")
Back to Journals » Journal of Inflammation Research » Volume 15
The Role of Clinical Features and Serum Biomarkers in Identifying Patients with Incomplete Lupus Erythematosus at Higher Risk of Transitioning to Systemic Lupus Erythematosus: Current Perspectives
Authors Sternhagen E, Bettendorf B , Lenert A, Lenert PS
Received 18 November 2021
Accepted for publication 2 February 2022
Published 18 February 2022 Volume 2022:15 Pages 1133—1145
DOI https://doi.org/10.2147/JIR.S275043
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Erin Sternhagen, Brittany Bettendorf, Aleksander Lenert, Petar S Lenert
Division of Immunology, Department of Internal Medicine, Carver College of Medicine, The University of Iowa, Iowa City, IA, 52242, USA
Correspondence: Petar S Lenert, Clinical Professor of Medicine, C428-2GH, 200 Hawkins Drive, Iowa City, Iowa City, 52242, USA, Email [email protected]
Abstract: Discovery of antinuclear antibodies (ANA) enabled earlier diagnosis of systemic lupus erythematosus (SLE) and other ANA+ connective tissue diseases (CTD). Rheumatologists increasingly encounter high referral volume of ANA+ patients. It has been estimated that only a small percentage of these patients will eventually transition to either SLE or other specified CTD. Incomplete lupus erythematosus (ILE) has been defined as a subset of patients who have some SLE-specific clinical manifestations but do not meet currently accepted classification criteria for SLE. Several studies have been performed with the goal of identifying clinical features, serum and tissue biomarkers that can distinguish those patients with ILE at risk of transitioning to SLE from those who will not. Increased autoantibody diversity, presence of anti-double-stranded DNA (dsDNA) antibodies, high expression of type I and type II interferon (IFN)-gene products, increased serum levels of B-cell-activating factor of the TNF family (BAFF), and certain serum cytokines and complement products have been identified as markers with positive predictive value, particularly when combined together. Once this patient population is better characterized biochemically, clinical trials should be considered with the primary objective to completely halt or slow down the transition from ILE to SLE. Hydroxychloroquine (HCQ) appears to be a promising agent due to its good tolerability and low toxicity profile and open-label studies in ILE patients have already shown its ability to delay the onset of SLE. Other therapeutics, like those targeting abnormal type I and type II IFN-signatures, B-cell specific signaling pathways, complement activation pathways and high BAFF levels should also be evaluated, but the risk to benefit ratio must be carefully determined before they can be considered.
Keywords: incomplete lupus erythematosus, systemic lupus erythematosus, hydroxychloroquine, type I interferon pathway, B cell activating factor of the TNF family
Introduction
SLE is a highly heterogeneous multisystemic polygenic autoimmune disease with unpredictable disease flares and highly variable organ involvement. The spectrum of SLE manifestations ranges from easily recognizable multiorgan disease involvement to cases of isolated central nervous system (CNS) or renal disease wherein a whole spectrum of differentials must be first excluded before a diagnosis of SLE can be entertained.
SLE is characterized by the presence of a myriad of autoantibodies, including those targeting a variety of nuclear antigens. It is estimated that between 100–200 different autoantibodies can be detected in SLE.1 SLE has a clear predilection for females compared to males (9:1 ratio) and can present in a very diverse way by affecting multiple organ systems. SLE disease activity tends to involve flares followed by periods of relative quiescence of disease/inactivity. At disease onset, the most observed clinical manifestations are those of mucocutaneous involvement, inflammatory polyarthritis, and hematologic abnormalities.2 However, some SLE-specific manifestations, such as malar rash or discoid rash, greatly vary from cohort to cohort and are observed only in a small percentage of newly diagnosed SLE patients, which makes the early diagnosis of SLE challenging. While at disease onset a high percentage of SLE patients (95–100%) test positive for anti-nuclear antibodies, early diagnosis of SLE remains a challenge.2,3
The exact etiology of SLE remains unknown. Multiple factors have been considered to play a role in susceptibility to SLE including genetic, epigenetic, and environmental risk factors (including but not limited to infections such as with Epstein-Barr virus, smoking, ultraviolet light, certain drugs, vitamin D deficiency, and hormonal factors).4,5 An abnormally high type I and type II IFN signature can be detected before SLE is diagnosed and this finding correlates with a break in tolerance resulting in emergence of autoreactive B cells6 (Figure 1).
