")
Back to Archived Journals » Hypoxia » Volume 2
The role of HIFs in ischemia-reperfusion injury
Received 22 March 2014
Accepted for publication 21 May 2014
Published 30 July 2014 Volume 2014:2 Pages 107—115
DOI https://doi.org/10.2147/HP.S49720
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Neil J Howell,1 Daniel A Tennant2
1Department of Cardiothoracic Surgery, University Hospital Birmingham, Edgbaston, Birmingham, UK; 2College of Medical and Dental Sciences, University of Birmingham, Birmingham, UK
Abstract: The reduction or cessation of the blood supply to an organ results in tissue ischemia. Ischemia can cause significant tissue damage, and is observed as a result of a thrombosis, as part of a disease process, and during surgery. However, the restoration of the blood supply often causes more damage to the tissue than the ischemic episode itself. Research is therefore focused on identifying the cellular pathways involved in the protection of organs from the damage incurred by this process of ischemia reperfusion (I/R). The hypoxia-inducible factors (HIFs) are a family of heterodimeric transcription factors that are stabilized during ischemia. The genes that are expressed downstream of HIF activity enhance oxygen-independent ATP generation, cell survival, and angiogenesis, amongst other phenotypes. They are, therefore, important factors in the protection of tissues from I/R injury. Interestingly, a number of the mechanisms already known to induce organ protection against I/R injury, including preconditioning, postconditioning, and activation of signaling pathways such as adenosine receptor signaling, converge on the HIF system. This review describes the evidence for HIFs playing a role in I/R protection mediated by these factors, highlights areas that require further study, and discuss whether HIFs themselves are good therapeutic targets for protecting tissues from I/R injury.
Keywords: hypoxia, adenosine, pre-conditioning, post-conditioning
Introduction
Ischemia, defined as an inadequate blood supply to an organ or part of the body,1 is a considerable clinical problem. It is observed in a number of chronic diseases including cardiovascular disease and diabetes, acute events such as stroke or myocardial infarctions, and during planned clinical procedures like organ transplantation and general surgery. Ischemia leads to a reduction in the supply of nutrients and oxygen as well as the removal of waste products from a tissue, and in doing this can lead to irreversible tissue damage and death.
Arguably more important is the subsequent reperfusion of the tissue or organ. Upon resumption of a normal blood supply, oxygen and nutrients flood back into the tissue. This can result in considerable damage to both the tissue in question and, depending on the scale of the ischemic insult, the rest of the body. If a severe ischemic episode has resulted in extensive necrotic damage, a wave of toxic cellular products can be released into the bloodstream upon reperfusion that, in the worst circumstances, can result in the death of the patient. However, even in acute ischemic events where this does not happen, the rush of nutrients and oxygen into the tissue can cause significant tissue damage, mainly through reactive oxygen species (ROS)-mediated oxidative damage of proteins, DNA, and lipids throughout the tissue.2,3
The objective of much research in this area is to find means of reducing infarct size and thereby preserving tissue function after ischemia–reperfusion (I/R) injury. This has involved the investigation of means of inducing a protective state in the tissue both before and after the ischemic insult. Although studies over many years have demonstrated the efficacy of a number of agents in eliciting protection of various organs from I/R injury, most often, this demonstration has not led to the agent becoming an integral part of clinical practice. This may well be because we do not yet fully understand the intracellular signaling pathways that are required to invoke protection and, therefore, cannot design agents that can specifically target these pathways without off-target effects.
This review will discuss the evidence suggesting that the family of hypoxia-inducible factor (HIF) transcription factors may well represent such a target. Data from studies of methods of protecting tissues from ischemia all suggest that HIFs may play a central role in mediating cellular protection. Future research strategies to specifically define how HIFs are best activated by either preconditioning (PreC) or postconditioning (PostC) approaches to generate a protected state may well lead to novel efficacious therapies to alleviate the clinical problems associated with I/R injury.
Control of the HIF transcription factors
The HIFs are heterodimeric transcription factors composed of an alpha and beta subunit. The beta subunit is considered to be constitutively expressed, while the alpha subunit is subject to a highly efficient oxygen-dependent system of control.4,5 There are three alpha subunits of HIF, of which 1α and 2α are the best described. Overall regulation of expression of the alpha subunits is through oxygen-induced proteolytic degradation, which gives a high degree of temporal control over the hypoxic response. Although stable under acute hypoxic conditions, alpha subunits are rapidly degraded in normoxia through the hydroxylation of target prolyl residues (P402 and P564 in human HIF1α) by prolyl hydroxylase enzymes (PHD1–3, encoded by the EGLN1-3 genes).6 These hydroxyprolyl residues are then recognized by the von Hippel–Lindau (pVHL) E3 ubiquitin ligase, which polyubiquitylates the HIFα subunit, targeting it for proteasomal degradation (Figure 1).7 This system provides exquisite control over the expression and activity of the HIF transcription factors, allowing them to be rapidly and reversibly upregulated upon reduction of the environmental oxygen.
