")
Back to Journals » Infection and Drug Resistance » Volume 8
The role of dolutegravir in the management of HIV infections
Authors Miller M, Liedtke MD, Lockhart SM, Rathbun RC
Received 14 November 2014
Accepted for publication 5 January 2015
Published 19 February 2015 Volume 2015:8 Pages 19—29
DOI https://doi.org/10.2147/IDR.S58706
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Suresh Antony
Misty M Miller,1 Michelle D Liedtke,1 Staci M Lockhart,2 R Chris Rathbun1
1Department of Pharmacy: Clinical and Administrative Sciences, University of Oklahoma College of Pharmacy, Oklahoma City, OK, USA; 2AbbVie Inc., Oklahoma City, OK, USA
Abstract: Dolutegravir is the most recent integrase strand transfer inhibitor approved for HIV-1 infection in both treatment-naïve and experienced patients. As a tricyclic carbamoyl pyridone analog, dolutegravir is rapidly absorbed and distributes through the cerebrospinal fluid. It is hepatically metabolized by uridine diphosphate glucuronosyl transferase 1A1; no inhibition or induction of cytochrome P450 enzymes is noted. As a substrate of CYP 3A4, dolutegravir is affected by rifampin, efavirenz, tipranavir/ritonavir, fosamprenavir/ritonavir, and dose increase is required. Dolutegravir inhibits the organic cation transporter 2, resulting in decreased creatinine clearance with no apparent decrease in renal function. Other adverse effects are minimal but include diarrhea, headache, and nausea. Clinical trials in treatment-naïve and experienced patients are ongoing and will be presented in this text.
Keywords: antiretroviral, integrase inhibitor, Tivicay®, treatment-naïve studies, treatment-experienced studies
Introduction to treatment developments in HIV
Treatment developments in HIV infection have been largely dominated by the integrase inhibitors (INSTIs) in recent years with the approval of elvitegravir (EVG) coformulated with tenofovir, emtricitabine, and cobicistat in 2012 and dolutegravir (DTG) in 2013. Additionally, 2014 brought the approval of EVG as an individual agent and DTG combined with abacavir and lamiviudine in a single-dose tablet. Both agents appear to follow the lead of raltegravir (RAL) with relatively few adverse effects and minimal drug interactions. This review will evaluate the literature on DTG and identify its potential placement in the therapy of HIV.
Current and emerging therapies for the management of HIV
Guidelines for the treatment of HIV continue to list two nucleoside reverse transcriptase inhibitors (NRTIs) as the backbone of an effective antiretroviral regimen. Depending upon baseline viral load and the presence of the HLA-B*5701 allele, the backbone combines tenofovir or abacavir with either emtricitabine or lamivudine. Either a non-nucleoside reverse transcriptase inhibitor (NNRTI; ie, efavirenz, rilpivirine), a protease inhibitor (ie, atazanavir or darunavir, each boosted by ritonavir), or an INSTI (ie, RAL, DTG, or EVG boosted by cobicistat) may complete the regimen.1 Selecting an appropriate regimen relies largely on patient factors, including renal and hepatic function, baseline resistance testing, and potential prescription, over-the-counter, and illicit drug interactions. Additionally, involvement of the patient in determining incorporation of antiretroviral therapy in their daily routine, willingness to endure potential adverse effects, and feasibility of adherence cannot be underscored. Researchers have made significant advancements in the treatment of HIV over the last 30 years and continue to explore new mechanisms, delivery methods, and chemical structures in an attempt to provide the safest and most efficacious therapy to a growing and aging HIV-infected population.
One of the more widely anticipated additions to the antiretroviral armamentarium is a new formulation of an older NRTI, tenofovir. Similar to tenofovir disoproxil fumurate (TDF), tenofovir alafenamide (TAF) is a prodrug requiring conversion to its active form. However, TAF primarily undergoes this process intracellularly, decreasing plasma concentrations up to 90%.2 With decreased plasma concentrations, theoretically, TAF could lead to fewer adverse effects on kidneys and bone mineral density. Phase III comparator studies are underway with TAF in fixed dose combination with either emtricitabine or emtricitabine, EVG, and cobicistat.3–6 Another development in the INSTI class centers on cabotegravir. As an analog of DTG, cabotegravir is currently in a number of Phase II studies evaluating its use as an oral or intramuscular agent. Additionally, its role in dual therapy when used intramuscularly, along with injectable rilpivirine, is being evaluated in treatment naïve adults.7,8 The possibility of using cabotegravir as a form of pre-exposure prophylaxis is also being explored.9
Pharmacology
Structurally, DTG is a tricyclic carbamoyl pyridone analog and has activity against wild-type HIV subtype 1 (HIV-1), with a protein-adjusted 90% inhibitory concentration (IC90) of 0.064 μg/mL (Figure 1).10,11 DTG also exhibits activity against clinical isolates of HIV subtype 2 (IC50 of 0.18 nM).11 DTG inhibits the strand transfer reaction of HIV integrase that is necessary for annealing proviral deoxyribonucleic acid (DNA) to host chromosomal DNA by binding to divalent cations (eg, magnesium) in HIV integrase within the host nucleus.10,12
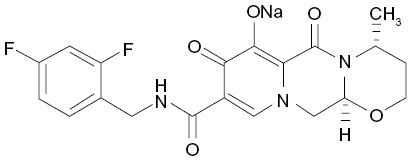 | Figure 1 Chemical structure of Tivicay® (dolutegravir 50 mg tablets). |
Pharmacokinetics
DTG is rapidly absorbed from the gastrointestinal tract (median time to maximum concentration [Tmax]: 2.1 hours) after oral administration of 50 mg under fasting conditions. The presence of food increases the extent and slows the rate of DTG absorption. A low-fat meal (300 kcal, 7% fat) increases DTG bioavailability by 33%, while a moderate-fat (600 kcal, 30% fat) and high-fat (870 kcal, 53% fat) meal increase bioavailability by 41% and 66%, respectively; Tmax is extended to 3, 4, and 5 hours, respectively, and suggests that DTG displays solubility-limited absorption.13 Administration of DTG with food is not required for INSTI-naïve patients but should be employed in patients with INSTI class resistance.11,14 The apparent volume of distribution (Vd/F) in HIV-infected subjects is 17–20 L based on population pharmacokinetics.11 DTG is greater than 99% bound to plasma proteins in a concentration-independent fashion.11 Distribution into the cerebrospinal fluid (CSF) and the male and female genital tract is evident; CSF concentrations (median: 18 ng/mL, range: 4–232 ng/mL) are similar to unbound serum concentrations (16.8 ng/mL); semen, rectal, and cervicovaginal tissue concentrations are 7%, 17%, and 7%–10% of steady-state serum concentrations.15–17