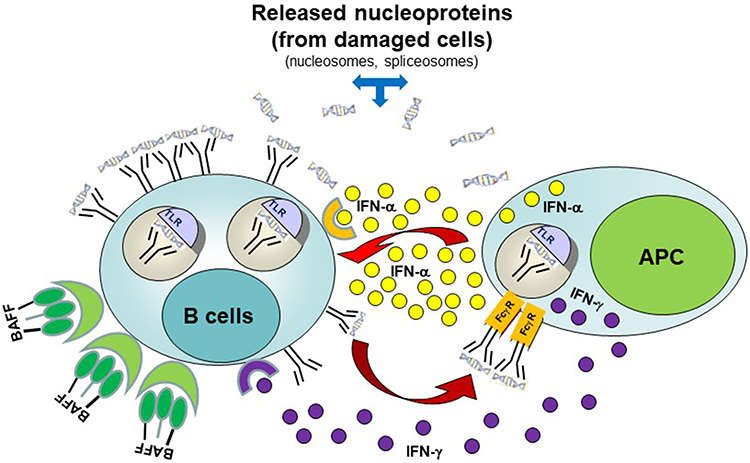 |
Figure 1 Interaction between autoreactive B-cells and antigen-presenting cells (APC) in ILE. Autoantigen (nucleoprotein)-specific B-cells may survive the selection processes in bone marrow. These cells are constantly exposed to autoantigens derived from either apoptotic cells, necrotic cells, or neutrophil extracellular traps. In genetically predisposed individuals, elevated levels of B-cell survival factors (BAFF or April) or APC (antigen-presenting cells)-derived type I (or type II) IFN, or abnormal intrinsic B-cell receptor signaling involving the Syk/BTK pathway may lead to upregulation of nucleic-acid sensing TLR and other innate receptors resulting in B-cell activation. In turn, this can lead to activation and maturation of autoreactive B-cells into anti-nucleoprotein secreting plasmablasts/plasma cells. Secreted autoantibodies can then combine with self-derived nucleoproteins and signal APCs through their Fc-γ receptors. Subsequent activation of innate receptors in APCs can trigger robust type I and II IFN response, which in turn can further decrease the threshold for B-cell activation by up-regulating IFN-inducible genes (“IFN-signature”). Released type I and type II IFN can also prime other immune cells and ultimately cause tissue inflammation and damage resulting in clinically overt SLE. Adapted fromJ Allergy Clin Immunol. 137(5), Singh N, Kumar B, Aluri V, Lenert P. Interfering with baffled B cells at the lupus tollway: promises, successes, and failed expectations. 1325–1333. Copyright 2016, with permission from Elsevier.91 |
Genetic Background of SLE
Regarding the possible role of genetics in SLE, it has been estimated that SLE rates range from 24–56% in monozygotic twins, while in dizygotic twins the concordance rate drops down to only 2–4%.7 Familial linkage studies have identified candidate genes involved in innate and adaptive immunity which were associated with increased risk of SLE.8 The first published SLE genome-wide association scan (GWAS) was performed on SLE patients of European ancestry9 and the results were later confirmed in 2 additional studies.10,11 The strongest associations were found between SLE and the Human leukocyte antigen (HLA) region on chromosome 6p21 and the IRF5 on chromosome 7q32. Several single-nucleotide polymorphisms (SNP) on chromosome 8p23.1, 8p21.1 and Lyn on 8q12 have also been shown to be associated with SLE. For example, on chromosome 8p23.1, the region upstream of the transcription-initiation site of the gene encoding C8orf13-B lymphocyte kinase (BLK) was associated with increased disease risk in both US and Swedish populations (rs13277113; odds ratio, 1.39; P=1x10 −10).11 The risk allele was in the promoter-region causing reduced expression of BLK and increased expression of C8orf12 in transformed B cell lines.12
However, common disease alleles identified by SNP-based GWAS studies only account for a small proportion of SLE heritability. GWAS studies have also frequently missed low-frequency functional risk alleles; for example, the recently identified Death-associated protein 1 regulatory haplotype has been linked to low transcription associated with higher autoantibody levels and enhanced autophagy which allows for the survival of autoreactive B-cells.13
Therefore, while inheritance definitively plays a role in susceptibility to SLE, the value of genetic testing in identifying patients who are at risk of developing SLE is limited and currently not recommended for either diagnostic, prognostic, or therapeutic purposes.
Pathophysiology of SLE
Discovery of Lupus Erythematosus cells (LE cells) followed by identification of antinuclear antibodies (ANA) by complement fixation assay or indirect immunofluorescence on cryogenically frozen tissue specimens (liver, kidney) or immortalized cell lines (such as Hep-2 cells) has tremendously helped in recognizing evolving SLE and other systemic rheumatologic diseases.14–16 Therefore, ANA has been included as the entry criterion for SLE classification in the 2019 European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) classification criteria.
However, ANA positivity is fairly common even in healthy individuals. Many patients are referred to rheumatology to evaluate for SLE in the setting of a positive ANA but lack any other critical (SLE-specific) diagnostic criteria. It has been estimated that despite its high sensitivity (98%), the utility of ANA for diagnosing SLE is limited by its much lower specificity (75–80%). Up to 20–25% of healthy subjects (women in particular) can have a positive ANA test and ANA positivity also increases with age. It has been suggested that the clinical utility of this test highly depends on its positive predictive value (PPV), for example ANA ordered by rheumatologists tends to have a higher PPV than ANA ordered by non-rheumatologists.17 One study evaluated the utility of ANA in 232 patients referred to Rheumatology for a positive ANA. The PPV for SLE in this cohort was 2.1% and for any ANA-associated rheumatic disease it was 9.1%; in other words, more than 90% of patients in the study who were referred to a tertiary rheumatology clinic for a positive ANA test had no evidence of ANA-associated rheumatic disease.18 Thus, the utility of the ANA test can be improved by selectively ordering it only in patients with higher PPV.
