")
Back to Journals » Cancer Management and Research » Volume 11
The role of Clusterin in cancer metastasis
Authors Peng M, Deng J , Zhou S, Tao T, Su Q , Yang X, Yang X
Received 4 December 2018
Accepted for publication 28 February 2019
Published 27 March 2019 Volume 2019:11 Pages 2405—2414
DOI https://doi.org/10.2147/CMAR.S196273
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chien-Feng Li
Mei Peng,1,2 Jun Deng,2 Sichun Zhou,2 Ting Tao,2 Qiongli Su,2 Xue Yang,1 Xiaoping Yang2
1Department of Pharmacy, Xiangya Hospital, Central South University, Changsha, Hunan 410008, People’s Republic of China; 2Key Laboratory of Study and Discovery of Small Targeted Molecules of Hunan Province, Department of Pharmacy, School of Medicine, Hunan Normal University, Changsha, Hunan 410013, People’s Republic of China
Abstract: Clusterin is a conserved glycoprotein that has been characterized from almost all human tissues and fluids and plays a key role in cellular stress response and survival. Recently, research efforts have been contributed to explore the function of Clusterin in cancer metastasis, which is particularly important to design the strategies for treating metastatic patients. Evidence collected has demonstrated that Clusterin is overexpressed in tumor metastatic patients and experimental metastasis models. Specifically, Clusterin has been shown to have the role in anti-apoptotic capacities, development of therapy resistance and induction of epithelial–mesenchymal transition, all associated with cancer metastasis. Inhibition of Clusterin is known to increase the cytotoxic effects of chemotherapeutic agents and improves advanced cancer patients survival in clinical trials. Our unpublished data have demonstrated that Clusterin is overexpressed in bladder cancer and metformin, a well-known metabolism modulator specifically targets Clusterin by inhibiting migration of bladder cancer cells. In this review, we provide a general view of how Clusterin modulates cancer metastasis and update current understanding of detailed molecular mechanisms underlying of Clusterin for developing cancer management in future.
Keywords: Clusterin, metastasis, EMT, apoptosis, drug resistance
Introduction
Cancer metastasis is the spread of cancer from one organ or part of the body to another without being directly connected with it. It is responsible for over 90% of cancer-associated mortality.1 Cancer metastasis could be regarded as a complicated cell-biological event and divided into a two-phase process: The first phase is the escape from the primary tumor tissues and the second phase is the survival to adapt to new microenvironments with rapid growth after successful adaption.2 In the last decade, it has been learned that cancer cell behavior can be modified by epithelial–mesenchymal transition (EMT), inducing non-cancer stem cells to enter into a cancer stem cell-like state, thereby escape from apoptosis in the first phase.3 Thus, it could be an efficient approach to prevent metastasis in patients who are diagnosed with early cancer lesions by finding various sensitive and feasible biomarkers which are correlated to the stages of tumor development and progresses.
Clusterin, a highly conserved glycoprotein, was firstly identified in ram rete testis fluid in 1983.4 Clusterin is a stress-induced molecular chaperone that performs its role in a manner similar to small heat shock proteins.5 It is encoded by the CLU gene on chromosome 8 and facilitates the folding of secreted proteins in an ATP-independent fashion.6 Clusterin participates in a series of biological processes, including cell adhesion and programmed cell death.7 It also exists in exosomes and helps cancer cells to survival in distant locations, which is necessarily required for tumor metastasis.8 Therefore, Clusterin may have multifaceted functions for carcinogenesis and cancer invasion. In addition, it is very well known that heat shock proteins have demonstrated an important effect on tumor metastasis.9 Thus, targeting Clusterin, one of recent characterized heat shock proteins to inhibit tumor metastasis, might be a valid strategy to block tumor progress and growth.
Emerging evidence showed that Clusterin is overexpressed in several metastatic cancer cells, such as colon, bladder, hepatocellular carcinoma and renal cell carcinoma.10,11 Overexpression of Clusterin in primary hepatocellular carcinoma increased cell migration by twofold in vitro and formation of metastatic tumor nodules in liver by eightfold in vivo.12,13 Introducing the Clusterin gene into human renal cell carcinoma cells enhances the formation of metastatic nodules in the lung by more than fivefold increases, an evidence of metastasis of cancer cells from kidney to lung enhanced by Clusterin.14 Shiota et al demonstrated that Clusterin promoted invasion and metastasis of prostate cancer via inducing EMT.15 Clusterin also facilitated metastasis by EIF3I/AKT/MMP13 signaling in hepatocellular carcinoma.16 These studies implied that Clusterin upregulation plays an important role in tumor invasiveness. Clarification of the specific mechanisms of Clusterin-induced metastasis is helpful to provide strategy for inhibiting metastasis in clinic.
In this minireview, we summarize the function of Clusterin in cancer metastasis, focusing on alterations of corresponding signaling pathways. We anticipate that this review will provide updated information with elucidating the mechanisms related to Clusterin serving a potential diagnosed biomarker or target to block cancer metastasis.
The pathological role of Clusterin on cancer metastasis
The identification of useful biomarkers for the detection and diagnosis of human cancer provides a promising advance for the management of this deadly disease. Clusterin antigen has been characterized as a sensitive and stable histological marker for human intestinal tumors.17 Interestingly, Kang et al firstly demonstrated that Clusterin overexpression is associated with poor Edmondson’s histological grade and high Tumor-Node-Metastasis (TNM) stage.18 Subsequently, Lau et al have analyzed the expression levels of Clusterin using a tissue microarray containing 104 pairs of primary hepatocellular carcinoma (HCC) tissues and their matched metastases. The frequency of Clusterin overexpression is significantly increased in metastatic tissues (59.1%) compared with that in primary tumors (32.6%).10 Furthermore, Chen et al investigated the dynamic expression patterns of identified proteins during early invasion of HCC. The results revealed that Clusterin was continuously upregulated during the entire invasion process.19
Recent study from Shimizu demonstrated Clusterin-positive regions in progressive phenotypes of Spasmolytic polypeptide-expressing metaplasia in gastric adenocarcinoma.20 These phenotypes could be considered as the neoplastic precursors with invasive characteristics. Differential expression of Clusterin in messenger RNA and protein levels was found between invasive and non-invasive non-functioning pituitary adenomas,21 using gene microarray technology and liquid chromatography-tandem mass spectrometry. Moreover, this work applied molecular and cellular function analysis tools to identify that Clusterin exerted the invasion function.