DTG is hepatically metabolized by uridine diphosphate glucuronosyl transferase 1A1 (UGT1A1) and is a minor substrate in vitro for UGT1A3, UGT1A9, and cytochrome P450 3A4 (CYP3A4).11,18 Four metabolites have been detected but represent less than 5% of circulating DTG and are not considered to contribute to efficacy or adverse event outcomes.19 DTG has a terminal elimination half-life of 12 hours in HIV-infected subjects.20 DTG clearance is 32% lower in patients who are poor metabolizers through UGT1A1.21 DTG pharmacokinetics are nonlinear, but dose-proportional increases in serum concentration are generally observed between 25 and 50 mg with the tablet formulation.11 DTG displays limited interpatient variability (coefficients of variation below 30% for area under the serum-concentration time curve [AUC], maximum serum concentration [Cmax], and minimum serum concentration [Cmin]).20 No difference in pharmacokinetics is evident according to age, race, or sex.11 Individuals with moderate hepatic disease (Child-Pugh class B) have a 1.5- to 2-fold increase in unbound DTG serum concentration but do not require a change in dose; the pharmacokinetics of DTG have not been studied in patients with severe hepatic impairment (Child-Pugh class C).11,22 Renal excretion accounts for 32% of the glucuronide metabolite elimination but less than 1% of unchanged DTG. DTG is also a substrate for the transport proteins p-glycoprotein and the breast cancer resistance protein.18
Drug interactions
DTG does not exhibit induction or inhibition of CYP450 enzymes or UGT1A1 at physiologic concentrations.18 DTG inhibits the organic cation transporter 2 (OCT2) (IC50=1.93 μM), and multidrug and toxin extrusion protein (MATE-1) (IC50=6.34 μM), resulting in a 10%–14% decrease in creatinine clearance.14,23 This may result in elevated plasma concentrations for drugs that are substrates for these transporters. Administration of DTG with dofetilide is contraindicated because dofetilide renal tubular secretion may be altered; metformin serum concentration may also be increased by this mechanism and warrants closer monitoring for metformin-related adverse events (eg, lactic acidosis; see Table 1).14 Inhibition of the renal uptake transporters, organic anion transporters 1 and 3 (OAT1, OAT3), is observed in vitro (IC50=2.12 and 1.97 μM, respectively), but the absence of effect on the pharmacokinetics of the OAT substrates tenofovir and para-aminohippurate suggest that in vivo inhibition is not clinically significant.11,14,24
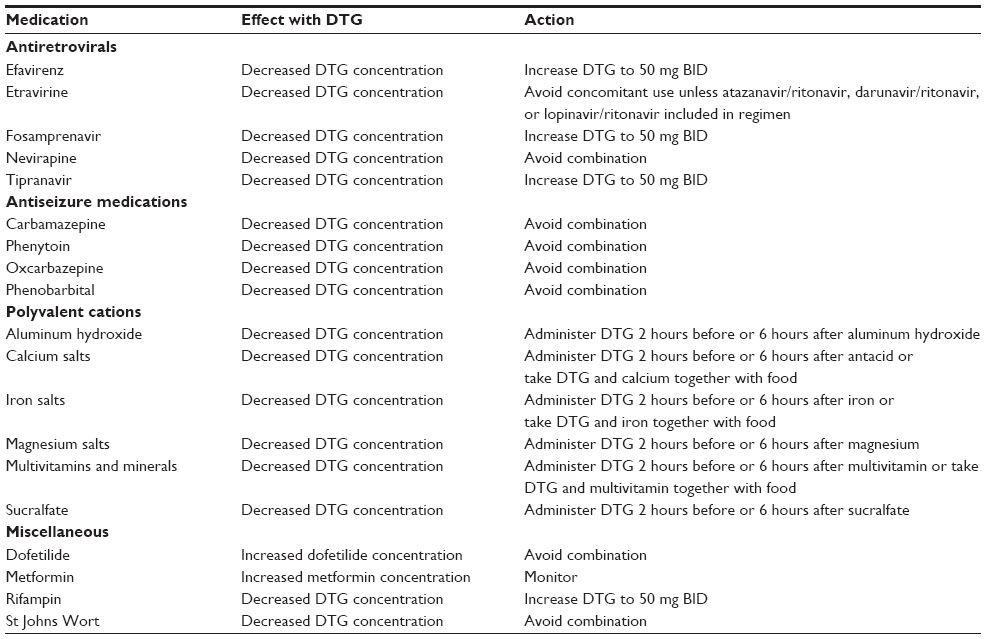 | Table 1 Clinically relevant drug interactions |
DTG does not interact with the HIV protease inhibitors atazanavir, atazanavir/ritonavir, darunavir/ritonavir, nelfinavir, and lopinavir/ritonavir and can be coadministered without dosage adjustment.14,25,26 Tipranavir/ritonavir decreases DTG AUC and Cmin by 59% and 76%, respectively. Therefore, the dose of DTG should be increased to 50 mg twice daily when coadministered with tipranavir/ritonavir in patients without INSTI resistance.11,27 Fosamprenavir/ritonavir similarly decreases DTG AUC and Cmin (35% and 49%, respectively) and requires DTG dosage adjustment to 50 mg twice daily based on US labeling when administered in patients without INSTI resistance; however, no dosage adjustment is recommended under European labeling.11,14,28 Administration of DTG with either tipranavir/ritonavir or fosamprenavir/ritonavir should be avoided when INSTI resistance is present.11 The NNRTI, efavirenz, reduces the DTG AUC and Cmin by 57% and 75%, respectively. In the absence of protease inhibitor coadministration, the dose of DTG should be increased to 50 mg twice daily when given with efavirenz in patients with no preexisting INSTI resistance.27 Administration of DTG with nevirapine is predicted to result in lower DTG serum concentrations but has not been studied; US labeling recommends avoiding this combination, whereas European labeling recommends increasing the DTG dose to 50 mg twice daily.11,14 Coadministration of DTG and etravirine should be avoided due to reduction in DTG serum concentrations (71% decline in AUC) unless given with either darunavir/ritonavir, atazanavir/ritonavir, or lopinavir/ritonavir.29 No dosage adjustment is required with rilpivirine coadministration.30 DTG can be administered with NRTIs at standard dosage.14,24
DTG serum concentrations may be altered when administered with some non-antiretroviral medications and dietary supplements. Rifampin (600 mg/d) reduces DTG AUC by 54%, but administration of DTG 50 mg twice daily with rifampin achieves DTG Cmin values that are 20%–30% higher than with standard once-daily dosing.14,31 DTG systemic exposure is decreased by 30% by rifabutin (300 mg/d) but is not predicted to require an increase in DTG dosage.31 DTG administration with antiepileptic medications (eg, phenytoin, phenobarbital, carbamazepine) has not been studied and should be avoided.11,14 St John’s wort (Hypericum perforatum) is predicted to lower DTG serum concentrations and should not be coadministered.11 Oral administration of magnesium/aluminum-containing antacids, iron and calcium supplements, and multivitamins can decrease DTG systemic exposure by 30%–75% and should be administered either 6 hours before or 2 hours after DTG to minimize the potential for decreased DTG absorption.32 Administration of DTG with sucralfate has not been studied but should be administered in the same manner to avoid an interaction.14 As an alternative, iron and calcium supplements and multivitamins can be administered simultaneously with DTG if taken with food.33
Resistance