Early mediators in SLE include IFN-inducible gene expression, IFN-γ induced protein 10 (IP-10) and BAFF. Treatment of mice predisposed to develop a lupus-like disease with type I IFN can accelerate the course of disease.19 Treatment of humans with type I IFN has been occasionally linked to the induction of a lupus-like disease.20,21 The question remains: where is IFN-α coming from in patients with SLE? Herpes simplex virus–containing immune complexes stimulated the production of type I IFN in rabbits.22 Similarly, nucleic acid–containing immune complexes could induce type I IFN secretion in mouse and human dendritic cells. This effect was believed to involve Toll-like receptors (TLR) 7 and 9.23 Once released, IFN-α could amplify this response by upregulating IFN-responsive genes (and thus creating an “IFN signature”). Ronnblom and his colleagues pioneered this work and observed that nucleic acid-containing immune complexes in sera of patients with SLE could induce type I IFN production in peripheral blood mononuclear cells obtained from healthy individuals.24,25 Furthermore, type I IFN from sera of patients with SLE could promote differentiation of normal monocytes into dendritic cells (DC).26 Interestingly, type I IFN further enhanced B-cell activation via BAFF.27
Transitioning from ILE/UCTD to SLE
Prompt diagnosis of SLE and timely institution of treatment is imperative to limit disease progression, organ damage and mortality. Failure to recognize early progressive forms of ILE may have potentially dangerous consequences, however, not all patients will progress to SLE. Due to a variety of presenting symptoms and inconsistent presence of biomarkers, early diagnosis of SLE still remains elusive.28
The term ILE has been proposed to describe patients who meet some criteria, but do not fulfill the SLE classification criteria. Similarly, the term Undifferentiated CTD (UCTD) was proposed by LeRoy in 1980 to describe patients with positive ANA and non-specific symptoms typically lasting for more than 12 months who do not meet criteria for diagnosis of any specific CTD (including SLE). Many of these patients never develop a definite CTD.29,30 For the purposes of this paper, we are considering the terms UCTD and ILE to be synonymous.
Patients with ILE may present with a wide spectrum of disease manifestations, ranging from photosensitivity and mild skin disease to severe life-threatening renal involvement. Moreover, the timing of symptom onset and the rate of progression of symptoms is highly variable between patients. For example, the time interval between the first manifestations attributable to SLE and final classification of SLE has been estimated to be anywhere between 24 and 50 months.2,31,32 Decreased time interval to diagnosis was associated with better quality of life and improved survival.33,34
Autoantibody production and other immune abnormalities may precede the development of clinically active disease for months to years, thus creating another layer of complexity when approaching patients referred to Rheumatology for positive ANA. Many of the patients who are referred for evaluation of a positive ANA will have nonspecific symptoms, often widespread myofascial pain, and will not meet the classification criteria for SLE (or other systemic autoimmune disease) diagnosis at initial assessment. Most of these patients will never go on to develop clinically significant rheumatologic disease. Unfortunately, there is a scarcity of studies addressing the transition of patients with high-positive ANA titers into clinically significant CTD. One study showed that only 5 out of 27 patients who responded to a survey and had their blood retested for ANA developed a CTD after 10 years of follow-up.35 Interestingly, 21 of 27 remained ANA positive. Retrospective studies have shown that 63–88% of SLE patients will have serum autoantibodies years before they can be classified as having SLE.36,37 Anti-Ro/SS-A has been identified as one of the earliest autoantibodies detectable in ILE patients, followed by anti-dsDNA and anti-spliceosome antibodies.38 Autoantibody diversity tends to increase closer to the time of SLE diagnosis. Anti-Ro/SS-A antibody particularly targeting the 60 kD antigen is relatively common in SLE and tends to be associated with photosensitivity, cutaneous lupus, and hematologic manifestations.39 An Italian inception cohort study on SLE patients with disease duration less than 12 months has established that SS-A antibody can be detected in 28% of early SLE patients.2 However, its sole presence is not sufficient to predict the future transition to SLE.
Multiple studies have attempted to identify symptoms and biologic markers which may be used to stratify risk for progression to SLE. It is estimated that between 5–55% of ILE patients will eventually transition to definite CTD or SLE, however, the majority of these patients will never transition to overt CTD or SLE.29,40–45 It is therefore extremely important to stratify the risk for disease progression from ILE to SLE. Low-risk patients may require less frequent follow-ups, while those in the high-risk category should be monitored more frequently, and even considered for earlier treatment.