Osteosarcoma is the most common bone tumor in young adults under the age of 20. Using an Affymetrix microarray, Clusterin was recognized as one of the critical genes which significantly overexpressed in the metastatic samples and mediated osteosarcoma metastasis to the lungs.22 DNA aptamers of Clusterin, as specific affinity probes binding to sites of overexpressed lung cancer tissue, were identified to characterize postoperative lung adenocarcinoma.23 The above findings show accumulating evidence that abnormal Clusterin expression was closely associated with tumor metastasis. Thus, Clusterin in either serum and/or tissue is expected to be a candidate of diagnosis biomarkers for detection of some early metastatic cancers.
The role of Clusterin on metastasis-related biological activities and cell signaling pathway
A broad range of functions have been ascribed to Clusterin, including tissue remodeling, adhesion, apoptosis, etc.24 As described earlier, Clusterin plays an important role in cancer metastasis. Recently, the mechanisms that Clusterin favors cancer metastasis have been explored.10,12 Upregulation of Clusterin promotes invasion via the induction of EMT process and plays a critical regulatory role in transforming growth factor-beta (TGF-β) signaling.15 In addition, Clusterin can also protect metastatic cells from apoptosis, leading to cell survival in adverse environment.25 Similar to other heat shock proteins, Clusterin is a stress-activated cytoprotective chaperone upregulated by many varied anticancer therapies to confer treatment resistance. Park et al demonstrated that Clusterin enhanced paclitaxel resistance in ovarian cancer cells by physically binding to paclitaxel, which may prevent paclitaxel from interacting with microtubules to induce apoptosis.26 Collectively, these studies highlighted that Clusterin mediated cancer invasion through distinct ways.
The role of Clusterin on metastasis-related EMT
EMT was first recognized as a feature of embryogenesis. It is a process by which epithelial cells lose their cell polarity and cell–cell adhesion, and gain migratory and invasive properties to become mesenchymal stem cells.27 Epithelial cells express high levels of E-cadherin, whereas mesenchymal cells express those of N-cadherin, fibronectin and vimentin.28 Beerling et al tested the existence and role of epithelial–mesenchymal plasticity in metastasis of mammary tumors without artificially modifying EMT regulators. Using microscopy, they detected that a portion of breast tumor cells undergoes EMT, become motile, disseminate and then reverse to the epithelial state upon metastatic outgrowth.29 Thus, it would be very interesting to investigate the connections between Clusterin and EMT.
Chou et al reported that Clusterin levels are positively correlated with the degree of invasiveness and Clusterin regulated EMT through modulating ERK signaling and slug expression.30 It was observed that Clusterin-rich cells displayed a spindle-shape morphology while cells with low Clusterin levels were cuboidal in shape, implying that Clusterin plays a role in modulating the phenotypes of cells. Silencing Clusterin induced a mesenchymal-to-epithelial transition (MET) evidenced by the spindle-to-cuboidal morphological change, increased E-cadherin expression and decreased fibronectin expression, inhibiting cell migration and invasion. Re-expression of Clusterin reversed the MET and restored the mesenchymal and invasive phenotypes. Knocking down Clusterin downregulated Slug, a zinc-finger-containing transcriptional repressor of E-cadherin.30 By analyzing serum samples collected from metastatic hepatocellular carcinoma patients and orthotopic xenograft tumor model, Wang et al found that Clusterin was significantly upregulated during cancer metastasis. ShRNA-mediated downregulation of Clusterin resulted in a reduced migratory capacity in HCC cell lines, as well as a reduction in pulmonary metastasis in vivo. In contrast, overexpression of Clusterin showed increased cell migratory ability.13
Profoundly, it was shown that TGF-β induces Clusterin expression in various cancer cells along with cancer development. In early stage, TGF-β suppresses tumor development by arresting proliferation and inducing cell death. However, in the late stage, TGF-β increases the Clusterin expression, promoting invasion and metastasis.30,31 This prometastatic role of TGF-β may be linked to its ability to induce tumor cell EMT through activating E-cadherin repressors as well. In mink lung cancer cells, TGF-β induced Clusterin expression which was repressed by overexpression of c-Fos proto-oncogene. TGF-β abrogated the repression of c-Fos through effect on c-Fos protein synthesis and/or stability.32 Using the approach of detecting the transcriptome changes in mammary tumor epithelial cells undergoing TGF-β-induced EMT, Lenferink et al found that Clusterin gene is the most highly upregulated one throughout entire TGF-beta-treated time course. Antibodies targeting Clusterin inhibited TGF-β-induced EMT, as well as the invasive phenotype of several breast and prostate tumor cell lines with monitoring hallmark features of EMT, indicating that Clusterin is a functionally important EMT mediator that lies downstream within TGF-β’s EMT-promoting transcriptional cascade. To further investigate the role of Clusterin in tumor metastasis, the effect of several anti-Clusterin monoclonal antibodies in vivo using a 4T1 syngeneic mouse breast cancer model was assessed and found that these antibodies significantly reduced lung metastasis.33 Together, these findings solidly confirmed that Clusterin prompted EMT-induced metastasis (Figure 1).