DTG is considered a second-generation INSTI and exhibits a higher barrier to the development of resistance than other currently-available INSTIs (RAL, EVG).34,35 DTG has a prolonged dissociation rate half-life (70 hours) that may contribute to its lower predilection for resistance development.36 DTG demonstrates activity against RAL- and EVG-resistant strains of HIV-1 with common resistance-associated mutations (RAMs) including Y143CHR, N155H, and Q148HKR in the absence of secondary mutations.34,37 Up to 4-fold decline in susceptibility to DTG has been observed during serial passage of wild-type HIV-1 strains in cell culture, leading to emergence of E92Q, G118R, S153FY, G193E, or R263K. Viruses harboring Q148HR selected additional mutations (T97A, E138K, G140S, and M154I) during serial passages and exhibited a 13- to 46-fold loss of DTG activity. The combination of Q148HR with G140S led to acquisition of additional RAMs (L74M, E92Q, N155H) during subsequent passages.14
No resistance to DTG has been reported in clinical studies involving antiretroviral-naïve patients (SPRING-1, SPRING-2, SINGLE, FLAMINGO).38–41 Less than 1% of antiretroviral-experienced, INSTI-naïve patients have developed treatment-emergent RAMs to DTG during clinical studies to date; however, 45% of patients with baseline INSTI resistance demonstrated treatment-emergent RAMs while receiving DTG.14,42,43 Findings from the VIKING-2 and -3 trials demonstrate that acquisition of multiple INSTI RAMs is generally necessary to significantly impair the activity of DTG. INSTI-experienced patients with Q148HR and two or more additional INSTI mutations (L74IM, E138ADKT, G140A/S, Y143HR, E157Q, G163EKQRS, G193ER) experienced the most substantial loss of DTG activity, with only 18% of patients achieving HIV-1 RNA below 50 copies/mL after 24 weeks.14,43,44 The most common combination of substitutions occurred at Q148+ G140+ E138. Substitutions at N155 or Y143 without Q148 or Q148HR with G140AS alone conferred less loss of activity, with viral suppression achieved in 56%–80% of patients.14,44 Resistance to DTG has been demonstrated by the G118R and F121Y mutations recently described in two patients failing RAL-based antiretroviral therapy, leading to greater than 100-fold decline in susceptibility.45 Results from national genotypic resistance test surveillance data suggest that 12% of patients with RAL- or EVG-resistant virus also have high-level DTG resistance, representing 2% of all patients undergoing genotypic testing between 2009–2012.46
Comparative efficacy of DTG
Treatment-naïve studies
The utility of DTG in HIV-infected, treatment-naïve individuals has been examined in three randomized, multicenter Phase III trials to establish safety, efficacy, and tolerability. An additional Phase III trial, exclusively in HIV-infected, treatment-naïve women, is currently underway. In each of the initial three trials, patients were stratified by baseline viral load ≤100,000 copies/mL or >100,000 copies/mL. Notable findings included demonstrated efficacy, good overall safety, minimal adverse effects, rapid virologic suppression, minimal virologic breakthrough, and lack of INSTI or other major mutations.
SPRING-2
SPRING-2 was the first Phase III trial in treatment-naïve patients that compared DTG 50 mg daily to RAL 400 mg twice daily in combination with an investigator selected NRTI-backbone of either tenofovir/emtricitabine or abacavir/lamivudine. The study design was a noninferiority, randomized, multicenter, placebo-controlled trial. An intent-to-treat population was used for analyses. The primary endpoint was the number of patients with viral load <50 copies/mL at week 48. Secondary objectives included change in CD4 cell counts, adverse effects, and resistance. Primary and secondary objectives were reassessed at week 96. Patient demographics included median age of 36 years, 85% men, 85% white race, 28% baseline viral load >100,000 copies/mL, 13% CD4 cell counts <200 cells/mm3, and less than 10% with hepatitis B or hepatitis C co-infection.
In total, 822 patients were randomized and received study drugs (411 DTG, 411 RAL). Sixty percent had tenofovir/emtricitabine chosen as the NRTI backbone. At week 48, 88% in the DTG group and 86% in the RAL group reached viral suppression and met the 10% noninferiority criteria. No significant difference in achieving the primary endpoint was found to be associated with NRTI-backbone or baseline viral load. Changes in CD4 cell counts were similar between DTG and RAL at week 48; however, there was a favorable trend toward DTG in patients with baseline CD4 cell counts less than 350 and less than 200 cells/mm3 (86% vs 80% and 78% vs 68%, respectively) but it did not reach statistical significance. Other similarities included safety profiles, adverse events leading to discontinuation, laboratory changes, and time to viral suppression. Of interest, more patients in the RAL group had protocol-defined virologic failure (PDVF) compared to the DTG group. Of those that met PDVF, five patients in the RAL group had genotypic mutations (1 INSTI, 4 NRTI) while none were noted in the DTG group.39
There were a total of 681 patients that completed 96 weeks of treatment. Findings were reflective of those at 48 weeks. Viral suppression occurred in 81% in the DTG group compared to 76% of those treated with RAL. Similar CD4 cell count increases and rates of discontinuation were noted. An additional three patients had PDVF between weeks 48 and 96 (2 DTG, 1 RAL); however, no resistance mutations were detected in either group.47
SINGLE
The SINGLE study compared once daily DTG 50 mg with fixed-dose abacavir/lamivudine to tenofovir/emtricitabine/efavirenz as a fixed-dose combination tablet through 144 weeks. Each patient received three tablets per day: study drugs plus matching placebo(s). The study design was a noninferiority, randomized, double-blind, multicenter trial. If noninferiority was met in both per-protocol and intent-to-treat analyses, superiority would be assessed. The primary endpoint was the number of patients with viral load <50 copies/mL at week 48. Secondary endpoints were time to viral suppression and change in CD4 cell count. Additional assessments were performed at week 96. An open-label phase through 144 weeks was extended to patients that met week 96 objectives. Patient demographics were similar to those in the SPRING-2 trial with a median age of 35 years, 84% men, 68% white race, 31% baseline viral load >100,000 copies/mL, 14% CD4 cell counts <200 cells/mm3, and less than 10% hepatitis B or hepatitis C co-infection.