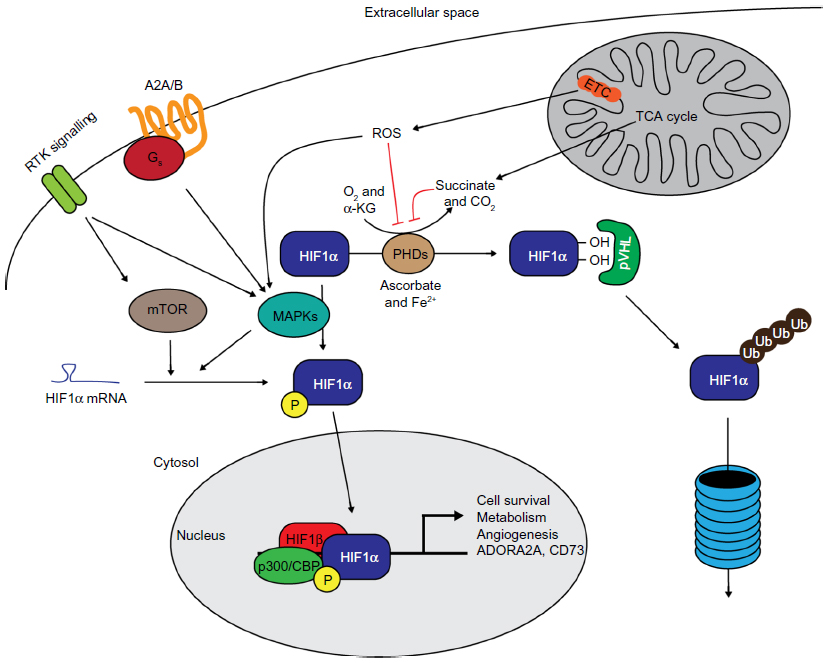 | Figure 1 The control of HIF expression and activity by factors involved in ischemic signaling. |
The PHD enzymes, which represent the oxygen sensors in this system, are 2-oxoglutarate-dependent dioxygenases, which use 2-oxoglutarate (alpha-ketoglutarate) and oxygen to hydroxylate target substrates. They are, therefore, less active in low-oxygen (and presumably low-2-oxoglutarate) environments, such as during ischemia. Although HIF-independent roles for the PHD enzymes are becoming clearer, their most understood role is through the regulation of the HIF transcription factors. PHD2 has been shown as the major hydroxylase involved in regulating HIF1α stability,8,9 while PHD1 and 3 activities have both been correlated with HIF2α stability.10–12 During ischemic conditions, where PHDs are less active, HIFα subunits are rapidly stabilized, leading to the expression of the HIF-induced transcriptome. Interestingly, PHD2 and 3 are both transcriptional targets of HIF1, providing a HIF1-specific negative feedback loop (PHD2 and 3 have not been shown as HIF2 target genes) where hypoxia-induced HIF1 activity leads to increased PHD2 and 3 levels and, as a result, decreased HIF1α (and potentially HIF2α) expression.13
Importantly, in terms of relevance to I/R injury, conditions that increase ROS generation, such as reoxygenation, have been shown to result in a longer-term stabilization of HIF1α, thereby inducing a prolonged hypoxic phenotype in reperfused cells.14 The mechanism by which this occurs has been suggested as the oxidative inactivation of the non-heme iron catalytic site of the PHD enzymes,15–17 although a further study suggested that the active site of the PHDs may not be accessible by ROS.18
Other signaling pathways are also known to increase HIF1α protein expression independently of oxygen tension. Activation of AKT has been shown to increase HIF1α expression in normoxic conditions (Figure 1).19 The downstream pathway involved in this regulation appears to be through activation of mechanistic target of rapamycin ([mTOR] also known as FKBP12 rapamycin-associated protein).19 mTOR and mitogen-activated protein kinases (MAPKs) can both phosphorylate 4E-BP1 to permit efficient cap-dependent translation of HIF1α messenger ribonucleic acid (mRNA).20 In ischemia, general translation is shut down through mechanisms including nutrient-sensitive inhibition of mTOR to conserve adenosine triphosphate (ATP) levels.21 However, cap-independent translation of some mRNA species, including HIF1 and 2α, is conserved.21 As previously mentioned, mTOR acts as a nutrient sensor – it is able to sense intracellular concentrations of the amino acids leucine and glutamine,22,23 thereby linking nutrient availability with anabolic processes. Other metabolic alterations that occur in ischemia are also capable of signaling to alter cellular phenotype. Succinate, a tricarboxylic acid cycle metabolite is often found at considerably higher concentrations in ischemic tissue,24,25 and is a well-described inhibitor of the PHD enzymes, leading to HIFα stabilization (Figure 1).26
MAPKs have also been shown to be required for the transactivational activity of HIF1 and 2. MAPKs are a large family of protein kinases consisting of three main groups: extracellular signal-regulated kinases; p38 MAPKs; and c-Jun N-terminal kinases.27–29 They are activated in response to diverse stimuli including growth factor signaling, cellular and environmental stress, and inflammatory cues. Once activated, members of this family have been shown to phosphorylate HIF1α, leading to enhanced binding of the coactivator p300/CBP-binding protein (Figure 1), although other independent pathways have also been suggested.30–32
Hence, although oxygen tension is the most potent modulator of HIFα stability, there are many diverse means of altering HIF activity that can occur in ischemic and reperfused tissues. Many of these stabilization stimuli are downstream of signaling pathways that are known to be important in the protection of tissues from I/R injury and, therefore, provide some evidence, albeit indirect, for an important role for HIF in protection.