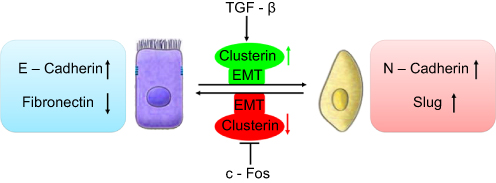 | Figure 1 Schematic illustration of roles of clusterin in the cancer metastasis with a function of EMT. TGF-β upregulated CLU expression which was repressed by overexpression of c-Fos proto-oncogene, resulting EMT and cancer invasion. |
The role of Clusterin on metastasis-related apoptosis
One of the important processes in efficient cancer metastasis is tumor cell survival during tumor transfer process. Studies have shown that Clusterin exerted cytoprotective properties. It prevents cell death through stabilizing the association between Ku70 and Bax, thereby inhibiting the release of cytochrome c from the mitochondria.34 This would apparently explain how progression towards high-grade and metastatic carcinoma leads to Clusterin overexpression, and that the role of Clusterin for tumor invasion may be related to an anti-apoptotic pattern.
Apoptotic pathways are mediated via multiple activators (eg, Bax, p53) and suppressors (eg, growth factors and Bcl-2). P53 has an important and essential role in responding to cell stress by either activating or repressing gene expression, controlling checkpoint responses, and inducing apoptosis. Interestingly, it was reported that p53 can suppress basal as well as ionizing radiation (IR)-induced Clusterin expression in both breast and colon cancer cells by repressing Clusterin promoter activity and transcription.35
Insulin-like growth factor-1(IGF-1)-Clusterin pathway is hypersensitive to IR and triggers microenvironment alternation which suppressed apoptosis and promoted tumor growth and metastasis.36 Boothman et al found that stimulation of IGF-1 and IGF-1R protein kinase signaling was required for Clusterin induction after IR exposure, and they further studied interaction among p53, IGF signaling and Clusterin expression. Their results revealed that DNA damage resulting from genetic instability produced by IR amplified IGF-1-Clusterin cell signaling pathway. This is due to loss of p53 and NF-YA occupancy on the IGF-1 promoter. Ataxia-telangiectasia-mutated Kinase (ATM), as one member of PIKKs family, is usually activated in response to the DNA damage. Further study revealed that ATM causes NF-YA modification, prevents or reverses its binding to IGF-1 promoter, depending on expression level of p53. Thus, expression level of p53 is a crucial adjustor which determines whether DNA damage accelerates or decelerates IGF-1-Clusterin pathway.37
In addition, Clusterin has nuclear factor kappa B (NF-κB) regulatory action. Modulation of NF-κB activity is thought to be important for cell survival, stress response, cell motility and transformation. Wang et al investigated the molecular mechanisms underlying the effect of Clusterin on TNF-alpha-induced apoptosis in breast cancer cells. Their results suggested that Clusterin confers breast cancer cells resistance to TNF-alpha-induced apoptosis through NF-κB activation and Bcl-2 overexpression. Silencing Clusterin abrogated TNF-alpha-mediated NF-κB activation and Bcl-2 overexpression and rendered more sensitive to TNF-alpha-mediated apoptosis. In contrast, overexpression of Clusterin has the opposite results, consistently confirming its role of Clusterin on TNF-alpha-mediated apoptosis.38 Surprisingly, contradictory result that Clusterin inhibits human neuroblastoma cell invasion appeared.39 The mechanistic investigations suggested that Clusterin stabilized IκB, a tumor necrosis factor-dependent increase in NF-κB activity, decreased transcription of the NF-κB target gene and up-modulated p53 protein. It was also observed that Clusterin suppressed the metastasis phenotype of neuroblastoma xenografts in immunocompromised mice. Regulation of NF-κB activity satisfactorily explained opposing effects of Clusterin in different systems (Figure 2).
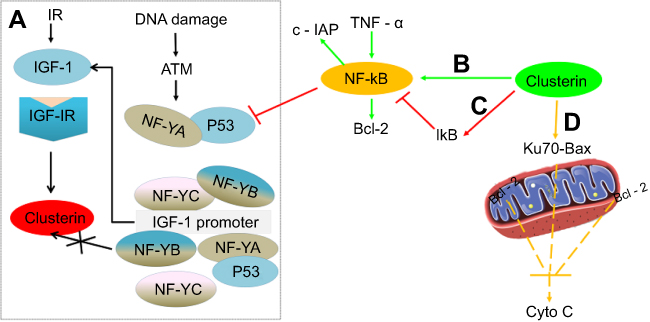 | Figure 2 Comprehensive overview of the role of Clusterin on apoptosis-related events in cancer cells. (A) (black): Ionization Radiation, and DNA damage stimulated ATM-IGF-1–MAPK–CLU signaling pathway in the absence or presence of p53 (reference 33); (B) (green): Clusterin promotes TNF-α-induced activation of NF-κB and increases the expression of Bcl-2; (C) (red): Clusterin stabilized IκB, decreased transcription of the NF-κB target gene and up-modulated p53 protein; and (D) (yellow): Clusterin prevents cell death through stabilizing the association between Ku70 and Bax, thereby inhibiting the release of Cytochrome c from the mitochondria. |
Clusterin also exhibits its anti-apoptotic property through other signaling. It was reported that Clusterin was positively regulated by B-MYB, a transcription factor involved in the regulation of cell survival, proliferation and differentiation.40 Small interfering RNA targeted to Clusterin blocks tumor growth, motility and invasion in breast cancer through increasing cell apoptosis, reducing ERK and MMP-9 protein levels.41 And a novel anticancer PEG-SMR-Clusterin peptide caused cell cycle G2/M phase arrest in both estrogen responsive and non-responsive breast cancer cells, preventing breast cancer metastasis.42 Interestingly, Clusterin can also protect hepatocellular carcinoma cells from endoplasmic reticulum stress-induced apoptosis through GRP78.43
The role of Clusterin on metastasis-related chemotherapeutic resistance
Resistance to existing or novel chemotherapies adversely affects outcomes in the management of patients, leading to cancer metastasis and death.44,45 Understanding the mechanisms of underlying resistance and developing new strategies that can overcome resistance would improve treatment outcomes and prolong survival. According to reported drug resistance mechanisms, induction of heat-shock proteins including Clusterin that inhibit apoptosis and promote cancer metastasis may be one of the critical events.