A total of 844 patients were randomized with 833 patients receiving study drug (414 DTG + ABC/3TC, 419 TDF/FTC/EFV). Patients were stratified according to baseline viral load and CD4 cell count (>200 cells/mm3 or ≤200 cells/mm3). At week 48, viral load <50 copies/mL was achieved in 88% of patients in the DTG + ABC/3TC group compared to 81% in the TDF/FTC/EFV group; the difference between the two groups met noninferiority criteria. Superiority analysis demonstrated a statistically significant difference between the DTG-treated patients and EFV-treated patients (P=0.003). Furthermore, no difference in virologic response was noted in baseline viral load or other subgroups. For secondary endpoints, a greater change in CD4 cell count was found to be statistically significant for DTG (267 cells/mm3 vs 208 cells/mm3, P<0.001) and time to viral suppression was shorter for DTG (28 days vs 84 days). PDVF was similar between the treatment arms and occurred in 4% of the patients. Consistent with other studies to date, no major NRTI or INSTI mutations were detected in the DTG arm, however, 1 NRTI and 4 NNRTI were noted in the TDF/FTC/EFV arm.40
All 844 patients from the 48-week study period continued through 96 weeks. Findings remained similar to those demonstrated at 48 weeks. The number of patients with viral load <50 copies/mL was higher in DTG arm compared to the TDF/FTC/EFV arm (80% vs 72%) and did reach statistical significance (P=0.006). Additionally CD4 cell count increases continued to be greater with DTG (325 cells/mm3 vs 281 cells/mm3, P=0.004). Rates of virologic failure between groups were similar. Virologic resistance was not observed between weeks 48 and 96 in the DTG group.48 However, resistance was detected in seven patients in the TDF/FTC/EFV arm (one NRTI, six NNRTI).
During the open-label phase from 96 weeks to 144 weeks, DTG continued to demonstrate superiority and long-term viral load suppression compared to TDF/FTC/EFV (71% vs 63%; P=0.010). Differences in rates of viral suppression were noted between groups for baseline viral load >100,000 copies/mL (69% DTG vs 61% EFV), women (69% DTG vs 48% EFV), and nonwhite race (71% DTG vs 47% EFV); however, statistical significance was not reported. Discontinuation of study drug occurred more frequently in the TDF/FTC/EFV arm (14% EFV vs 4% DTG). PDVF was similar between groups (9% DTG vs 8% EFV); however, genotypic resistance was not detected in the DTG arm. Seven patients in the EFV arm had detectable genotypic resistance (one NRTI, six NNRTI).49
FLAMINGO
FLAMINGO was a 96 week trial that compared DTG 50 mg daily to darunavir/ritonavir (DRV/RTV) 800 mg/100 mg daily. The NRTI backbone consisted of either ABC/3TC or TDF/FTC and was left to the discretion of the investigator. The study design was a noninferiority, open-label, randomized, multicenter trial. A modified intent-to-treat population was used for analyses. Additionally, if noninferiority was met, superiority would be assessed for primary and secondary objectives. The primary endpoint was the number of patients with viral load <50 copies/mL at week 48. Secondary endpoints were more extensive than previous trials and included change in CD4 cell counts, time to virologic suppression, adverse events, rates of discontinuation, laboratory changes, virologic resistance, disease progression, and health outcomes measures. Patient demographics were again similar to the SPRING-2 and SINGLE trials with median age of 34 years, 85% men, 23% African American, 25% baseline viral load >100,000 copies/mL, 8% CD4 cell counts <200 cells/mm3, and <10% hepatitis B or hepatitis C co-infection.
A total of 484 patients received study drug (243 DTG, 242 DRV/RTV). Thirty three percent of patients had abacavir/lamivudine selected for the NRTI backbone. Patients were stratified by baseline viral load (≤100,000 copies/mL or >100,000 copies/mL) and NRTI backbone. At week 48, 90% in the DTG group and 83% in the DRV/RTV group achieved viral load <50 copies/mL; the difference met noninferiority criteria as well as superiority (P=0.025). Furthermore, time to viral suppression at week 8 was notably shorter for DTG vs DRV/RTV (87% vs 31%). Secondary endpoints that were similar between groups through week 48 included CD4 cell count increases, PDVF, and adverse events. In patients with PDVF, no primary NRTI, INSTI, or PI mutations were detected.41 Ninety-six week results have yet to be published.
ARIA
ARIA is a Phase III, 48-week, open-label trial exclusive to HIV-infected, treatment-naïve women. Recruitment is currently underway.50
Treatment-experienced studies
There are currently three published studies examining the use of DTG in HIV-infected, treatment experienced patients. The SAILING study currently has published 48-week results and compares DTG to RAL in antiretroviral experienced patients who are INSTI naïve. The VIKING and VIKING 3 trials were conducted in antiretroviral and INSTI experienced patients who had evidence of INSTI resistance. Table 2 summarizes primary outcome and safety.
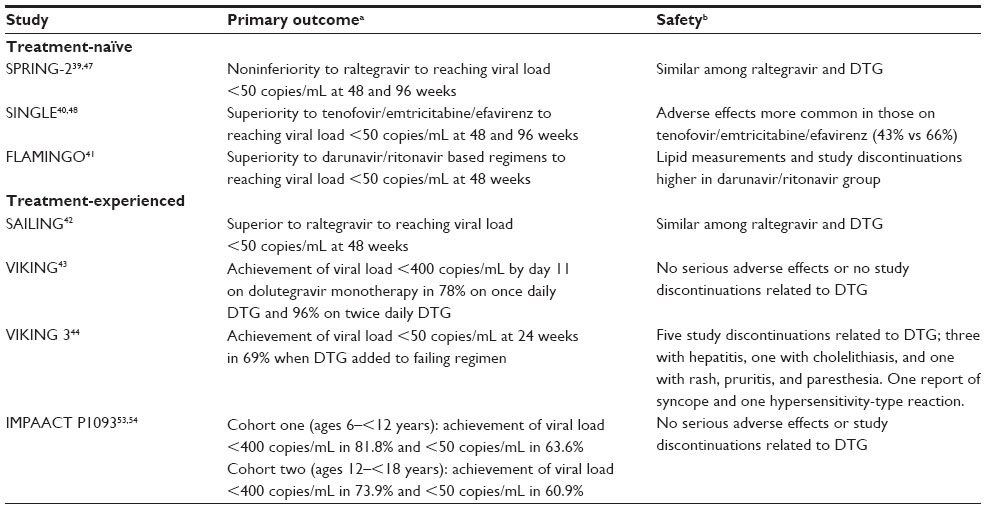 | Table 2 Summary of dolutegravir (DTG) efficacy studies |
SAILING
SAILING is an ongoing Phase III clinical trial conducted in adult patients with HIV-1 who are antiretroviral experienced but INSTI naïve.42 It is a randomized, double-blind, active-controlled, double-placebo study conducted at 156 sites across several countries. Ultimately 719 patients were treated, 357 in the DTG arm and 362 in the RAL arm.
Patients eligible for the study had to have prior antiretroviral treatment experience with documented resistance to two or more classes of antiretroviral agents but one or two fully active agents available for the background therapy. At the time of screening, patients had to have two consecutive plasma viral loads ≥400 copies/mL.