The effect of HIF stabilization on I/R injury
HIF1α was originally identified as a hypoxia-inducible nuclear factor that bound the EPO gene locus and induced its expression.33 Its gene targets have since been extensively characterized by a number of groups, and the hypoxia responsive elements (HREs) to which HIFs bind have been identified in numerous gene loci. More recently, it has been suggested that HIF1 may also have more wide-ranging effects on the ischemic transcriptome through its influence on promoter-bound RNA polymerase II, even at loci without associated HREs.34 HIF2α, also known as endothelial PAS protein 1 was cloned in 1997 by a number of groups.35–38 Although its regulation is less well-described than that of HIF1a, it also binds to HRE sequences and transactivates its target genes. Although HIF1α is ubiquitously expressed, HIF2α is more limited and has been observed in a subset of cell types, including hepatocytes, cardiac myocytes, and endothelial cells.39 The different phenotypes observed in the HIF1α−/− and HIF2α−/− mice, as well as a number of other studies, strongly suggest that, although each shares a set of genes that they regulate, they also each control the expression of unique target genes.40–43 Studies performed in different cell types have shown that HIF2α specifically regulates hypoxic upregulation of gene products including cyclin D1, transforming growth factor α, POU5F1, and matrix metalloproteinase 2, whereas HIF1 regulates a large number of glycolytic enzymes including glyceraldehyde 3-phosphate dehydrogenase, phosphoglycerate kinase 1, Bnip3, and carbonic anhydrase 9.43–48 Importantly, the precise gene set altered by HIF1 and 2 is tissue specific, as it depends on the prevailing chromatin structure.49
HIFs are therefore stabilized rapidly upon the loss of oxygen supply, resulting in a concerted transcriptional response to modulate cellular phenotype. This transcriptional response has wide-ranging effects that are beneficial to ischemic and recently reperfused tissue. As the HIF transcription factors regulate the expression of such a considerable number of genes, they are likely to exert their protective influence on I/R tissues through pleiotropic effects.50 A large number of upregulated HIF1 target genes are associated with glycolysis and, therefore, permit higher rates of ATP production from glucose, which is an oxygen-independent process. This is important both under ischemic conditions and upon reperfusion as it allows ATP levels to be sustained further into the ischemic episode, as well as permitting rapid ATP production after resumption of the blood supply. For tissues with considerable ATP demand, such as the myocardium, the rapid restoration of ATP generation capacity is key to retaining function post-I/R. Avoiding dependency on oxidative phosphorylation for ATP production after reperfusion is important as many mitochondrial metabolic enzymes can be inhibited or inactivated by the ROS generated by reperfusion, and efficient oxidative metabolism may therefore have to wait for new protein production by the cell. In addition, HIF stabilization also increases the production of angiogenic factors by the tissue, such as vascular endothelial growth factor, which can aid the restoration of an efficient blood supply through the damaged tissue.51
HIFs are a central part of PreC and PostC mechanisms
Ischemic PreC (IPC) and ischemic PostC have been shown to be effective in reducing infarct size in a number of models and tissues.52–57 Both conditioning protocols use acute cycles of I/R, with the ischemic episode lasting a few minutes.58–61 IPC results in a protected phenotype in the tissue that has two phases: one that occurs within minutes and can last a few hours, and another that begins at around 24–48 hours and lasts a number of days.60,62,63 There are several lines of evidence that places HIF as a central mediator of IPC-mediated protection. IPC and PreC protocols have both been demonstrated to elicit HIF1α stabilization and transactivational activity – an effect that was shown in one study to be specifically PHD2 (and not PHD1 or 3) mediated.55 Evidence from a heart model of IPC in mice also demonstrated that HIF1α+/− mice have reduced IPC-mediated protection from I/R.53 Indeed, further evidence in a renal ischemic model showed that the inhibition of PHDs and stabilization of HIF1α and 2α was sufficient to produce a PreC phenotype.54,64 However, the rapidity of induction of the early IPC phase is most likely quicker than a HIF-mediated transcriptional response can be observed. Therefore, it is possible that, instead, the upstream oxygen sensors that control HIFs – the PHDs – can induce this biphasic response through direct hydroxylation of as-yet unknown targets. There is also strong evidence to suggest that PHD1 inhibition, which is observed downstream of dimethyloxalylglycine (DMOG) treatment, can produce a strong protective effect in a number of organ systems, which is HIF1α-independent (although it may be HIF2α-dependent).10,65 However, considering that PHDs themselves can be directly inactivated by increased cellular ROS levels, as would occur with repeated I/R cycles, both IPC and PostC protocols appear ideal to inactivate these enzymes, thereby altering their capacity to hydroxylate target substrates. It is possible, therefore, that the PHDs themselves may play an important role in protection from I/R injury through the loss of hydroxylation of as-yet unknown substrates to directly and acutely alter cell signaling. The late phase of IPC, although also downstream of PHD signaling, has a profile that is consistent with HIF-mediated protection. Although hypoxia-mediated gene transcription downstream of HIF can be observed within 24 hours, full stabilization of this transcriptome can take up to 48 hours, which is very similar to the profile of the late IPC response.66
Interestingly, it appears that HIF1 activation may be key in the remote IPC scenario, where HIF1-mediated upregulation of interleukin-10 as well as stromal cell-derived factor 1 have both been shown as capable of inducing remote IPC in the heart.67–69 Interleukin-10 provides protection from I/R injury through activation of AKT signaling during oxygen–glucose deprivation of cortical neurons, suggesting that this mechanism may be pertinent in a number of different tissues.