Much evidence has indicated that overexpression of Clusterin is correlated with tumor progression in various tumor types during chemotherapeutic treatment. An increase in Clusterin expression was observed in prostate cancer following androgen therapy alone or in combination with docetaxel.46,47 And Clusterin levels determined in biopsy specimens taken prior to neoadjuvant hormone therapy were significantly lower than those in corresponding prostatectomy specimens, and a 17-fold increase in expression levels could be observed within 4 weeks of therapy. Overexpression of Clusterin was also associated with resistance to preoperative neoadjuvant chemotherapy in breast cancer and pancreatic cancer.48,49 These researches imply Clusterin expression appears to be induced in response to therapy. Therefore, targeting Clusterin may be an effective approach to reverse resistance and control cancer.
Due to the extensive posttranslational processing and heterogeneous oligomerization of Clusterin, there is no definitive structure so far. This means that targeting Clusterin with small molecule inhibitors is challenging. Therefore, inhibiting Clusterin at the gene-expression level using siRNA or antisense is a valid approach to inhibit its function. In pancreatic cancer, it was found that knockdown of Clusterin sensitized MIA-PaCa-2 cells to gemcitabine chemotherapy via modulating NF-κB/Bcl-2 pathway.50 Interestingly, Melittin, a Chinese traditional medicine, decreased gemcitabine resistance by targeting Clusterin based on microarray analyses.51 siRNA-directed Clusterin downregulation promotes cisplatin antitumor activity in both in vitro human non-small cell lung cancer cells and in vivo xenografts through decreasing phosphorylation of AKT and ERK1/2.52,53
It was also observed that abnormal Clusterin expression is related to multidrug resistance (MDR) of hepatocellular carcinoma. Clusterin mediates Oxaliplatin resistance via decreasing Gadd45a expression and activating PI3K/Akt signaling pathway.54 Inhibition of Clusterin expression in HepG2/ADM cells obviously enhanced cells chemo-sensitivity, increased doxorubicin-induced apoptosis and decreased the drug efflux pump activity. Exploration of the mechanisms showed that knocking-down Clusterin significantly suppressed MDR1/P-glycoprotein and β-catenin, suggesting that downregulation of Clusterin might reverse multidrug resistance of HepG2/ADM cells.50 Silencing Clusterin also potentiates the lethality of sorafenib in association with inhibition ERK1/2 signals.55 Zoledronic acid (ZOL) is a promising adjuvant molecule to chemotherapy to treat osteosarcoma, limiting the osteolytic component of bone tumors. Unexpectedly, ZOL triggers the elevation of Clusterin, which could enhance tumor cell survival and treatment resistance, leaving the unexpired side effect. Lamoureux et al investigated the combined effects of OGX-011, a second-generation antisense oligonucleotide designed to block Clusterin mRNA and ZOL in osteosarcoma. The vitro and vivo results showed OGX-011 and ZOL synergistically inhibited tumor growth and prolonged survival through decreasing Clusterin, MDR1 and Heat shock factor 1 transcriptional activity.56 In castrate-resistant prostate cancer, cotargeting androgen receptor and Clusterin prolong survival by inhibiting adaptive stress and AR stability.57 In human renal cell carcinoma, sorafenib treatment increased Clusterin expression. OGX-011 synergistically enhanced the sensitivity to sorafenib through marked downregulation of phosphorylated Akt and p44/42 MAPK pathways58 (Figure 3).
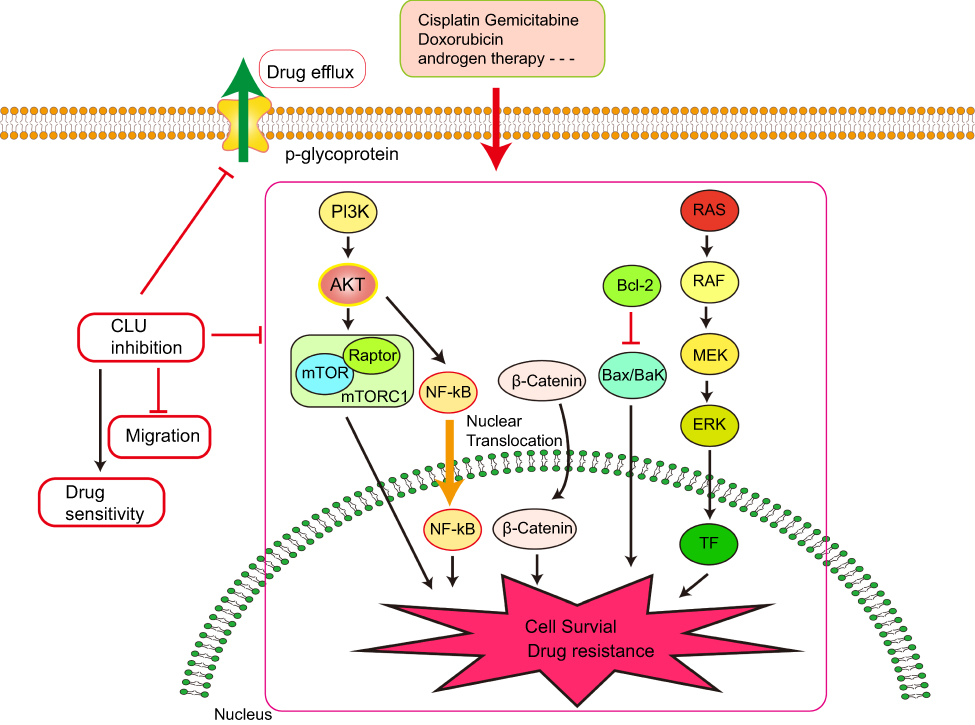 | Figure 3 Knocking down Clusterin sensitizes the therapeutic effects of several conventional administrations via affecting cell signaling pathways and drug efflux pump, etc. |
Clinical trials combined Clusterin inhibitor (Custirsen) with chemotherapy drugs in patients with advanced cancer have been conducted successively. The effects of Custirsen combining with gemcitabine and platinum were evaluated on chemotherapy-naive stage IIIB/IV non-small cell lung cancer (NSCLC).59 The results showed that Custirsen treatment decreased serum Clusterin level and achieved a median survival of 27.1 compared with 16.1 months for patients who did not. Based on these results, a multinational, randomized, open-label study to compare overall survival in advanced or metastatic NSCLC is recruiting.60 In a randomized phase II study of patients with metastatic castration-resistant prostate cancer (CRPC), first-line docetaxel and prednisone in combination with Custiren treatment has significant survival benefit compared with those received docetaxel and prednisone alone (median progression-free survival: 7.3 months vs 6.1 months; overall survival: 23.8 months vs 16.9 months).61
Although the above phase II results are exciting, the phase III synergy trial reported on Lancet Oncol in 2017 was not that optimistic.62 The overall survival in patients with metastatic castration-resistant prostate cancer was not improved (23.4 months vs 22.0 months). The possible reasons are 1) The intratumoral Clusterin level was not assessed in the patients enrolled in the trial. Moreover, the Clusterin gene—located on chromosome 8p proximal to the prostate cancer tumor-suppressor gene NKX3-1—might be homozygously deleted in about 20% of the men with castration-resistant prostate cancer. Thus, this subpopulation whose Clusterin expression is low or even absent may affect the synergy trial results. 2) The dose of Custirsen. The addition of Custirsen to first-line docetaxel and prednisone was well tolerated, so it may be feasible to increase the dose reasonably. 3) Overall survivals obtained from Phase II and phase III were clearly different. It may be attributed to two aspects: the right time to administer the drug in combination and a rapid change in the treatment of CRPC with newly approved drugs, such as Enzalutamide and Abiraterone, which extended the patients survival time.