It remains challenging to distinguish patients who will evolve into having a definitive CTD or SLE versus those who will remain with clinically “stable, non-progressive ILE”. Non-progressing patients with ILE tend to have absence of severe organ involvement and have a relatively simple serologic profile. In contrast, those who tend to progress to SLE characteristically not only have a positive ANA, but frequently will have multiple other specific autoantibodies (such as anti-dsDNA, anti-proliferating cell nuclear antigen, anti-C1q, anti-cardiolipin, etc.). These patients also tend to have more clinical symptoms of SLE and tend to have higher ANA titers at baseline.46 Autoreactive IgM autoantibodies are more commonly observed in patients with ILE than in SLE.46 Disease transition rate is higher in ANA+ African American women compared to ANA+ European American (white) women. It has been estimated that 20–60% of those patients with ILE who will ultimately evolve into SLE will do so within 1–5 years of follow up.47 Some studies have shown oral ulcers, the presence of anti-dsDNA antibodies and proteinuria are independent predictors of developing future SLE, but unfortunately other studies have failed to identify any disease features that are predictive of eventual progression to SLE.29,41,48 Among other factors, the diagnosis of ILE vs SLE also depends on which classification criteria is used. The 2012 Systemic Lupus Erythematosus International Collaborating Clinics (SLICC) criteria appear to be more sensitive for diagnosing SLE (and thus more inclusive for clinical trials) than the widely used EULAR/ACR criteria.49 Regardless of classification criteria used, patients with SLE when compared to ILE have been shown to have higher incidence of malar rash, photosensitivity, oral ulcers, serositis, hematologic disorders, and immunologic disorders. Additionally, SLE patients have higher rates of renal and neurologic dysfunction (Table 1).
 |
Table 1 Comparison of Features in SLE versus ILE in ACR-1997 Criteria and SLICC Criteria (Olsen NJ et al, 201689) |
Biomarkers of ILE/SLE
It appears that patients with SLE vs ILE display different immunopathogenic patterns. Various biomarkers have been studied, including complement levels, soluble mediators, urinary mediators, and interferons (IFN) to better distinguish these two categories of patients. One potential biomarker of interest identified is the B-lymphocyte stimulator, also known as B-cell-activating-factor (BAFF), a soluble mediator belonging to the TNF cytokine family, which plays a crucial role in B cell survival and differentiation to plasma cells. Elevated levels of BAFF have been linked to survival of anergic autoreactive B-cells, thus prompting a breach in tolerance. This has been clearly implicated in autoantibody production and subsequent development of autoimmune disease in various mouse models.50 BAFF has been shown to be elevated at the time of diagnosis of SLE and has been used to predict possible future SLE flares. Specifically, in relation to ILE and SLE, BAFF levels in ILE patients have been shown to be higher than the general population, but lower than in SLE patients.49 The question remains whether targeting ILE patients expressing higher levels of BAFF with monoclonal anti-BAFF therapeutics, such as belimumab or blisibimod, may be used to prevent their evolution into full-blown SLE (assuming high benefit to risk ratio).51 Belimumab has been approved in 2011 for treatment of SLE, but at this time no clinical study is planned to address this important question.52
Common immune characteristics of SLE patients are increased expression of both type I and II IFN pathways along with evidence of T cell expansion. ANA+ healthy African Americans tend to express higher levels of T cell activation markers and plasma IL-6 concentrations compared to ANA+ healthy European American (white) counterparts. In contrast, CD11c+autoimmunity-associated B-cells were found to be lower in healthy ANA+ European Americans.53
A study of the healthy blood relatives of patients with SLE (n=409) who were followed for 6.4 years identified 45 patients (11%) who transitioned to classified SLE.54 Those patients who transitioned to SLE tended to have more SLE-associated antibodies at baseline, higher SLE activity scores, elevated inflammatory cytokines (BAFF, stem cell factor, etc.) and IFN-associated chemokines. Not unexpectedly, regulatory cytokines such as IL-10 and TGF-β tended to be lower over time. Plasma levels of stem cell factor and TGF-β appeared to be significant and independent predictors of SLE transition.