The concept of ischemic PostC has received much attention recently due to the potential for ameliorating ischemic damage in patients received into emergency departments after an ischemic incident. PostC is carried out using a series of short I/R cycles immediately upon reperfusion after the original ischemic event.61 A large number of signaling pathways have been proposed as being involved in this process, including adenosine receptor signaling, alterations in Ca2+ dynamics, and inhibition of the mitochondrial permeability transition pore.70–72 Two recent papers reported a role for HIF1α in the PostC protection phenomenon.57,73 In the first, which investigated PostC of the myocardium, PostC was elicited through DMOG-mediated inhibition of the PHDs, thereby stabilizing HIF1α (and presumably HIF2α) expression.73 In the second, a mechanism for sevoflurane-mediated protection from cerebral ischemia was investigated.57 The authors found a phosphatidylinositol 3-kinase (PI-3K)-dependent effect of sevoflurane on HIF1α expression, which resulted in increased heme oxygenase 1 mRNA, a HIF1 target gene. However, the data in these papers are also consistent with a HIF1α-independent mechanism that could be through PHD signaling or other PI-3K targets. Indeed, PI-3K activation has many protective effects on cell viability independently of HIF-1, including through the phosphorylation of the proapoptotic protein BAD and the enzyme glycogen synthase kinase 3β. It is worth noting that the demonstration of sevoflurane-mediated protection by Ye et al is part of a growing body of evidence that suggests that the HIF signaling axis is a potential mechanism by which volatile anesthetics can protect from I/R injury.57,74,75 Much more investigation into this area is required in order for efficacious clinically relevant therapeutics to be developed to be used as PostC agents.
Interplay between adenosine signaling and HIFs
Signaling by the metabolite adenosine has long been linked to protection from ischemia in the heart, brain, and kidney.76–78 Extracellular adenosine is observed as a result of cellular damage, release of adenosine from intracellular sources, or through the breakdown of extracellular adenosine monophosphate (AMP) by 5′-nucleotidases. The increase in adenosine levels observed during ischemia in the myocardium are thought to be due to the relative increase in AMP concentrations due to the action of adenylate kinase on adenosine diphosphate, producing ATP and AMP.76 Adenosine is recognized by plasma membrane-spanning adenosine receptors A1, 2A, 2B, and 3 (encoded by ADORA1, 2A, 2B, and 3 genes), all of which are G protein-coupled receptors.79 Types A1 and A3 are Go/I linked, meaning that their activation leads to a reduction in adenylyl cyclase activity (and therefore cAMP levels) and increased phospholipase C (PLC) activity. PLC action on phosphoinositol-containing lipids produces inositol phosphates and diacylglycerol, both of which signal to downstream effector pathways, which includes the activation of protein kinase C (PKC). Conversely, A2A and B subtypes are coupled to stimulatory G proteins and result in increased cAMP levels and protein kinase A activation.79 Both subtypes are able to activate MAPK signaling. Adenosine signaling through its receptors has been strongly linked with modification of myocardial function and survival,76 as well as modulation of I/R injury in a different organs.80–84 It was initially shown that adenosine infusion alone or as part of a cardioplegic solution could be used to increase ATP levels during and after short ischemic episodes.85 However, adenosine was also shown to play a role in myocardial PreC, as treatment with either adenosine or an A1 agonist were shown to recapitulate a bona fide IPC stimulus, while an adenosine-receptor antagonist reversed the effect of conventional IPC.86,87
It is likely that some of the protection from I/R injury through the action of HIFs is mediated through this signaling cascade. HIF1 has been shown to increase the expression of the extracellular 5′-nucleotidase CD73 during intestinal ischemia,56 which would be predicted to increase extracellular adenosine concentrations. The authors also showed that a DMOG induced an increase in the A2B receptor that was abolished in HIF1α−/− mice, suggesting that HIF1-dependent protection may be through activation of this receptor (Figure 1). HIF1-dependent induction of A2B has also been observed in vascular endothelial cells, where it was shown to play a role in hypoxia-induced tubule formation as well as barrier protection.88
The other stimulatory G protein-linked adenosine receptor, A2A, has been proposed to modulate HIF1 activity. Activation of this receptor in macrophages has been shown to increase HIF1α expression and HRE binding in normoxic conditions.89 Using inhibitor studies, the authors suggested that this stabilization was through AKT and PKCδ activation downstream of the A2A receptor. Both these kinases have been previously shown to stabilize HIF1α independently of oxygen tension; AKT activation is known to result in enhanced HIF1α expression through increased translation90 and PKCδ activation (which could also be downstream of AKT activation) was shown to increase HIF1 binding to HREs by an as-yet undescribed mechanism.91 It is possible that the regulation of HIF1α by PKCδ could involve phosphorylation of the same site as MAPKs, which is known to enhance heterodimer formation.30
Interestingly, it has been shown that ADORA2A is a HIF2 target gene in pulmonary endothelial cells.92 Endothelial cells from different sources have been shown to express different levels of the A2A and A2B receptors,92–94 and it has been reported that the relative expression of each changes upon induction of hypoxia.94 However, signaling through A2B and A2A receptors have both been shown to induce angiogenesis.92,94 It is also important to note that A2A receptor signaling during ischemia is not always protective; data from cerebral ischemic models suggest that blockade of A2A receptors protects against associated damage in adult rodents.84,95 The effect of this on the ischemic stabilization of HIF was not established. Finally, the A3 receptor has also been shown to synergize with hypoxia to induce further stabilization of HIF1α.96
It is therefore clear that there is a reciprocal relationship between the HIFs and adenosine receptors that varies with the expression of both the adenosine receptors and HIFα subunits. In the majority of cases, it appears that this signaling axis is protective, but with tissue specific outcomes; for example, signaling elicits an angiogenic response in endothelial cells whereas, in the myocardium, it appears to be prosurvival.