Study participants were randomly assigned (1:1) to receive either DTG 50 mg once daily or RAL 400 mg twice daily plus an investigator selected background therapy. The background regimen had to consist of at least one fully active antiretroviral agent and could include a second agent with or without full activity. In order to assist in isolating DTG’s contribution to viral suppression, the number of patients whose regimen included DRV/RTV (without primary protease inhibitor resistance) was capped at 170 patients. Patients were stratified based on HIV-1 viral load ≤50,000 or >50,000 copies/mL, inclusion of DRV/RTV (with or without primary protease inhibitor resistance), and number of active agents in the background regimen (two vs fewer than two fully active background agents). Study sites were to remain masked and matching placebo given up to week 48.
The treatment arms were well balanced based on disease characteristics and treatment history. Notable demographics include median age 43 years, 49% white, 42% African American, baseline viral load 4.18 log10 copies/mL, CD4 200 cells/mm3, and 5% Hepatitis B and 11% Hepatitis C co-infection. Secondary to the broad geographical participation, the population was diverse and included a high percentage of female patients (32%) and a high number of patients listed as nonwhite/Caucasian/European heritage (51%). Patients in the study also generally had more advanced disease with almost half of the population having a previous AIDS diagnosis and resistance to 3 or more classes of antiretrovirals.
The primary endpoint of the study was the proportion of patients with plasma HIV-1 RNA <50 copies/mL at week 48. There were several additional secondary endpoints looking at efficacy and safety of DTG. At week 48, 251 (71%) of 354 patients in the DTG group and 230 (64%) of 361 patients in the RAL group achieved a plasma viral load <50 copies/mL. The adjusted treatment difference was determined to be 7.4% (95% CI: 0.7–14.2). The prespecified treatment difference for noninferiority was set at -12%. Based on the large difference, a predetermined superiority test was applied, and DTG was found to be superior to RAL (P=0.03). PDVF occurred more frequently in the RAL group (12% vs 6%) by week 48. Of these virologic failures, 19 (42%) of the RAL patients and only two (10%) of the DTG patients were found to be nonresponders as opposed to failures due to virologic rebound. CD4 cell count increased in both groups with mean changes of 162 cells/mm3 in the DTG arm and 153 cells/mm3 in the RAL arm. In the subgroup analysis, DTG also performed better than RAL in the more advanced patients, including those with plasma viral loads >50,000 copies/mL and those not on DRV/RTV or those on DRV/RTV but with primary protease inhibitor mutations.
Fewer patients in the DTG group were found to have treatment-emergent genotypic or phenotypic evidence of INSTI resistance at week 48 (4 of 354 DTG patients vs 17 of 361 RAL patients, P=0.003). Of these patients, one in each group had RAL primary resistance at baseline. The remaining three DTG failures had a less than twofold change in susceptibility to DTG and as such, the study concluded that no treatment-emergent phenotypic resistance to DTG or RAL was reported from any DTG treated patient. Sixteen RAL treated patients with virologic failure had high fold-change to RAL but limited cross-resistance to DTG. Twelve RAL vs four DTG patients had treatment-emergent resistance to their background regimen at week 48.
VIKING
VIKING was a 24-week, Phase IIb pilot study designed to examine safety and efficacy of DTG in treatment-experienced subjects with genotypic evidence of RAL resistance.43 It was a multicenter study including 25 study sites. Initially patients were treated with DTG 50 mg daily; however, the protocol was amended to include a second cohort (DTG 50 mg twice daily) due to low viral load response among some patients in cohort one. Patients in cohort two were also required to have one fully active antiretroviral agent in the optimized background regimen; this was not required in cohort one. The study consisted of a 10-day functional monotherapy where DTG was used in place of RAL in the patients’ failing regimen. On day 11, the background regimen was optimized using resistance information. Fifty-one patients were enrolled with 27 patients in cohort one and 24 patients in cohort two.
The primary endpoint at day eleven was a reduction in plasma viral load of ≥0.7 log10 copies/mL below baseline or a viral load <400 copies/mL. Seventy-eight percent (21/27) of patients in cohort one and 96% (23/24) in cohort two achieved the primary outcome. At week 24, 11/27 (41%) patients in cohort one and 18/24 (75%) patients in cohort two achieved a viral load <50 copies/mL. The median cell/mm3 increase in CD4 cells was 54 in cohort one and 60 in cohort two.
VIKING 3
VIKING 3 is an ongoing Phase III study that was designed to build on the results of VIKING.44,51,52 Researchers intended to determine outcomes in DTG treated patients with documented resistance to either RAL and/or EVG. This study is a single-arm, open-label, study conducted at 65 sites in the US, Canada, and Europe. It was designed with a 7-day functional monotherapy period in which DTG 50 mg twice daily was substituted for RAL or EVG, but the patient’s background regimen remained the same. After those 7 days, the background regimen was optimized according to resistance data. The study was designed to report primary outcome data at week 24, though patients were allowed to continue the study and some of the secondary outcomes were set for 48 weeks. Currently only the 24-week data is published though 48-week data has been presented in abstract form.
The study included adult patients with screening or documented history of resistance to RAL and/or EVG and to ≥2 other antiretroviral classes. Patients had to have plasma viral load ≥500 copies/mL and had to have at least one active antiretroviral available for the background regimen. Patients treated with efavirenz or nevirapine within 14 days of DTG start were excluded. Patients on etravirine were only included if it was coadministered with LPV/RTV or DRV/RTV. Patients treated with TPV/RTV or FMP/RTV were only allowed to start on day 8 and only if subjects did not have Q148+≥2 associated mutations.
A total of 183 patients were enrolled and received at least one dose of DTG. Median age was 48 years, 77% male, and 71% white race. Average baseline viral load was 4.38 log10 copies/mL, and CD4 140 cells/mm3. Similar to other studies, Hepatitis B co-infection rate was 5% but Hepatitis C was 14%. Seventy-three percent had baseline genotypic and/or phenotypic INSTI resistance; the remaining 27% had historic evidence only. The median fold-change to RAL was 47.5 while the median DTG fold-change was 1.29. Seventy-three percent of patients had ≥3 NRTI major mutations, 70% had ≥2 major PI mutations, and 59% had ≥2 NNRTI major mutations.
The primary endpoints for the study were the mean change from baseline in plasma viral load at day 8 and the proportion of subjects with <50 copies/mL at week 24. At day 8, the mean change in plasma viral load from baseline was 1.43 log10 copies/mL. At week 24, 69% (126/183) of subjects achieved a plasma viral load <50 copies/mL. Subgroup analysis showed the least response among patients with a DTG fold-change >10 and those harboring Q148+≥2 mutations. The group with no Q148 mutation present had the highest response rates. Increasing activity of the background regimen did not significantly improve day 8 or week 24 response. In analysis, baseline INSTI resistance and baseline viral load were highly significant predictors for week 24 response. For each two-fold increase in DTG fold-change, a 63% lower chance of achieving viral load <50 copies/mL was found. For every 10-fold increase in baseline viral load, the odds of achieving viral load <50 copies/mL were 80% lower. The median change in CD4 cell count at week 24 was 61 cells/mm3. Secondary outcomes included efficacy at week 48 and have only been presented in abstract form thus far. At week 48, 116/183 (63%) patients maintained a viral load <50 copies/mL. Again the subgroup with no Q148 mutation present had the highest response rate with 71% achieving <50 copies/mL at week 48.