However, the exploratory post-hoc analysis suggests that Custirsen might have had a survival benefit in patients who had poor prognostic features, which deserves further study. All these studies suggest the strong rationale of treating cancers with a combination of the chemotherapy drug and a drug which inhibits Clusterin expression.
Metformin, a well-known metabolism modulator, attracts extensive researches on investigating its anticancer activity and shows much potential for cancer therapy while combining with chemo- and targeted drugs.63,64 Interestingly, we found that metformin inhibits bladder cancer cells migration and decreases Clusterin expression [Figure 4A and B]. The classic anticancer mechanism of metformin is activation of AMPK signaling pathway, and new mechanisms are being explored. Our recent unpublished data show that Clusterin is overexpressed in human bladder cancer tissues, especially muscle-invasive bladder cancer patients [Figure 4C]. Metformin significantly decreases Clusterin expression, suggesting that inhibiting Clusterin expression could be a novel mechanism of metformin’s anticancer activities which warrants further investigation.
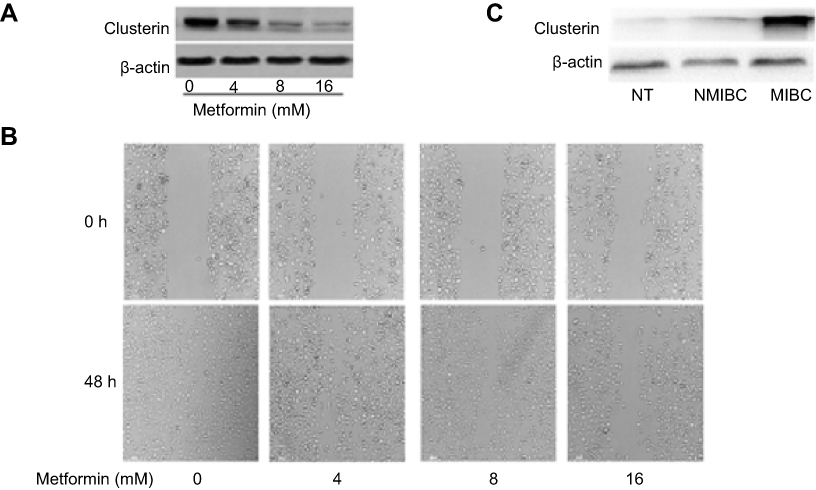 | Figure 4 (A) Metformin significantly decreases Clusterin expression. Murine bladder cancer cells (MB49) were treated with metformin for 48 hrs and Clusterin expression was detected using Western blot, ß-actin as a loading control. (B) metformin inhibited cells migration in wound healing assays. (C) Clusterin expression in tissues was evaluated using immunohistochemistry. Three groups of specimens (N=3 for adjacent normal tissues (NT), non-muscle invasive bladder cancer (NMIBC) and muscle-invasive bladder cancer (MIBC) tissues, respectively) were paraffin-embedded, sectioned and immunohistochemically stained with anti-Clusterin. |
Discussion
To date, while surgical resection and adjuvant therapy can cure well-confined primary tumors, metastatic disease is largely incurable because of the resistance to existing therapeutic agents. Thus, over 90% of mortality from cancer is attributable to metastases.65,66 Although a number of fundamental questions concerning the basic nature of carcinoma metastasis remain incompletely understood, recent research has succeeded in implicating specific molecules in the regulation of discrete cell-biological aspects of the invasion-metastasis cascade.
In this mini-review, we emphasize on the crucial role of Clusterin in cancer metastasis. Compelling evidence showed that Clusterin is overexpressed in various advanced cancers and correlates with cancer grades. Upregulation of Clusterin promotes cancer invasion and metastasis. Detecting tissue or serum Clusterin level is a potential way to diagnose cancer early metastasis. As described earlier, preclinical data showed that inhibition of Clusterin delays cancer metastasis and enhances the sensitivity of cytotoxic chemotherapy. In addition, clinical phase I/II studies demonstrated that inhibition of Clusterin significantly improves patients overall survival. Major molecular mechanisms of Clusterin-mediated metastasis could be classified into activation of EMT, anti-apoptotic property and induction of chemotherapeutic resistance. However, phase III trial of combining Custiren with first-line docetaxel and prednisone in metastatic CRPC patients did not improve overall survival. These complicated findings prompt us to develop novel therapeutic routes to decrease the risk of metastasis and to enhance the tumoral sensitivity to chemotherapy or radiotherapy, especially in those cases with high levels of Clusterin expression. In addition, further efforts are needed to increase the current understanding of the role of Clusterin in specific situation of cancer metastasis. Along with this strategy, we could improve clinical patient survival more effectively through targeting Clusterin signaling.