ILE and SLE patients showcase distinct patterns of biomarker positivity. High serum levels of IFN-α and evidence of high expression of type I IFN related genes (IFN signature) are found in approximately 50% of SLE patients.55–64 ILE patients, on the contrary, tend to display lower levels of type I IFN-inducible genes (specifically, IP-10, MCP-1, and MIP1-alpha) compared to patients with SLE. IFN-γ and BAFF have been found to increase around the time of diagnosis of SLE. Healthy first-degree relatives of patients with SLE, interestingly, also display high type I IFN activity probably related to IFN-regulatory factor (IRF)-5 and IRF-7 expression.65,66 The odds of progression to well defined autoimmune disease (SLE or primary Sjogren’s syndrome) was higher in patients with a family history of autoimmune rheumatic disease and in those with IFN-stimulated gene expression score (IFN-Score B).67 Others have confirmed that IFN score was elevated in ILE (in 50% of ILE patients compared to 46% of SLE patients) and its expression correlated with the Myxovirus-resistance protein A expression in blood and in unaffected skin (found in 29% of ILE skin biopsies vs 31% of SLE skin samples).68
Disease severity in SLE also tends to correlate with high vs low serum levels of IFN-α. A rare haplotype involving the promoter region of EFNA5 gene particularly, and to some degree PPM1H, PTPRM and NRGN regions, has been associated with high IFN-α levels.69
Single cell RNA sequencing applied to renal biopsies in patients with SLE nephritis helped identify potential new targets for treatment in SLE further confirming the role of type I IFN-response signatures in tubular cells.70
In addition to serum IFN-α and Interferon-γ inducible protein 10 (IP-10) levels, monocyte-specific surface sialic acid binding Ig-like lectin 1 (SIGLEC-1 protein) and its circulating soluble form correlate well with type I interferon-regulated gene expression in patients with SLE71 and appear to be good biomarkers for monitoring SLE disease activity.72 Higher concentrations of soluble SIGLEC-1 were particularly seen in patients of European origin with lower serum C3 levels and with higher incidence of renal disease.73
Immune Dysregulation Preceding the Development of SLE (Table 2)
Amplified crosstalk between innate and adaptive immunity is felt to cause a break of tolerance leading to survival and subsequent activation of autoreactive B cells.74,75 Numerous cytokines have been implicated as key mediators of this crosstalk including IFN-γ (Th1), interleukin (IL)-4 and IL-5 (Th2), IL-17 and IL-21 (Th17) which help facilitate formation of germinal centers and pathogenic autoantibody production with help from T-follicular cells.62
 |
Table 2 Biomarkers Associated with Higher Risk of Progression to SLE |
IFN-γ becomes elevated prior to SLE-associated autoantibodies, and thus may be a nice biomarker of early disease.62 As already discussed above, BAFF is upregulated in response to type I and type II IFNs and further propagates autoantibody production.50 Not only do these mediators drive the production of pathogenic autoantibodies, but they also contribute to inflammation preceding a flare of active SLE and contribute to organ damage in SLE.76,77 Age-associated B-cells (defined as CD19+CD21lowCD11c+) correlated with IgG levels and together with serum BAFF and IFN scores were elevated in patients with ILE compared with healthy controls.78 However, we still do not fully understand the role of these mediators in transition to SLE.62
Lu et al found that patients who went on to develop SLE, exhibited increased inflammatory mediators from multiple immune pathways more than 3.5 years pre-diagnosis. Implicated inflammatory mediators included IL-4, IL-5, IL-6, IL-12p70, IFN-γ, and the IFN-associated chemokine IFN-γ-inducible protein 10.62 Levels of IL-2 (p=0.008), IL-5 (p=0.001), and IL-21 (p=0.007) increased throughout the pre-classification period in case patients, while healthy controls showed low and stable levels of these mediators.62 These mediators remained dysregulated throughout the preclinical period with additional immune dysregulation observed as patients approached disease diagnosis. Additionally, investigators found lower levels of the regulatory mediator TGF-β in cases who went on to develop SLE compared to controls at all time periods, with no significant longitudinal changes in either group.62 Notably, cases who moved toward SLE classification gained an average of 0.5 dysregulated mediators per year, compared to 0.06 in controls (p<0.001).62 IFN activity was particularly notable with cases showing evidence of expanding IFN activity and increasing levels of the IFN-associated mediators IP-10 (p<0.001) and monokine-induced by IFN-γ (MIG) (p<0.001).62 TNFRI, TNFRII, BAFF, and APRIL were dysregulated only as patients approached SLE classification, suggesting these are later mediators of the evolving pathologic process.62 Lu et al implicated dysregulated innate and adaptive mediators superimposed on a background of deficient regulatory mechanisms in the pathogenesis of early preclinical SLE.62 In random forest models, a combination of autoantibody positivity against ANA and SS-A along with elevated levels of IL-5, IL-6 and MIG was able to distinguish future SLE with 92% accuracy (compared to 78% accuracy with ANA alone).62
Attempts to stratify SLE patients according to inflammatory pathways and neutrophilic vs lymphocytic gene clusters involved in active disease may help characterize those patients who tend to have more progressive disease. For example, some data suggest that patients in the neutrophil-driven clusters have an increased risk of developing proliferative forms of lupus nephritis.79 The same strategy may help identify those patients who may benefit from anti-type I IFN receptor therapies versus those who better respond to B cell targeted therapies.
Complement System
Complement activation by in situ formed or circulating immune complexes plays an important role in the immunopathogenesis of SLE. In a retrospective study in patients with ILE meeting 1–3 ACR criteria, decreased C3 complement levels were more commonly observed in ILE progressors (25%) than in non-progressors (3%) with the caveat that only 8 patients showed evidence of progression from ILE to SLE.42 This observation was not confirmed in a different ILE cohort studied by Md Yusof MY.67
A recent study by Ramsey-Goldman et al found that cell-bound complement activation products in red blood cells and B-cells aided in diagnosis of SLE when combined with anti-dsDNA and other serum autoantibodies in multianalyte assay scores. In addition, patients with ILE who expressed these cell-bound products tended to more commonly evolve into SLE.80
Classification Criteria for Identifying Incomplete and Complete Cases: SLICC Criteria for SLE Classification May Identify a Subset of Patients Transitioning from ILE to SLE
Primary care physicians have an extremely important role in identifying patients not only with full blown SLE but also those with suspected ILE and referring them to practicing rheumatologists, nephrologists, or other specialists (eg, neuroimmunologists) for further work-up that may include organ biopsy when necessary to rule out possible SLE mimickers such as drug-induced lupus-like disease or infections.