IMPAACT P1093
The IMPAACT P1093 study examines the use of DTG in a pediatric patient population.53,54 Currently DTG is approved for use in children ≥12 years old and weighing at least 40 kg.14 These patients can be treatment-experienced, but must be INSTI naïve. The P1093 study patient population is broken down into five cohorts based on age. To date, data has been presented in abstract form for cohort one consisting of patients ≥12 years old but <18 years old and cohort two consisting of patients ≥6 years old and <12 years old.53,54 Patients had to have a viral load >1,000 copies/mL and have at least one active agent available for their background regimen. Demographics in cohort one include median age of 10 years, 63.6% male, 36% African American, baseline viral load of 5 log10 copies/mL, and CD4 645 cells/mm3. Cohort two displayed demographics contrasting from most other trials with 78.3% female and 52% African American patients. In addition, median age was 15 years, baseline viral load 4.3 log10 copies/mL, and CD4 466 cells/mm3; no patients in either cohort were infected with Hepatitis B or C.
In cohort one, 23 patients were enrolled and 22 had follow-up through 48 weeks.54 DTG dose was weight based with patients ≥40 kg receiving 50 mg a day and patients between 30 and 40 kg receiving 35 mg a day. Nineteen patients in the cohort received the 50 mg dose while only four patients received the 35 mg dose. A viral load <400 copies/mL was achieved in 73.9% (17/23) of patients at week 48; a viral load <50 copies/mL was achieved by 60.9% (14/23) of patients. The median gain in CD4 cells was 84 cells/mm3.
In cohort two, eleven children were enrolled and completed 24 weeks of therapy.53 Again DTG dose was based on weight. Five patients were on 50 mg daily (≥40 kg), two patients received 35 mg daily (30–<40 kg), and four patients received 25 mg daily (20–<30 kg). Virologic success defined as viral load <400 copies/mL or viral load decrease >1 log10 was achieved in 81.8% (9/11) patients. A viral load of <50 copies/mL was achieved in 63.6% (7/11) of patients. Median increase in CD4 count at week 24 was 209 cells/mm3.
Overall safety
Patients receiving DTG experienced relatively few adverse effects during trials. Consistent through each of the naïve trials in patients taking DTG was a rise in serum creatinine, reflective of the inhibition of renal tubular creatinine secretion by OCT2. Other commonly reported effects included diarrhea, headache, nausea, and upper respiratory tract infections. Discontinuation rates were similar between DTG and comparator antiretrovirals. No deaths related to study drug occurred in any trial.
Patient focused perspectives
Adherence to an antiretroviral regimen is imperative to achieve virologic control. A number of factors contribute to adherence, including number of pills per day, frequency of doses, and adverse effect profile.55–58 Virologic control can contribute to improved quality of life in patients infected with HIV; this in turn can improve antiretroviral adherence.1,55 With available once daily dosing, a new fixed dose combination with abacavir and lamivudine, and a relatively low adverse effect profile, DTG meets these characteristics.
While the majority of information concerning patients’ satisfaction with DTG is anecdotal evidence, the FLAMINGO study did include patient satisfaction outcomes as part of the secondary endpoint. The HIV treatment satisfaction questionnaire (HIVTSQ), HIV Symptoms Index (HSI), and the European Quality of Life-5 Dimensions (ED-5D) were administered at weeks 4, 24, 48, and 96. No differences were observed in the HSI and EQ-5D surveys. There was a statistically significant difference at weeks 24 and 48 for the HIVTSQ questionnaire, in favor of DTG (P=0.003; 0.48).59
Conclusion
To date, clinical trials evaluating the safety and efficacy of DTG justify its role as first line therapy for treatment-naïve patients infected with HIV. In treatment-experienced patients, the quantity and type of resistance mutations must be considered prior to initiating therapy. Among the INSTIs, DTG has the benefit of once daily administration as in EVG, but with relatively few drug interactions, similar to RAL. Additionally, it offers the only single tablet regimen for patient with or at risk for renal dysfunction. Providers should consider the benefit of sequencing first- and second-generation INSTIs, but this should not preclude the use of DTG in a patient. Lack of significant drug interactions, minimal adverse effects, and the availability of a single tablet regimen make DTG a valuable and viable option for both patients and providers.
Disclosure
The authors report no conflicts of interest in this work.
References
Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Washington, DC: Department of Health and Human Services. Available from: http://aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf. Updated May 1, 2014. Accessed October 8, 2014. | |
Sax PE, Zolopa A, Brar I, et al. Tenofovir alafenamide vs tenofovir disoproxil fumarate in single tablet regimens for initial HIV-1 therapy: a randomized phase 2 study. J Acquir Immune Defic Syndr. 2014;67(1):52–58. | |
Gilead Sciences. Study to evaluate the safety and efficacy of elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide versus elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate in HIV-1 positive, antiretroviral treatment-naïve adults. Available from: http://clinicaltrials.gov/ct2/show/NCT01797445. NLM Identifier: NCT01797445. Accessed January 5, 2015. | |
Gilead Sciences. Phase 3 open-label study to evaluate switching from optimized stable antiretroviral regimens containing darunavir to elvitegravir/cobicistat/emtricitabine/tenofovir alafenamide single tablet regimen plus darunavir in treatment experienced HIV-1 positive adults. Available from: http://clinicaltrials.gov/ct2/show/NCT01968551. NLM Identifier: NCT01968551. Accessed January 5, 2015. | |
Gilead Sciences. Open-label study to evaluate switching from a TDF-containing combination regimen to a TAF-containing combination single table regimen in virologically-suppressed, HIV-1 positive subjects. Available from: http://clinicaltrials.gov/ct2/show/NCT01815736. NLM Identifier: NCT01815736. Accessed January 5, 2015. | |
Gilead Sciences. Switch study to evaluate F/TAF in HIV-1 positive participants who are virologically suppressed on regimens containing TDF/FTC. Available from: http://clinicaltrials.gov/ct2/show/NCT02121795. NLM Identifier: NCT02121795. Accessed January 5, 2015. | |