In conclusion, Clusterin plays an important role in cancer metastasis. New strategies to effectively inhibit Clusterin level in cancer patients are in the development phase. Although we highlighted novel findings regarding the role of Clusterin on tumor metastasis in this mini-review, more evidence targeting Clusterin is needed for exploring detailed molecular mechanisms. We believe that reducing Clusterin levels with feasible approaches will provide benefit to delay cancer metastasis, based on a complete understanding of Clusterin function in cancer progression in near future.
Abbreviations list
AMPK, adenosine 5ʹ-monophosphate-activated protein kinase; ATP, adenosine triphosphate; AKT, Protein kinase B; ATM, ataxia-telangiectasia-mutated kinase; EIF3I, eukaryotic translation initiation factor 3 subunit I; ERK, extracellular signal-regulated kinase; EMT, epithelial-to-mesenchymal transition; IR, ionizing radiation; IGF-1, insulin-like growth factor-1; IGF-1R, insulin-like growth factor-1 receptor; MET, mesenchymal-to-epithelial transition; MMP, matrix metalloproteinase; PIKKs, phosphatidylinositol 3-kinase-related kinases; TGF-β, transforming growth factor-beta; TNF, tumor necrosis factor; HCC, hepatocellular carcinoma; TNM, Tumor-Node-Metastasis; MDR, multidrug resistance; ZOL, zoledronic acid; HSF1, heat shock factor 1; NSCLC, non-small cell lung cancer; CRPC, castration-resistant prostate cancer; NF-YA, nuclear transcription factor Y subunit alpha.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81703008), Hunan Natural Science Foundation (2018JJ3831), Institutional Fund of Xiangya Hospital Central South University (No 2016Q02) and National Natural Science Foundation of China (81874212), Hunan Natural Science Foundation (2016JJ2187), Key Project of Hunan Province 2016 (2016JC2036), Xiaoxiang Endowed University Professor Fund of Hunan Normal University (No. 840140-008) and Opening Fund for Key Laboratory of Study and Discovery of Small Targeted Molecules of Hunan Province (2017TP1020).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–292. doi:10.1016/j.cell.2011.09.024
2. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. doi:10.1126/science.1203543
3. Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi:10.1016/j.cell.2008.03.027
4. Blaschuk O, Burdzy K, Fritz IB. Purification and characterization of a cell-aggregating factor (Clusterin), the major glycoprotein in ram rete testis fluid. J Biol Chem. 1983;258(12):7714–7720.
5. Ischia J, So AI. The role of heat shock proteins in bladder cancer. Nat Rev Urol. 2013;10(7):386–395. doi:10.1038/nrurol.2013.108
6. Nelson AR, Sagare AP, Zlokovic BV. Role of Clusterin in the brain vascular clearance of amyloid-β. Proc Natl Acad Sci U S A. 2017;114(33):8681–8682. doi:10.1073/pnas.1711357114
7. Oh SB, Kim MS, Park S, et al. Clusterin contributes to early stage of Alzheimer’s disease pathogenesis. Brain Pathol. 2018. doi:10.1111/bpa.12660
8. Guitart K, Loers G, Buck F, et al. Improvement of neuronal cell survival by astrocyte-derived exosomes under hypoxic and ischemic conditions depends on prion protein. Glia. 2016;64(6):896–910. doi:10.1002/glia.22963
9. Fang F, Chang R, Yang L. Heat shock factor 1 promotes invasion and metastasis of hepatocellular carcinoma in vitro and in vivo. Cancer. 2012;118(7):1782–1794. doi:10.1002/cncr.26482
10. Shapiro B, Tocci P, Haase G, et al. Clusterin, a gene enriched in intestinal stem cells, is required for L1-mediated colon cancer metastasis. Oncotarget. 2015;6(33):34389–34401. doi:10.18632/oncotarget.5360
11. Makridakis M, Roubelakis MG, Bitsika V, et al. Analysis of secreted proteins for the study of bladder cancer cell aggressiveness. J Proteome Res. 2010;9(6):3243–3259. doi:10.1021/pr100189d
12. Lau SH, Sham JS, Xie D, et al. Clusterin plays an important role in hepatocellular carcinoma metastasis. Oncogene. 2006;25(8):1242–1250. doi:10.1038/sj.onc.1209141
13. Wang C, Jiang K, Kang X, et al. Tumor-derived secretory Clusterin induces epithelial-mesenchymal transition and facilitates hepatocellular carcinoma metastasis. Int J Biochem Cell Biol. 2012;44(12):2308–2320. doi:10.1016/j.biocel.2012.09.012
14. Miyake H, Gleave ME, Arakawa S, et al. Introducing the Clusterin gene into human renal cell carcinoma cells enhances their metastatic potential. J Urol. 2002;167(5):2203–2208.
