fact or fantasy? Semin Nephrol. 2013;33(4):363–374. doi:10.1016/j.semnephrol.2013.05.008
12. Zhou X, Zang X, Guan Y, et al. Targeting enhancer of zeste homolog 2 protects against acute kidney injury. Cell Death Dis. 2018;9(11):1067. doi:10.1038/s41419-018-1012-0
13. Liang H, Huang Q, Liao MJ, et al. EZH2 plays a crucial role in ischemia/reperfusion-induced acute kidney injury by regulating p38 signaling. Inflamm Res. 2019;68(4):325–336. doi:10.1007/s00011-019-01221-3
14. Ni J, Hou X, Wang X, et al. 3-deazaneplanocin A protects against cisplatin-induced renal tubular cell apoptosis and acute kidney injury by restoration of E-cadherin expression. Cell Death Dis. 2019;10(5):355. doi:10.1038/s41419-019-1589-y
15. Gao L, Liu MM, Zang HM, et al. Restoration of E-cadherin by PPBICA protects against cisplatin-induced acute kidney injury by attenuating inflammation and programmed cell death. Lab Invest. 2018;98(7):911–923. doi:10.1038/s41374-018-0052-5
16. Herranz N, Pasini D, Diaz VM, et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol. 2008;28(15):4772–4781. doi:10.1128/MCB.00323-08
17. Dong C, Wu Y, Wang Y, et al. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene. 2013;32(11):1351–1362. doi:10.1038/onc.2012.169
18. Zeybel M, Luli S, Sabater L, et al. A proof-of-concept for epigenetic therapy of tissue fibrosis: inhibition of liver fibrosis progression by 3-deazaneplanocin A. Mol Ther. 2017;25(1):218–231. doi:10.1016/j.ymthe.2016.10.004
19. Chen SQ, Li JQ, Wang XQ, et al. EZH2-inhibitor DZNep enhances apoptosis of renal tubular epithelial cells in presence and absence of cisplatin. Cell Div. 2020;15(1):8. doi:10.1186/s13008-020-00064-3
20. Liu H, Chen Z, Weng X, et al. Enhancer of zeste homolog 2 modulates oxidative stress-mediated pyroptosis in vitro and in a mouse kidney ischemia-reperfusion injury model. FASEB J. 2020;34(1):835–852. doi:10.1096/fj.201901816R
21. Liu Y, Yu Y, Zhang J, Wang C. The therapeutic effect of dexmedetomidine on protection from renal failure via inhibiting KDM5A in lipopolysaccharide-induced sepsis of mice. Life Sci. 2019;239:116868. doi:10.1016/j.lfs.2019.116868
22. Li J, Qiu Y, Li L, et al. Histone methylation inhibitor DZNep ameliorated the renal ischemia-reperfusion injury via inhibiting TIM-1 mediated T cell activation. Front Med. 2020;7:305. doi:10.3389/fmed.2020.00305
23. Johnson AC, Ware LB, Himmelfarb J, Zager RA. HMG-CoA reductase activation and urinary pellet cholesterol elevations in acute kidney injury. Clin J Am Soc Nephrol. 2011;6(9):2108–2113. doi:10.2215/CJN.02440311
24. Naito M, Bomsztyk K, Zager RA. Renal ischemia-induced cholesterol loading: transcription factor recruitment and chromatin remodeling along the HMG CoA reductase gene. Am J Pathol. 2009;174(1):54–62. doi:10.2353/ajpath.2009.080602
25. Zhou X, Zang X, Ponnusamy M, et al. Enhancer of zeste homolog 2 inhibition attenuates renal fibrosis by maintaining Smad7 and phosphatase and tensin homolog expression. J Am Soc Nephrol. 2016;27(7):2092–2108. doi:10.1681/ASN.2015040457
26. Yu C, Xiong C, Tang J, et al. Histone demethylase JMJD3 protects against renal fibrosis by suppressing TGFbeta and Notch signaling and preserving PTEN expression. Theranostics. 2021;11(6):2706–2721. doi:10.7150/thno.48679
27. Majumder S, Thieme K, Batchu SN, et al. Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J Clin Invest. 2018;128(1):483–499. doi:10.1172/JCI95946
28. Guo Y, Xiong Z, Guo X. Histone demethylase KDM6B regulates human podocyte differentiation in vitro. Biochem J. 2019;476(12):1741–1751. doi:10.1042/BCJ20180968
29. Siddiqi FS, Majumder S, Thai K, et al. The histone methyltransferase enzyme enhancer of zeste homolog 2 protects against podocyte oxidative stress and renal injury in diabetes. J Am Soc Nephrol. 2016;27(7):2021–2034. doi:10.1681/ASN.2014090898
30. Zhou X, Xiong C, Tolbert E, Zhao TC, Bayliss G, Zhuang S. Targeting histone methyltransferase enhancer of zeste homolog-2 inhibits renal epithelial-mesenchymal transition and attenuates renal fibrosis. FASEB J. 2018;fj201800237R. doi:10.1096/fj.201800237R
31. Jia Y, Reddy MA, Das S, et al. Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-beta1-induced gene expression in mesangial cells and diabetic kidney. J Biol Chem. 2019;294(34):12695–12707. doi:10.1074/jbc.RA119.007575
32. Shi Y, Xu L, Tao M, et al. Blockade of enhancer of zeste homolog 2 alleviates renal injury associated with hyperuricemia. Am J Physiol Renal Physiol. 2019;316(3):F488–F505. doi:10.1152/ajprenal.00234.2018
33. Han X, Sun Z. Epigenetic regulation of KL (Klotho) via H3K27me3 (Histone 3 Lysine [K] 27 Trimethylation) in renal tubule cells. Hypertension. 2020;75(5):1233–1241. doi:10.1161/HYPERTENSIONAHA.120.14642
34. Hu MC, Moe OW. Klotho as a potential biomarker and therapy for acute kidney injury. Nat Rev Nephrol. 2012;8(7):423–429. doi:10.1038/nrneph.2012.92