Viiv Healthcare. Dose ranging study of GSK1265744 plus nucleoside reverse transcriptase inhibitors for induction of Human Immunodeficiency Virus-1 virologic suppression followed by virologic suppression maintenance by GSK1265744 plus rilpivirine. Available from: http://clinicaltrials.gov/ct2/show/NCT01641809. NLM Identifier: NCT01641809. Accessed January 5, 2015. | |
Viiv Healthcare. A phase 2b study to evaluate a long-acting intramuscular regimen for maintenance of virologic suppression (following induction with an oral regimen of GK1265744 and abacavir/lamivudine) in Human Immunodeficiency Virus Type 1 infected, antiretroviral therapy-naïve adult subjects. Available from: http://clinicaltrials.gov/ct2/show/NCT02120352. NLM Identifier: NCT02120352. Accessed January 5, 2015. | |
Andrews CD, Spreen WR, Mohri, et al. Long-acting integrase inhibitor protects macaques from intrarectal simian/human immunodeficiency virus. Science. 2014;343:1151–1154. | |
Johns BA, Kawasuji T, Weatherhead JG, et al. Carbamoyl pyridine HIV-1 integrase inhibitors 3: a diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J Med Chem. 2013;56:5901–5916. doi:10.1021/jm400645w. | |
European Medicines Agency. Product information for Tivicay, 2014. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002753/WC500160680.pdf. Accessed November 6, 2014. | |
Hare S, Smith SJ, Métifiot M, et al. Structural and functional analysis of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol Pharmacol. 2011;80:565–572. doi:10.1124/mol.111.073189. | |
Song I, Borland J, Chen S, et al. Effect of food on the pharmacokinetics of the integrase inhibitor dolutegravir. Antimicrob Agents Chemother. 2012;56:1627–1629. doi:10.1128/AAC.05739-11. | |
Tivicay (dolutegravir) tablet [product information]. Research Triangle Park, NC: GlaxoSmithKline; 2014. | |
Letendre SL, Mills AM, Tashima KT, et al. ING116070: A study of the pharmacokinetics and antiviral activity of dolutegravir in cerebrospinal fluid in HIV-1-infected, ART-naïve subjects. Clin Infect Dis. 2014;59:1032–1037. doi:10.1093/cid/ciu477. | |
Greener BN, Patterson KB, Prince HM, et al. Dolutegravir pharmacokinetics in the genital tract and colorectum of HIV-negative men after single and multiple dosing. J Acquir Immune Defic Syndr. 2013;64:39–44. doi:10.1097/QAI.0b013e31829ed7a4. | |
Adams JL, Patterson KB, Prince HM, et al. Single and multiple dose pharmacokinetics of dolutegravir in the genital tract of HIV-negative women. Antivir Ther. 2013;18:1005–1013. doi:10.3851/IMP2665. | |
Reese MJ, Savina PM, Generaux GT, et al. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab Dispos. 2013;41:353–361. doi:10.1124/dmd.112.048918. | |
Castellino S, Moss L, Wagner D, et al. Metabolism, excretion, and mass balance of the HIV-1 integrase inhibitor dolutegravir in humans. Antimicrob Agents Chemother. 2013;57:3536–3546. doi:10.1128/AAC.00292-13. | |
Min S, Sloan L, DeJesus E, et al. Antiviral activity, safety, and pharmacokinetics/pharmacodynamics of dolutegravir as 10-day monotherapy in HIV-1-infected adults. AIDS. 2011;25:1737–1745. doi:10.1097/QAD.0b013e32834a1dd9. | |
Chen S, St Jean P, Borland J, et al. Evaluation of the effect of UGT1A1 polymorphisms on dolutegravir pharmacokinetics. Pharmacogenomics. 2014;15:9–16. doi:10.2217/pgs.13.190. | |
Song IH, Borland J, Savina PM, et al. Pharmacokinetics of single-dose dolutegravir in HIV-seronegative subjects with moderate hepatic impairment compared to healthy matched controls. Clin Pharmacol Drug Dev. 2013;2:342–348. doi:10.1002/cpdd.55. | |
Koteff J, Borland J, Chen S, et al. A phase 1 study to evaluate dolutegravir’s effect on renal function via measurement of iohexol and para-aminohippurate clearance in healthy subjects. Br J Clin Pharmacol 2013;75:990–996. doi:10.1111/j.1365-2125.2012.0440.x. | |
Song I, Min SS, Borland J, et al. Lack of interaction between the HIV integrase inhibitor S/GSK1349572 and tenofovir in healthy subjects. J Acquir Immune Defic Syndr. 2010;55:365–367. doi:10.1097/QAI.0b013e3181e67909. | |
Song I, Borland J, Chen S, et al. Effect of atazanavir and atazanavir/ritonavir on the pharmacokinetics of the next-generation HIV integrase inhibitor, S/GSK1349572. Br J Clin Pharmacol. 2011;72:103–108. doi:10.1111/j.1365-2125.2011.03947.x. | |
Song I, Min SS, Borland J, et al. The effect of lopinavir/ritonavir and darunavir/ritonavir on the HIV integrase inhibitor S/GSK1349572 in healthy participants. J Clin Pharmacol. 2011;51:237–242. doi:10.1177/0091270010371113. | |
Song I, Borland, J, Lou Y, et al. Effects of the enzyme inducers, tipranavir and efavirenz, on the pharmacokinetics of the integrase inhibitor, dolutegravir (S/GSK1349572) [abstract O-02]. Paper presented at: 12th International Workshop on Clinical Pharmacology of HIV Therapy; April 13–15, 2011; Miami, FL. | |
Song I, Borland J, Chen S, et al. Effect of fosamprenavir/ritonavir on the pharmacokinetics of the integrase inhibitor, dolutegravir, in healthy subjects [abstract A1-1727]. Paper presented at: 51st Annual Interscience Conference on Antimicrobial Agent and Chemotherapy; September 17–20, 2011; Chicago, IL. | |
Song I, Borland J, Min S, et al. Effects of etravirine alone and with ritonavir-boosted protease inhibitors on the pharmacokinetics of dolutegravir. Antimicrob Agents Chemother. 2011;55:3517–3521. doi:10.1128/AAC.00073-11. | |
Ford SL, Gould E, Chen S, et al. Lack of pharmacokinetic interaction between rilpivirine and integrase inhibitors dolutegravir and GSK1265744. Antimicrob Agents Chemother. 2014;57(11):5472–5477. doi:10.1128/AAC.01235-13. | |
Dooley KE, Sayre P, Borland J, et al. Safety, tolerability, and pharmacokinetics of the HIV integrase inhibitor dolutegravir given twice daily with rifampin or once daily with rifabutin: results of a phase 1 study among health subjects. J Acquir Immune Defic Syndr. 2013;62:21–27. doi:10.1097/QAI.0b013e318276cda9. | |
Patel P, Song I, Borland J, et al. Pharmacokinetics of the HIV integrase inhibitor S/GSK1349572 co-administered with acid-reducing agents and multivitamins in healthy volunteers. J Antimicrob Chemother. 2011;66:1567–1572. doi:10.1093/jac/dkr139. | |
Song I, Borland J, Arya N, Wynne B, Piscitelli S. The effect of calcium and iron supplements on the pharmacokinetics of dolutegravir in healthy subjects. Paper presented at: 15th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy; May 19–21, 2014; Washington, DC. Abstract P_13. | |