15. Shiota M, Zardan A, Takeuchi A, et al. Clusterin mediates TGF-β-induced epithelial-mesenchymal transition and metastasis via Twist1 in prostate cancer cells. Cancer Res. 2012;72(20):5261–5272. doi:10.1158/0008-5472.CAN-12-0254
16. Wang C, Jin G, Jin H, et al. Clusterin facilitates metastasis by EIF3I/Akt/MMP13 signaling in hepatocellular carcinoma. Oncotarget. 2015;6(5):2903–2916. doi:10.18632/oncotarget.3093
17. Chen X, Halberg RB, Ehrhardt WM, et al. Clusterin as a biomarker in murine and human intestinal neoplasia. Proc Natl Acad Sci U S A. 2003;100(16):9530–9535. doi:10.1073/pnas.1233633100
18. Kang YK, Hong SW, Lee H, et al. Overexpression of Clusterin in human hepatocellular carcinoma. Hum Pathol. 2004;35(11):1340–1346. doi:10.1016/j.humpath.2004.07.021
19. Chen RX, Song HY, Dong YY, et al. Dynamic expression patterns of differential proteins during early invasion of hepatocellular carcinoma. PLoS One. 2014;9(3):e88543. doi:10.1371/journal.pone.0088543
20. Shimizu T, Choi E, Petersen CP, et al. Characterization of progressive metaplasia in the gastric corpus mucosa of Mongolian gerbils infected with Helicobacter pylori. J Pathol. 2016;239(4):399–410. doi:10.1002/path.4735
21. Yu SY, Hong LC, Feng J, et al. Integrative proteomics and transcriptomics identify novel invasive-related biomarkers of non-functioning pituitary adenomas. Tumour Biol. 2016;37(7):8923–8930. doi:10.1007/s13277-015-4767-2
22. Liu K, He Q, Liao G, et al. Identification of critical genes and gene interaction networks that mediate osteosarcoma metastasis to the lungs. Exp Ther Med. 2015;10(5):1796–1806. doi:10.3892/etm.2015.2767
23. Zamay GS, Kolovskaya OS, Zamay TN, et al. Aptamers selected to postoperative lung adenocarcinoma detect circulating tumor cells in human blood. Mol Ther. 2015;23(9):1486–1496. doi:10.1038/mt.2015.108
24. Zhang F, Kumano M, Beraldi E, et al. Clusterin facilitates stress-induced lipidation of LC3 and autophagosome biogenesis to enhance cancer cell survival. Nat Commun. 2014;5:5775. doi:10.1038/ncomms5972
25. Zhang H, Kim JK, Edwards CA, et al. Clusterin inhibits apoptosis by interacting with activated Bax. Nat Cell Biol. 2005;7(9):909–915. doi:10.1038/ncb1291
26. Park DC, Yeo SG, Wilson MR, et al. Clusterin interacts with Paclitaxel and confer Paclitaxel resistance in ovarian cancer. Neoplasia. 2008;10(9):964–972.
27. Chen X, Bode AM, Dong Z, Cao Y. The epithelial-mesenchymal transition (EMT) is regulated by oncoviruses in cancer. FASEB J. 2016;30(9):3001–3010. doi:10.1096/fj.201600388R
28. Mao XY, Li QQ, Gao YF, Zhou HH, Liu ZQ, Jin WL. Gap junction as an intercellular glue: emerging roles in cancer EMT and metastasis. Cancer Lett. 2016;381(1):133–137. doi:10.1016/j.canlet.2016.07.037
29. Beerling E, Seinstra D, de Wit E, et al. Plasticity between epithelial and mesenchymal states unlinks EMT from metastasis-enhancing stem cell capacity. Cell Rep. 2016;14(10):2281–2288. doi:10.1016/j.celrep.2016.02.034
30. Chou TY, Chen WC, Lee AC, et al. Clusterin silencing in human lung adenocarcinoma cells induces a mesenchymal-to-epithelial transition through modulating the ERK/Slug pathway. Cell Signal. 2009;21(5):704–711. doi:10.1016/j.cellsig.2009.01.008
31. Jolly MK, Ware KE, Gilja S, et al. EMT and MET: necessary or permissive for metastasis? Mol Oncol. 2017;11(7):755–769. doi:10.1002/1878-0261.12083
32. Jin G, Howe PH. Transforming growth factor beta regulates Clusterin gene expression via modulation of transcription factor c-Fos. Eur J Biochem. 1999;263(2):534–542.
33. Lenferink AE, Cantin C, Nantel A, et al. Transcriptome profiling of a TGF-beta-induced epithelial-to-mesenchymal transition reveals extracellular Clusterin as a target for therapeutic antibodies. Oncogene. 2010;29(6):831–844. doi:10.1038/onc.2009.399
34. Trougakos IP, Lourda M, Antonelou MH, et al. Intracellular Clusterin inhibits mitochondrial apoptosis by suppressing p53-activating stress signals and stabilizing the cytosolic Ku70-Bax protein complex. Clin Cancer Res. 2009;15(1):48–59. doi:10.1158/1078-0432.CCR-08-1805
35. Criswell T, Klokov D, Beman M, et al. Repression of IR-inducible Clusterin expression by the p53 tumor suppressor protein. Cancer Biol Ther. 2003;2(4):372–380.
36. Klokov D, Leskov K, Araki S, et al. Low dose IR-induced IGF-1 sClusterin expression: a p53-repressed expression cascade that interferes with TGFβ1 signaling to confer a pro-survival bystander effect. Oncogene. 2013;32(4):479–490. doi:10.1038/onc.2012.64
37. Goetz EM, Shankar B, Zou Y, et al. ATM-dependent IGF-1 induction regulates secretory Clusterin expression after DNA damage and in genetic instability. Oncogene. 2011;30(35):3745–3754. doi:10.1038/onc.2011.92
38. Wang Y, Wang X, Zhao H, et al. Clusterin confers resistance to TNF-alpha-induced apoptosis in breast cancer cells through NF-kappaB activation and Bcl-2 overexpression. J Chemother. 2012;24(6):348–357. doi:10.1179/1973947812Y.0000000049
39. Santilli G, Aronow BJ, Sala A. Essential requirement of apolipoprotein J (Clusterin) signaling for Ilapak expression and regulation of NF-kappaB activity. J Biol Chem. 2003;278(40):38214–38219. doi:10.1074/jbc.C300252200
40. Cervellera M, Raschella G, Santilli G, et al. Direct transactivation of the anti-apoptotic gene apolipoprotein J (Clusterin) by B-MYB. J Biol Chem. 2000;275(28):21055–21060. doi:10.1074/jbc.M002055200
41. Li J, Jia L, Zhao P, et al. Stable knockdown of Clusterin by vectorbased RNA interference in a human breast cancer cell line inhibits tumour cell invasion and metastasis. J Int Med Res. 2012;40(2):545–555. doi:10.1177/147323001204000216
42. Huang MB, Gonzalez RR, Lillard J, et al. Secretion modification region-derived peptide blocks exosome release and mediates cell cycle arrest in breast cancer cells. Oncotarget. 2017;8(7):11302–11315. doi:10.18632/oncotarget.14513
43. Wang C, Jiang K, Gao D, et al. Clusterin protects hepatocellular carcinoma cells from endoplasmic reticulum stress induced apoptosis through GRP78. PLoS One. 2013;8(2):e55981. doi:10.1371/journal.pone.0055981
44. Mansoori B, Mohammadi A, Davudian S, et al. The different mechanisms of cancer drug resistance: a brief review. Adv Pharm Bull. 2017;7(3):339–348. doi:10.15171/apb.2017.041
45. Tang L, Wei F, Wu Y, et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J Exp Clin Cancer Res. 2018;37(1):87. doi:10.1186/s13046-018-0758-7
46. July LV, Akbari M, Zellweger T, et al. Clusterin expression is significantly enhanced in prostate cancer cells following androgen withdrawal therapy. Prostate. 2002;50(3):179–188.