The concepts of lupus diagnosis and classification are often used interchangeably in clinical practice and classification criteria are frequently used for making SLE diagnosis even though these two concepts are clearly separate entities. Classification criteria typically combine a very limited number of clinical features and laboratory tests with the primary aim of identifying a relatively homogenous group with a particular disease that can be compared in clinical trials, and thus not meant to be used for diagnostic purposes. In contrast, the diagnosis of SLE is individualized to a particular patient and accounts for a wider spectrum of information which frequently includes items not considered specific enough for classification criteria and thus tends to be more sensitive, but less specific.81 There are no agreed upon diagnostic criteria for SLE, and thus classification criteria are often used for diagnostic purposes.
Patients with SLE were traditionally classified based on fulfillment of the 1997 American College of Rheumatology (ACR) criteria, which required meeting 4 or more out of 11 clinical and/or serologic criteria.82,83 SLICC classification criteria were then developed and validated in 2012. SLICC criteria are more sensitive and equally specific for SLE compared to 1997-revised ACR criteria.84 Therefore, they tend to be more inclusive as they classify more patients with SLE but can still distinguish those with ILE.49 Of 440 patients meeting 3 ACR criteria, 149 patients met criteria for SLICC-SLE, while 291 did not meet SLICC classification criteria. This additional number was based on complement and individual autoantibody criteria. SLICC also classified more African American patients based on leukopenia/lymphopenia criterion. Those patients who had 3 ACR criteria (ILE) were further compared with those classified as having SLE based on SLICC criteria. Patients with ILE tended to be slightly older and presented with milder symptoms. They also rarely had ANA titers > 1280, had fewer autoantibody specificities (1.3 vs 2.6) and had lower serum BAFF levels.
Another study by Mosca et al used newly revised criteria for SLE and systemic sclerosis to classify 91 patients diagnosed with UCTD.47 They found 12 patients could be re-classified with SLE and 3 with systemic sclerosis.
Recently, a new European Alliance of Associations for Rheumatology (EULAR)/ACR classification criteria have been released with the aim to further improve sensitivity and specificity for SLE. In a validation cohort these criteria reached sensitivity of 96% and specificity of 93%. Entry criterion was the presence of ANA at >1:80 titer, while clinical and immunological features were organized in 10 domains and the total score had to be at least 10, with at least one clinical criterion required for classification. It yet remains to be determined whether this new SLE classification can better identify patients with ILE at increased risk of transitioning to SLE.85
Potential Treatment of ILE
An interesting study of anti-malarial use in patients with ILE (SMILE) is ongoing. It aims at understanding whether hydroxychloroquine (HCQ) can slow down the progression from ILE to full-blown SLE and will hopefully characterize candidate biomarkers that may help to make earlier treatment decisions in the future (ClinTrial.gov NCT03030118).86
HCQ has been used in an open-label study in the past to abrogate autoantibody production and delay progression of ILE into full blown SLE.87 It is an attractive medication due to its low risk of toxicity associated with long-term use. A recent small sample study in patients with ILE and new-onset SLE showed that HCQ lowered levels of IFN-inducible genes and serum BAFF level.88 There was also a trend toward lowering IP-10 levels. Another study by Olsen et al showed that treatment of ILE with HCQ resulted in better self-reported health status scores and lower expression profiles of IFN-inducible genes compared to ILE patients not treated with HCQ.89 This correlates with studies on the in vivo and in vitro mechanism of action of HCQ which has been associated with an overall reduction in IFN-α levels in patients with SLE.90,91 Patients treated with HCQ additionally had lower levels of anti-C1q antibodies and cytokine IL-9.89 However, a subset of patients who present with symptoms of ILE will never go on to develop severe disease or full-blown SLE. In these patients, the risks of long-term HCQ treatment may outweigh the benefits and therefore this subgroup remains to be identified and better characterized before the treatment is initiated.
Targeting type I and type II IFN pathways, neutralizing B-cell stimulatory and survival molecules BAFF and a proliferation-inducing ligand (APRIL), targeting B-cell intrinsic signaling pathways (eg, Bruton’s tyrosine kinase), blocking TLR 7/9 signaling or neutralizing IL-6 by IL-6 receptor blockade are all attractive targets not only for full-blown SLE, but potentially also for those ILE patients at risk of transitioning. However, one must carefully weigh the risks and benefits of these medications particularly in those ILE patients with non-specific musculoskeletal complaints, as all these agents can cause profound immunosuppression and increase the risk for life-threatening opportunistic infections. Additional concern comes from the ability of these agents to interfere with the post-vaccinal immune response, for example with the induction of neutralizing antibodies against the COVID-19 virus and other emerging pathogens, potentially blunting the response and causing inadequate immunoprotection.