35. Qin Feng HW, Huck Hui NG, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr Biol. 2002;12(12):1052–1058.
36. Barry ER, Krueger W, Jakuba CM, et al. ES cell cycle progression and differentiation require the action of the histone methyltransferase Dot1L. Stem Cells. 2009;27(7):1538–1547. doi:10.1002/stem.86
37. Kim W, Kim R, Park G, Park JW, Kim JE. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J Biol Chem. 2012;287(8):5588–5599. doi:10.1074/jbc.M111.328138
38. Wu H, Chen L, Zhou Q, et al. Aqp2-expressing cells give rise to renal intercalated cells. J Am Soc Nephrol. 2013;24(2):243–252. doi:10.1681/ASN.2012080866
39. Wang F, Ngo J, Li Y, et al. Targeted disruption of the histone lysine 79 methyltransferase Dot1L in nephron progenitors causes congenital renal dysplasia. Epigenetics. 2020;16:1–16.
40. Liu L, Zou J, Guan Y, et al. Blocking the histone lysine 79 methyltransferase DOT1L alleviates renal fibrosis through inhibition of renal fibroblast activation and epithelial-mesenchymal transition. FASEB J. 2019;33(11):11941–11958. doi:10.1096/fj.201801861R
41. Yang C, Chen Z, Yu H, Liu X. Inhibition of disruptor of telomeric silencing 1-like alleviated renal ischemia and reperfusion injury-Induced fibrosis by blocking PI3K/AKT-mediated oxidative stress. Drug Des Devel Ther. 2019;13:4375–4387. doi:10.2147/DDDT.S224909
42. Lefevre GM, Patel SR, Kim D, Tessarollo L, Dressler GR. Altering a histone H3K4 methylation pathway in glomerular podocytes promotes a chronic disease phenotype. PLoS Genet. 2010;6(10):e1001142. doi:10.1371/journal.pgen.1001142
43. Soofi A, Kutschat AP, Azam M, Laszczyk AM, Dressler GR. Regeneration after acute kidney injury requires PTIP-mediated epigenetic modifications. JCI Insight. 2020;5(3):3. doi:10.1172/jci.insight.130204
44. Sun G, Reddy MA, Yuan H, Lanting L, Kato M, Natarajan R. Epigenetic histone methylation modulates fibrotic gene expression. J Am Soc Nephrol. 2010;21(12):2069–2080. doi:10.1681/ASN.2010060633
45. Guo Q, Li X, Han H, et al. Histone lysine methylation in TGF-beta1 mediated p21 gene expression in rat mesangial cells. Biomed Res Int. 2016;2016:6927234. doi:10.1155/2016/6927234
46. Sasaki K, Doi S, Nakashima A, et al. Inhibition of SET domain-containing lysine methyltransferase 7/9 ameliorates renal fibrosis. J Am Soc Nephrol. 2016;27(1):203–215. doi:10.1681/ASN.2014090850
47. Fujino T, Hasebe N. Alteration of histone H3K4 methylation in glomerular podocytes associated with proteinuria in patients with membranous nephropathy. BMC Nephrol. 2016;17(1):179. doi:10.1186/s12882-016-0390-8
48. Irifuku T, Doi S, Sasaki K, et al. Inhibition of H3K9 histone methyltransferase G9a attenuates renal fibrosis and retains klotho expression. Kidney Int. 2016;89(1):147–157. doi:10.1038/ki.2015.291
49. Liu H, Wang W, Weng X, et al. The H3K9 histone methyltransferase G9a modulates renal ischemia reperfusion injury by targeting Sirt1. Free Radic Biol Med. 2021;172:123–135. doi:10.1016/j.freeradbiomed.2021.06.002
50. Gao D, Zuo Z, Tian J, et al. Activation of SIRT1 attenuates klotho deficiency-induced arterial stiffness and hypertension by enhancing AMP-activated protein kinase activity. Hypertension. 2016;68(5):1191–1199. doi:10.1161/HYPERTENSIONAHA.116.07709
51. Chen Y, Huang C, Zhu SY, Zou HC, Xu CY, Chen YX. Overexpression of HOTAIR attenuates Pi-induced vascular calcification by inhibiting Wnt/beta-catenin through regulating miR-126/Klotho/SIRT1 axis. Mol Cell Biochem. 2021;476(10):3551–3561. doi:10.1007/s11010-021-04164-8
52. Liu L, Liu F, Guan Y, et al. Critical roles of SMYD2 lysine methyltransferase in mediating renal fibroblast activation and kidney fibrosis. FASEB J. 2021;35(7):e21715. doi:10.1096/fj.202000554RRR
53. Zhang SF, Mao XJ, Jiang WM, Fang ZY. Qian Yang Yu Yin Granule protects against hypertension-induced renal injury by epigenetic mechanism linked to Nicotinamide N-Methyltransferase (NNMT) expression. J Ethnopharmacol. 2020;255:112738. doi:10.1016/j.jep.2020.112738
54. Zhu Y, Yu C, Zhuang S. Protein arginine methyltransferase 1 mediates renal fibroblast activation and fibrogenesis through activation of Smad3 signaling. Am J Physiol Renal Physiol. 2020;318(2):F375–F387. doi:10.1152/ajprenal.00487.2019
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.