47. Wang D, Liang H, Mao X, et al. Changes of transthyretin and Clusterin after androgen ablation therapy and correlation with prostate cancer malignancy. Transl Oncol. 2012;5(2):124–132.
48. Niu ZH, Wang Y, Chun B, et al. Secretory Clusterin (sClusterin) overexpression is associated with resistance to preoperative neoadjuvant chemotherapy in primary breast cancer. Eur Rev Med Pharmacol Sci. 2013;17(10):1337–1344.
49. Marchegiani G, Paulo JA, Sahora K, et al. The proteome of postsurgical pancreatic juice. Pancreas. 2015;44(4):574–582. doi:10.1097/MPA.0000000000000304
50. Xu M, Chen X, Han Y, et al. Clusterin silencing sensitizes pancreatic cancer MIA-PaCa-2 cells to gemcitabine via regulation of NF-kB/Bcl-2 signaling. Int J Clin Exp Med. 2015;8(8):12476–12486.
51. Wang X, Xie J, Lu X, et al. Melittin inhibits tumor growth and decreases resistance to gemcitabine by downregulating cholesterol pathway gene CLU in pancreatic ductal adenocarcinoma. Cancer Lett. 2017;399:1–9. doi:10.1016/j.canlet.2017.04.012
52. Ma G, Cai H, Gao L, et al. sClusterin regulates cisplatin chemosensitivity of lung cancer cells in vivo. World J Surg Oncol. 2015;13:80. doi:10.1186/s12957-015-0501-1
53. Zhang B, Zhang K, Liu Z, et al. Secreted Clusterin gene silencing enhances chemosensitivity of a549 cells to cisplatin through AKT and ERK1/2 pathways in vitro. Cell Physiol Biochem. 2014;33(4):1162–1175. doi:10.1159/000358685
54. Wang X, Zou F, Zhong J, et al. Secretory Clusterin mediates oxaliplatin resistance via the Gadd45a/PI3K/Akt signaling pathway in hepatocellular carcinoma. J Cancer. 2018;9(8):1403–1413. doi:10.7150/jca.23849
55. Zhong J, Yu X, Dong X, et al. Downregulation of secreted Clusterin potentiates the lethality of sorafenib in hepatocellular carcinoma in association with the inhibition of ERK1/2 signals. Int J Mol Med. 2018;41(5):2893–2900. doi:10.3892/ijmm.2018.3463
56. Lamoureux F, Baud’huin M, Ory B, et al. Clusterin inhibition using OGX-011 synergistically enhances zoledronic acid activity in osteosarcoma. Oncotarget. 2014;5(17):7805–7819. doi:10.18632/oncotarget.2308
57. Matsumoto H, Yamamoto Y, Shiota M, et al. Cotargeting androgen receptor and Clusterin delays castrate-resistant prostate cancer progression by inhibiting adaptive stress response and AR stability. Cancer Res. 2013;73(16):5206–5217. doi:10.1158/0008-5472.CAN-13-0359
58. Kususda Y, Miyake H, Gleave ME, et al. Clusterin inhibition using OGX-011 synergistically enhances antitumour activity of sorafenib in a human renal cell carcinoma model. Br J Cancer. 2012;106(12):1945–1952. doi:10.1038/bjc.2012.209
59. Laskin JJ, Nicholas G, Lee C, et al. Phase I/II trial of custirsen (OGX-011), an inhibitor of Clusterin, in combination with a gemcitabine and platinum regimen in patients with previously untreated advanced non-small cell lung cancer. J Thorac Oncol. 2012;7(3):579–586. doi:10.1097/JTO.0b013e31823f459c
60. Oncogenex Pharmaceuticals. TA Multinational, Randomized, Open-Label Study of Custirsen In Patients With Advanced or Metastatic (Stage IV) Non-Small Cell Lung Cancer. Available from:
61. Chi KN, Hotte SJ, Yu EY, et al. Randomized phase II study of docetaxel and prednisone with or without OGX-011 in patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(27):4247–4254. doi:10.1200/JCO.2009.26.8771
62. Chi KN, Higano CS, Blumenstein B, et al. Custirsen in combination with docetaxel and prednisone for patients with metastatic castration-resistant prostate cancer (SYNERGY trial): a phase 3, multicentre, open-label, randomised trial. Lancet Oncol. 2017;18(4):473–485. doi:10.1016/S1470-2045(17)30168-7
63. Peng M, Darko KO, Tao T, et al. Combination of metformin with chemotherapeutic drugs via different molecular mechanisms. Cancer Treat Rev. 2017;54:24–33. doi:10.1016/j.ctrv.2017.01.005
64. Deng J, Peng M, Wang Z, et al. Novel application of metformin combined with targeted drugs on anticancer treatment. Cancer Sci. 2018. doi:10.1111/cas.13849
65. Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi:10.1016/j.cell.2006.11.001
66. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895–904. doi:10.1038/nm1469
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.