The role of HIFs in the link between circadian rhythm and I/R injury
There has been recent interest in the observation that incidence of cardiovascular events, including myocardial infarctions and stroke, varies with the natural circadian rhythm.97 Interestingly, the circadian rhythm proteins, and most strikingly Per2, were recently identified as altered in the hearts of mice deficient for the A2B receptor (ADORA2B−/−).98 These mice not only lost the circadian control of Per2, but also lost an IPC-induced increase in Per2 expression. Previous studies have demonstrated that A2B-receptor activation results in MAPK activity, which has been demonstrated to regulate Per2.99,100 As the mice also lost the ischemic induction of glycolytic enzymes and lactate production in Per2−/− mice, the authors investigated the ischemic stabilization of HIF1α and found that it was abolished.98 These data introduce a novel and exciting link between adenosine signaling, control of circadian rhythms, and the cellular hypoxic response. Interestingly, there are further links between hypoxia and the control of circadian rhythm that may also play a role in I/R injury as DEC1 (also known as SHARP1 and Stra13), which has been shown to play a role in the regulation of the molecular clock, is strongly induced by hypoxia in a HIF-dependent mechanism.101,102 However, the role of DEC1 in I/R injury has not yet been explored.
Summary – the potential for therapeutic targeting of the HIF axis
Evidence from a number of tissues and in vitro studies suggests that activation of the HIF axis can protect from I/R injury. Therapeutic approaches that target this system, whether directly or indirectly, are likely to have beneficial effects on patient outcomes. Indeed, a number of studies described above support this, and used iron chelators or direct competitive inhibitors of the PHD enzymes to stabilize HIF in models of stroke and renal and myocardial infarction.55,103–105 It is not surprising that there are PHD-inhibiting compounds in both preclinical testing and clinical trials for use in acute ischemia. Interestingly, the use of iron chelators in neurological diseases such as stroke actually predates the discovery of HIFs and PHDs, and were used for their ability to preserve cellular metabolism (reviewed in Karuppagounder et al106). Therefore, it would not be unexpected if targets for iron chelators other than the PHDs and HIFs were found that help preserve tissue viability during I/R. Finally, therapies that indirectly target the HIF axis are also likely to aid tissue recovery as part of a multifactorial approach. Activators of AKT or MAPK signaling, such as the use of insulin during surgery, as well as adenosine receptor agonists could be used in combination to increase HIF activity before or during ischemia.
However, we need to understand more about the role of HIFs in I/R injury, in particular in two areas. First, studies are required that dissociate the role of PHD inhibition from HIF activation in I/R injury as it is not yet clear whether HIF is only one (albeit central) part of the protective effects of PHD inhibition. Second, we need to understand the temporal aspects of modulating this system. Direct effects of PHD inhibition may well be very rapid as their effect is posttranslational, whereas those responses downstream of HIFs can take much longer – even hours – to manifest themselves. It is possible that PHD inhibition can acutely protect tissues, while HIF activation is required post-reperfusion.
Regardless, it is clear that this oxygen-sensing system is central to the protection of tissues from I/R injury, and further work to elucidate the signaling mechanisms surrounding this phenomenon is likely to lead to better options for therapeutic interventions to improve clinical outcomes.
Acknowledgments
We thank Doctors Giulio Laurenti, David Bartlett, and Gavin McNee for their critical reading of the manuscript and constructive comments.
Disclosure
The authors report no conflicts of interest in this work.