Neutralizing Type I IFN as a Treatment for SLE
Based on evidence of increased type I IFN activity in patients with ILE and particularly in those with full blown SLE it is not a surprise that targeting this pathway with neutralizing antibodies has been attempted in several recent clinical trials.68 Rontalizumab is an antibody with activity against all type I IFN subtypes. Results of a Phase 1 study in patients with SLE showed good tolerability but a subsequent Phase 2 study (NCT00962832) did not reach the primary end point.92,93 Genentech discontinued further development of rontalizumab. Sifalimumab is another anti–IFN-α antibody that neutralizes most IFN-α subtypes. Results from a phase 1a study showed a dose-dependent inhibition of type I IFN–related mRNA expression associated with a trend towards improved clinical activity (NCT00299819).94 There was also a trend towards improvement in disease activity, but despite promising results, development of sifalimumab for SLE was stopped for strategic business reasons in 2015.95 IFN-α kinoid (IFN-K) is an interesting construct that contains inactivated IFN-α coupled to a protein keyhole limpet hemocyanin. The safety, immunogenicity, and biologic effects of active immunization with IFN-K in patients with SLE was studied and showed that this approach could result in decreased expression of IFN-inducible genes but no improvement in clinical activity has been observed.96 It remains to be determined if earlier treatment with any of these agents in patients with ILE showing evidence of increased type I or II IFN signature may prevent the future progression to SLE.
At this time, the only agent targeting the IFN pathway approved for treatment of SLE is anifrolimumab (SaphneloR) which targets IFN alpha receptors (IFNAR1). It is indicated for patients with moderate to severe SLE who are on standard therapy. It has not been studied in SLE patients with severe active SLE nephritis or severe CNS disease. Clinical trials have shown that anifrolimumab can significantly reduce the SLE disease activity (measured with BICLA score) at week 52 (TULIP-2 study, NCT02446899), primarily by improving the mucocutaneous and musculoskeletal symptomatology and decreasing the total corticosteroid dose.97 It would be of interest to see if the drug-manufacturer plans clinical trials in those ILE patients with high IFN signatures at higher risk of transitioning to SLE.
Conclusions
Multiple facets of SLE can make it difficult to diagnose, especially in cases with a relatively slow evolution. Identification of patients with ILE who are at higher risk of transition to SLE is critical so that these patients can be considered for earlier treatment before organ damage ensues. Over the last few decades, several groups have studied the potential predictive value of numerous immune biomarkers. While ANA has high sensitivity for SLE, its relatively low specificity generates a potential problem of over-diagnosing patients with non-specific musculoskeletal complaints of possibly having SLE. Therefore, a better understanding of lupus specific clinical features together with identification of disease specific biomarkers may help identify patients with ILE at risk of transitioning to SLE. Increased autoantibody diversity, presence of anti-dsDNA and other autoantibodies, increased expression of type I and type II IFN-gene products, BAFF serum levels, and other serum cytokines, together with the evidence of complement activation (including detection of membrane bound complement activation products) have been identified as markers with positive predictive value, particularly when combined. These biomarkers should be studied in a longitudinal manner in larger cohorts of ILE patients who are at increased risk of transitioning towards SLE. As immune biomarkers for transitioning from ILE to SLE are better understood, this will guide risk-benefit decisions about initiating early therapy.
Abbreviations
ACR, American College of Rheumatology; ANA, anti-nuclear antibodies; APC, antigen-presenting cells; APRIL, A proliferation-inducing ligand; BAFF, B cell activating factor of the TNF family; BLK, B lymphocyte kinase; CNS, central nervous system; CTD, connective tissue disease; EULAR, European Alliance of Associations for Rheumatology; GWAS, genome-wide association scan; HCQ, hydroxychloroquine; HLA, human leukocyte antigen; IFN, interferon; IFN-K, IFN-α kinoid; IL, interleukin; ILE, incomplete lupus erythematosus; IRF, interferon-regulatory factor; LE cells, lupus erythematosus cells; MIG, monokine induced by IFN-γ; PPV, positive predictive value; SIGLEC-1, sialic acid binding Ig-like lectin 1; SLE, Systemic Lupus Erythematosus; SLICC, Systemic Lupus Erythematosus International Collaborating Clinics; SNP, single-nucleotide polymorphism; TLR, toll-like receptors; UCTD, undifferentiated connective tissue disease.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Choi MY, Fritzler MJ. Autoantibodies in SLE: prediction and the p value matrix. Lupus. 2019;28(11):1285–1293. doi:10.1177/0961203319868531