Canducci F, Ceresola ER, Boeri E, et al. Cross-resistance profile of the novel Integrase inhibitor dolutegravir (S/GSK1349572) using clonal viral variants selected in patients failing raltegravir. J Infect Dis. 2011;204:1811–1815. doi:10.1093/infdis/jir636. | |
Underwood MR, Johns BA, Sato A, et al. The activity of the integrase inhibitor dolutegravir against HIV-1 variants isolated from raltegravir-treated adults. J Acquir Immune Defic Syndr. 2012;3:297–301. doi:10.1097/QAI.0b013e31826bfd02. | |
DeAnda F, Hightower KE, Nolte RT, et al. Dolutegravir interactions with HIV-1 Integrase-DNA: structural rationale for drug resistance and dissociation kinetics. PLoS One. 2013;8:e77448. doi:10.1371/journal.pone.0077448. | |
Kobayashi M, Yoshinaga T, Seki T, et al. In vitro antiretroviral properties of S/GSK1349572, a next generation HIV integrase inhibitor. Antimicrob Agents Chemother. 2011;55:813–821. doi:10.1128/AAC.01209-10. | |
van Lunzen J, Maggiolo F, Arribas JR, et al. Once-daily dolutegravir (S/GSK1349572) in combination therapy in antiretroviral-naïve adults with HIV: planned interim 48 week results from SPRING-1, a dose-ranging, randomised, phase 2b trial. Lancet Infect Dis. 2012;12:111–118. doi:10.1016/S1473-3099(11)70290-0. | |
Raffi F, Rachlis A, Stellbrink HJ, et al. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, non-inferiority SPRING-2 study. Lancet. 2013;381:735–743. doi:10.1016/S0140-6736(12)61853-4. | |
Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Eng J Med. 2013;369:1807–1818. doi:10.1056/NEJMoa1215541. | |
Clotet B, Feinberg J, van Lunzen J, et al. Once-daily dolutegravir versus darunavir plus ritonavir in antiretroviral-naïve adults with HIV-1 infection (FLAMINGO):48 week results from the randomised open-label phase 3b study. Lancet. 2014;383;2222–2231. doi:10.1016/S0140-6736(14)60084-2. | |
Cahn P, Pozniak AL, Mingrone H, et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naïve adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet. 2013;382:700–708. doi:10.1016/S0140-6736(13)61221-0. | |
Eron JJ, Bonaventua C, Durant J, et al. Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING study. J Infect Dis. 2013;207:740–748. doi:10.1093/infdis/jis750. | |
Castagna A, Maggiolo F, Penco G, et al. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the phase 3 VIKING-3 Study. J Infect Dis. 2014;210:354–362. doi: 10.1093/infdis/jiu051. | |
Malet I, Gimferrer Arriaga L, Artese A, et al. New raltegravir resistance pathways induce broad cross-resistance to all currently used integrase inhibitors. J Antimicrob Chemother. 2014;69:2118–2122. doi:10.1093/jac/dku095. | |
Hurt CB, Sebastian J, Hick CB, Eron JJ. Resistance to HIV integrase strand transfer inhibitors among clinical specimens in the United States, 2009–2012. Clin Infect Dis. 2014;58:423–431. doi:10.1093/cid/cit697. | |
Raffi F, Jaeger H, Quiros-Roldan E, et al. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naïve adults with HIV-1 infection (SPRING-2 study):96 week results from randomized, double-blind, non-inferiority trial. Lancet. 2013;13:927–935. | |
Walmsley S, Berenguer J, Khuong-Josses M, et al. Dolutegravir regimen statistically superior to efavirenz/tenofovir/emtricitabine: 96-week results from the SINGLE study (ING114467). Paper presented at: 21st Conference on Retroviruses and Opportunistic Infections; March 3–6, 2014; Boston, MA. | |
Pappa K, Baumgarten A, Felizarta F, et al. Doltegravir + abacavir/lamivudine once dialy superior to tenofovir/emtricitabine/efavirenz in treatment naïve HIV subjects: 144-week results from SINGLE (INH114467). Presented at: 54th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; September 5–9, 2014; Washington DC. Abstract H-647a. | |
Viiv Healthcare. A study to determine safety and efficacy of dolutegravir/abacavir/lamivudine in Human Immunodeficiency Virus-1 infected antiretroviral therapy naïve women. Available from: http://clinicaltrials.gov/ct2/show/NCT01910402. NLM Identifier: NCT01910402. Accessed January 5, 2015. | |
Nichols G, Lazzarin A, Maggiolo F, et al. Phase 3 assessment of dolutegravir (DTG) 50 mg twice daily (BID) in HIV-1-infected subjects with raltegraivr (RAL) and/or elvitegravir (EVG) resistance in VIKING-3: week 24 results of all 183 subjects enrolled (abstract TULBPE19). Presented at: 7th IAS Conference on HIV Pathogenesis, Treatment and Prevention; June 30–July 3, 2013; Kuala Lumpur, Malaysia. | |
Vavro CL, Huang J, Avatapally C, MinS, and Ait-Khaled M. Durable efficacy and limited integrase resistance evolution in subjects receiving dolutegravir after failing prior integrase inhibititor (INI) regimens: week 48 results from VIKING-3 (abstract O_10). Presented at: 12th European Meeting on HIV and Hepatitis – Treatment Strategies and Antiviral Drug Resistance; March 26–28, 2014; Barcelona, Spain. | |
Viani RM, Alvero C, Fenton T, et al. Safety, pharmacokintetics, and efficacy of dolutegravir in treatment experienced HIV positive children. Poster presented at: 21st Conference on Retroviruses and Opportunistic Infections; March 3–6, 2014; Boston, MA. | |
Viani RM, Alvero C, Fenton T, et al. Safety and efficacy of dolutegravir in HIV treatment-experienced adolescents: 48-week results. Poster presented at: 21st Conference on Retroviruses and Opportunistic Infections; March 3–6, 2014; Boston, MA. | |
Airoldi M, Zaccarelli M, Bisi L, et al. One-pill once-a-day HAART: a simplification strategy that improves adherence and quality of life in HIV-infected subjects. Patient Prefer Adherence. 2010;4:115–125. | |
Parienti JJ, Bangsberg DR, Verdon R, Gardner EM. Better adherence with once-daily antiretroviral regimens: a meta-analysis. Clin Infect Dis. 2009;48:484–488. | |
Stone VE, Jordan J, Tolson J, Miller R, Pilon T. Perspectives on adherence and simplicity for HIV-infected patients on antiretroviral therapy. J Acquir Immuni Defic Syndr. 2004;36(3):808–816. | |
Willig JH, Abroms S, Westfall AO, et al. Increased regimen durability in the era of once daily fixed-dose combination antiretroviral therapy. AIDS. 2008;22(15):1951–1960. | |
Murray M, Goodwin B, Hagins D, et al. Measuring patient views of HIV treatments: comparing dolutegravir with darunavir/r in the FLAMINGO study. Presented at: 20th International AIDS Conference; July 20–25, 2014; Melbourne, Australia. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.