References
Oxford English Dictionary. Oxford University Press. Available from http://www.oxforddictionaries.com/. Accessed July 8, 2014. | |
Hearse DJ, Humphrey SM, Chain EB. Abrupt reoxygenation of the anoxic potassium-arrested perfused rat heart: a study of myocardial enzyme release. J Mol Cell Cardiol. 1973;5:395–407. | |
Ambrosio G, Becker LC, Hutchins GM, Weisman HF, Weisfeldt ML. Reduction in experimental infarct size by recombinant human superoxide dismutase: insights into the pathophysiology of reperfusion injury. Circulation. 1986;74:1424–1433. | |
Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996;271:17771–17778. | |
Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. | |
Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–354. | |
Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–5206. | |
Berra E1, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22:4082–4090. | |
Tennant DA, Frezza C, MacKenzie ED, et al. Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene. 2009;28:4009–4021. | |
Aragonés J, Schneider M, Van Geyte K, et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40:170–180. | |
Taniguchi CM, Finger EC, Krieg AJ, et al. Cross-talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat Med. 2013;19:1325–1330. | |
Bishop T, Gallagher D, Pascual A, et al. Abnormal sympathoadrenal development and systemic hypotension in PHD3−/− mice. Mol Cell Biol. 2008;28:3386–3400. | |
Ginouves A, Ilc K, Macias N, Pouyssegur J, Berra E. PHDs overactivation during chronic hypoxia “desensitizes” HIFalpha and protects cells from necrosis. Proc Natl Acad Sci U S A. 2008;105:4745–4750. | |
Martinive P, Defresne F, Bouzin C, et al. Preconditioning of the tumor vasculature and tumor cells by intermittent hypoxia: implications for anticancer therapies. Cancer Res. 2006;66:11736–11744. | |
Brunelle JK, Bell EL, Quesada NM, et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–414. | |
Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–408. | |
Mansfield KD, Guzy RD, Pan Y, et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1:393–399. | |
Masson N, Singleton RS, Sekirnik R, et al. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 2012;13:251–257. | |
Treins C, Giorgetti-Peraldi S, Murdaca J, Semenza GL, Van Obberghen E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J Biol Chem. 2002;277:27975–27981. | |
Sonenberg N, Hinnebusch AG. New modes of translational control in development, behavior, and disease. Mol Cell. 2007;28:721–729. | |
Yee Koh M, Spivak-Kroizman TR, Powis G. HIF-1 regulation: not so easy come, easy go. Trends Biochem Sci. 2008;33:526–534. | |
Durán RV, Hall MN. Glutaminolysis feeds mTORC1. Cell Cycle. 2012;11:4107–4108. | |
Kimball SR, Shantz LM, Horetsky RL, Jefferson LS. Leucine regulates translation of specific mRNAs in L6 myoblasts through mTOR-mediated changes in availability of eIF4E and phosphorylation of ribosomal protein S6. J Biol Chem. 1999;274:11647–11652. | |
Goldberg ND, Passonneau JV, Lowry OH. Effects of changes in brain metabolism on the levels of citric acid cycle intermediates. J Biol Chem. 1966;241:3997–4003. | |
Folbergrová J, Ljunggren B, Norberg K, Siesjö BK. Influence of complete ischemia on glycolytic metabolites, citric acid cycle intermediates, and associated amino acids in the rat cerebral cortex. Brain Res. 1974;80:265–279. | |
Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. | |
Cobb MH. MAP kinase pathways. Prog Biophys Mol Biol. 1999;71:479–500. | |
Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–18. | |
Davies C, Tournier C. Exploring the function of the JNK (c-Jun N-terminal kinase) signalling pathway in physiological and pathological processes to design novel therapeutic strategies. Biochem Soc Trans. 2012;40:85–89. | |
Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem. 2003;278:14013–14019. | |
Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem. 1999;274:32631–32637. | |
Sodhi A, Montaner S, Patel V, et al. The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 2000;60:4873–4880. | |
Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88:5680–5684. | |
Choudhry H, Schödel J, Oikonomopoulos S, et al. Extensive regulation of the non-coding transcriptome by hypoxia: role of HIF in releasing paused RNApol2. EMBO Rep. 2014;15:70–76. | |
Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997;94:4273–4278. | |
Flamme I, Fröhlich T, von Reutern M, Kappel A, Damert A, Risau W. HRF, a putative basic helix-loop-helix-PAS-domain transcription factor is closely related to hypoxia-inducible factor-1 alpha and developmentally expressed in blood vessels. Mech Dev. 1997;63:51–60. | |
Hogenesch JB, Chan WK, Jackiw VH, et al. Characterization of a subset of the basic-helix-loop-helix-PAS superfamily that interacts with components of the dioxin signaling pathway. J Biol Chem. 1997;272:8581–8593. | |
Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11:72–82. | |
Wiesener MS, Jürgensen JS, Rosenberger C, et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003;17:271–273. | |
Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. | |
Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998;12:3320–3324. | |
Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. | |
Raval RR, Lau KW, Tran MG, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. | |
Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. 1994;269:23757–23763. | |
Krick S, Eul BG, Hänze J, et al. Role of hypoxia-inducible factor-1alpha in hypoxia-induced apoptosis of primary alveolar epithelial type II cells. Am J Respir Cell Mol Biol. 2005;32:395–403. | |
Grabmaier K, A de Weijert MC, Verhaegh GW, Schalken JA, Oosterwijk E. Strict regulation of CAIX(G250/MN) by HIF-1alpha in clear cell renal cell carcinoma. Oncogene. 2004;23:5624–5631. | |
Hu CJ, Sataur A, Wang L, Chen H, Simon MC. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol Biol Cell. 2007;18:4528–4542. | |
Covello KL, Kehler J, Yu H, et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–570. | |
Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2011;117:e207–e217. | |
Loor G, Schumacker PT. Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. 2008;15:686–690. | |
Lee SH, Wolf PL, Escudero R, Deutsch R, Jamieson SW, Thistlethwaite PA. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med. 2000;342:626–633. | |
Whitlock NA, Agarwal N, Ma JX, Crosson CE. Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest Ophthalmol Vis Sci. 2005;46:1092–1098. | |
Sarkar K, Cai Z, Gupta R, et al. Hypoxia-inducible factor 1 transcriptional activity in endothelial cells is required for acute phase cardioprotection induced by ischemic preconditioning. Proc Natl Acad Sci U S A. 2012;109:10504–10509. | |
Bernhardt WM, Câmpean V, Kany S, et al. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol. 2006;17:1970–1978. | |
Eckle T, Kohler D, Lehmann R, El Kasmi K, Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008;118:166–175. | |
Hart ML, Grenz A, Gorzolla IC, Schittenhelm J, Dalton JH, Eltzschig HK. Hypoxia-inducible factor-1alpha-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J Immunol. 2011;186:4367–4374. | |
Ye Z, Guo Q, Xia P, Wang N, Wang E, Yuan Y. Sevoflurane postconditioning involves an up-regulation of HIF-1alpha and HO-1 expression via PI3K/Akt pathway in a rat model of focal cerebral ischemia. Brain Res. 2012;1463:63–74. | |
Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. | |
Pérez-Pinzón MA1, Xu GP, Mumford PL, Dietrich WD, Rosenthal M, Sick TJ. Rapid ischemic preconditioning protects rats from cerebral anoxia/ischemia. Adv Exp Med Biol. 1997;428:155–161. | |
Rodrigo GC, Samani NJ. Ischemic preconditioning of the whole heart confers protection on subsequently isolated ventricular myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H524–H531. | |
Zhao ZQ, Corvera JS, Halkos ME, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285:H579–H588. | |
Pérez-Pinzón MA, Xu GP, Dietrich WD, Rosenthal M, Sick TJ. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J Cereb Blood Flow Metab. 1997;17:175–182. | |
Perez-Pinzon MA, Mumford PL, Rosenthal M, Sick TJ. Anoxic preconditioning in hippocampal slices: role of adenosine. Neuroscience. 1996;75:687–694. | |
Mahfoudh-Boussaid A, Zaouali MA, Hadj-Ayed K, et al. Ischemic preconditioning reduces endoplasmic reticulum stress and upregulates hypoxia inducible factor-1alpha in ischemic kidney: the role of nitric oxide. J Biomed Sci. 2012;19:7. | |
Schneider M, Van Geyte K, Fraisl P, et al. Loss or silencing of the PHD1 prolyl hydroxylase protects livers of mice against ischemia/reperfusion injury. Gastroenterology. 2010;138:1143–1154. e1–e2. | |
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. | |
Cai Z, Luo W, Zhan H, Semenza GL. Hypoxia-inducible factor 1 is required for remote ischemic preconditioning of the heart. Proc Natl Acad Sci U S A. 2013;110:17462–17467. | |
Davidson SM, Selvaraj P, He D, et al. Remote ischaemic preconditioning involves signalling through the SDF-1alpha/CXCR4 signalling axis. Basic Res Cardiol. 2013;108:377. | |
Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858–864. | |
Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M. Postconditioning inhibits mitochondrial permeability transition. Circulation. 2005;111:194–197. | |
Argaud L, Gateau-Roesch O, Augeul L, et al. Increased mitochondrial calcium coexists with decreased reperfusion injury in postconditioned (but not preconditioned) hearts. Am J Physiol Heart Circ Physiol. 2008;294:H386–H391. | |
Philipp S, Yang XM, Cui L, Davis AM, Downey JM, Cohen MV. Postconditioning protects rabbit hearts through a protein kinase C-adenosine A2b receptor cascade. Cardiovasc Res. 2006;70:308–314. | |
Zhao HX, Wang XL, Wang YH, et al. Attenuation of myocardial injury by postconditioning: role of hypoxia inducible factor-1alpha. Basic Res Cardiol. 2010;105:109–118. | |
Raphael J, Zuo Z, Abedat S, Beeri R, Gozal Y. Isoflurane preconditioning decreases myocardial infarction in rabbits via up-regulation of hypoxia inducible factor 1 that is mediated by mammalian target of rapamycin. Anesthesiology. 2008;108:415–425. | |
Wang C, Weihrauch D, Schwabe DA, et al. Extracellular signal-regulated kinases trigger isoflurane preconditioning concomitant with upregulation of hypoxia-inducible factor-1alpha and vascular endothelial growth factor expression in rats. Anesth Analg. 2006;103:281–288. | |
Mubagwa K, Mullane K, Flameng W. Role of adenosine in the heart and circulation. Cardiovasc Res. 1996;32:797–813. | |
Schubert P, Kreutzberg GW. Cerebral protection by adenosine. Acta Neurochir Suppl (Wien). 1993;57:80–88. | |
Yap SC, Lee HT. Adenosine and protection from acute kidney injury. Curr Opin Nephrol Hypertens. 2012;21:24–32. | |
Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5:247–264. | |
Sharma AK, Linden J, Kron IL, Laubach VE. Protection from pulmonary ischemia-reperfusion injury by adenosine A2A receptor activation. Respir Res. 2009;10:58. | |
Tracey WR, Magee W, Masamune H, Oleynek JJ, Hill RJ. Selective activation of adenosine A3 receptors with N6-(3-chlorobenzyl)-5′-N-methylcarboxamidoadenosine (CB-MECA) provides cardioprotection via KATP channel activation. Cardiovasc Res. 1998;40:138–145. | |
Mozzicato S, Joshi BV, Jacobson KA, Liang BT. Role of direct RhoA-phospholipase D1 interaction in mediating adenosine-induced protection from cardiac ischemia. FASEB J. 2004;18:406–408. | |
Von Lubitz DK, Lin RC, Popik P, Carter MF, Jacobson KA. Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol. 1994;263:59–67. | |
Chen JF, Huang Z, Ma J, et al. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19:9192–9200. | |
Isselhard W, Eitenmúller J, Mäurer W, et al. Increase in myocardial adenine nucleotides induced by adenosine: dosage, mode of application and duration, species differences. J Mol Cell Cardio. 1980;12:619–634. | |
Deutsch E, Berger M, Kussmaul WG, Hirshfeld JW Jr, Herrmann HC, Laskey WK. Adaptation to ischemia during percutaneous transluminal coronary angioplasty. Clinical, hemodynamic, and metabolic features. Circulation. 1990;82:2044–2051. | |
Liu GS, Thornton J, Van Winkle DM, Stanley AW, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–356. | |
Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J. 2006;20:2242–2250. | |
De Ponti C, Carini R, Alchera E, et al. Adenosine A2a receptor-mediated, normoxic induction of HIF-1 through PKC and PI-3K-dependent pathways in macrophages. J Leukoc Biol. 2007;82:392–402. | |
Zhong H, Chiles K, Feldser D, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–1545. | |
Baek SH, Lee UY, Park EM, Han MY, Lee YS, Park YM. Role of protein kinase Cdelta in transmitting hypoxia signal to HSF and HIF-1. J Cell Physiol. 2001;188:223–235. | |
Ahmad A, Ahmad S, Glover L, et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc Natl Acad Sci U S A. 2009;106:10684–10689. | |
Olanrewaju HA, Qin W, Feoktistov I, Scemama JL, Mustafa SJ. Adenosine A(2A) and A(2B) receptors in cultured human and porcine coronary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2000;279:H650–H656. | |
Feoktistov I, Ryzhov S, Zhong H, et al. Hypoxia modulates adenosine receptors in human endothelial and smooth muscle cells toward an A2B angiogenic phenotype. Hypertension. 2004;44:649–654. | |
Monopoli A, Lozza G, Forlani A, Mattavelli A, Ongini E. Blockade of adenosine A2A receptors by SCH 58261 results in neuroprotective effects in cerebral ischaemia in rats. Neuroreport. 1998;9:3955–3959. | |
Merighi S, Benini A, Mirandola P, et al. A3 adenosine receptors modulate hypoxia-inducible factor-1alpha expression in human A375 melanoma cells. Neoplasia. 2005;7:894–903. | |
Cohen MC, Rohtla KM, Lavery CE, Muller JE, Mittleman MA. Meta-analysis of the morning excess of acute myocardial infarction and sudden cardiac death. Am J Cardiol. 1997;79:1512–1516. | |
Eckle T, Hartmann K, Bonney S, et al. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat Med. 2012;18:774–782. | |
Feoktistov I, Goldstein AE, Biaggioni I. Role of p38 mitogen-activated protein kinase and extracellular signal-regulated protein kinase kinase in adenosine A2B receptor-mediated interleukin-8 production in human mast cells. Mol Pharmacol. 1999;55:726–734. | |
Petrzilka S, Taraborrelli C, Cavadini G, Fontana A, Birchler T. Clock gene modulation by TNF-alpha depends on calcium and p38 MAP kinase signaling. J Biol Rhythms. 2009;24:283–294. | |
Nakashima A, Kawamoto T, Honda KK, et al. DEC1 modulates the circadian phase of clock gene expression. Mol Cell Biol. 2008;28:4080–4092. | |
Wykoff CC, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Identification of novel hypoxia dependent and independent target genes of the von Hippel-Lindau (VHL) tumour suppressor by mRNA differential expression profiling. Oncogene. 2000;19:6297–6305. | |
Prass K, Ruscher K, Karsch M, et al. Desferrioxamine induces delayed tolerance against cerebral ischemia in vivo and in vitro. J Cereb Blood Flow Metab. 2002;22:520–525. | |
Nangaku M, Izuhara Y, Takizawa S, et al. A novel class of prolyl hydroxylase inhibitors induces angiogenesis and exerts organ protection against ischemia. Arterioscler Thromb Vasc Biol. 2007;27:2548–2554. | |
Hill P, Shukla D, Tran MG, et al. Inhibition of hypoxia inducible factor hydroxylases protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2008;19:39–46. | |
Karuppagounder SS, Ratan RR. Hypoxia-inducible factor prolyl hydroxylase inhibition: robust new target or another big bust for stroke therapeutics? J Cereb Blood Flow Metab. 2012;32:1347–1361. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.