2. Sebastiani GD, Prevete I, Piga M, et al. Early Lupus Project - A multicentre Italian study on systemic lupus erythematosus of recent onset. Lupus. 2015;24(12):1276–1282. doi:10.1177/0961203315585817
3. Nossent J, Kiss E, Rozman B, et al. Disease activity and damage accrual during the early disease course in a multinational inception cohort of patients with systemic lupus erythematosus. Lupus. 2010;19(8):949–956. doi:10.1177/0961203310366572
4. Poole BD, Templeton AK, Guthridge JM, Brown EJ, Harley JB, James JA. Aberrant Epstein-Barr viral infection in systemic lupus erythematosus. Autoimmun Rev. 2009;8(4):337–342. doi:10.1016/j.autrev.2008.12.008
5. Ritterhouse LL, Crowe SR, Niewold TB, et al. Vitamin D deficiency is associated with an increased autoimmune response in healthy individuals and in patients with systemic lupus erythematosus. Ann Rheum Dis. 2011;70(9):1569–1574. doi:10.1136/ard.2010.148494
6. Crow MK, Ronnblom L. Type I interferons in host defence and inflammatory diseases. Lupus sci med. 2019;6(1):e000336. doi:10.1136/lupus-2019-000336
7. Guerra SG, Vyse TJ, Cunninghame graham DS. The genetics of lupus: a functional perspective. Arthritis Res Ther. 2012;14(3):211. doi:10.1186/ar3844
8. Dai C, Deng Y, Quinlan A, Gaskin F, Tsao BP, Fu SM. Genetics of systemic lupus erythematosus: immune responses and end organ resistance to damage. Curr Opin Immunol. 2014;31:87–96. doi:10.1016/j.coi.2014.10.004
9. Harley JB, Alarcón-Riquelme ME, Criswell LA, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40(2):204–210. doi:10.1038/ng.81
10. Demirci FY, Wang X, Morris DL, et al. Multiple signals at the extended 8p23 locus are associated with susceptibility to systemic lupus erythematosus. J Med Genet. 2017;54(6):381–389. doi:10.1136/jmedgenet-2016-104247
11. Hom G, Graham RR, Modrek B, et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008;358(9):900–909. doi:10.1056/NEJMoa0707865
12. Guthridge JM, Lu R, Sun H, et al. Two functional lupus-associated BLK promoter variants control cell-type- and developmental-stage-specific transcription. Am J Hum Genet. 2014;94(4):586–598. doi:10.1016/j.ajhg.2014.03.008
13. Raj P, Song R, Zhu H, et al. Deep sequencing reveals a DAP1 regulatory haplotype that potentiates autoimmunity in systemic lupus erythematosus. Genome Biol. 2020;21(1):281. doi:10.1186/s13059-020-02184-z
14. Hargraves MM, Richmond H, Morton R. Presentation of two bone marrow elements; the tart cell and the L.E. cell. Proc Staff Meet Mayo Clin. 1948;23(2):25–28.
15. Holborow EJ, Weir DM, Johnson GD. A serum factor in lupus erythematosus with affinity for tissue nuclei. Br Med J. 1957;2(5047):732–734. doi:10.1136/bmj.2.5047.732
16. Robbins WC, Holman HR, Deicher H, Kunkel HG. Complement fixation with cell nuclei and DNA in lupus erythematosus. Proce Soc Exp Biol Med Soc Exp Biol Med. 1957;96(3):575–579. doi:10.3181/00379727-96-23545
17. Slater CA, Davis RB, Shmerling RH. Antinuclear antibody testing. A study of clinical utility. Arch Intern Med. 1996;156(13):1421–1425. doi:10.1001/archinte.1996.00440120079007
18. Abeles AM, Abeles M. The clinical utility of a positive antinuclear antibody test result. Am J Med. 2013;126(4):342–348. doi:10.1016/j.amjmed.2012.09.014
19. Heremans H, Billiau A, Colombatti A, Hilgers J, de Somer P. Interferon treatment of NZB mice: accelerated progression of autoimmune disease. Infect Immun. 1978;21(3):925–930. doi:10.1128/iai.21.3.925-930.1978
20. Rönnblom LE, Alm GV, Oberg KE. Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J Intern Med. 1990;227(3):207–210. doi:10.1111/j.1365-2796.1990.tb00144.x
21. Wilson LE, Widman D, Dikman SH, Gorevic PD. Autoimmune disease complicating antiviral therapy for hepatitis C virus infection. Semin Arthritis Rheum. 2002;32(3):163–173. doi:10.1053/sarh.2002.37277
22. Fujibayashi T, Hooks JJ, Notkins AL. Production of interferon by immune lymphocytes exposed to herpes simplex virus-antibody complexes. J Immunol. 1975;115(5):1191–1193.
23. Green NM, Marshak-Rothstein A. Toll-like receptor driven B cell activation in the induction of systemic autoimmunity. Semin Immunol. 2011;23(2):106–112. doi:10.1016/j.smim.2011.01.016
24. Vallin H, Blomberg S, Alm GV, Cederblad B, Rönnblom L. Patients with systemic lupus erythematosus (SLE) have a circulating inducer of interferon-alpha (IFN-alpha) production acting on leucocytes resembling immature dendritic cells. Clin Exp Immunol. 1999;115(1):196–202. doi:10.1046/j.1365-2249.1999.00772.x
25. Vallin H, Perers A, Alm GV, Rönnblom L. Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-alpha inducer in systemic lupus erythematosus. J Immunol. 1999;163(11):6306–6313.
26. Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294(5546):1540–1543. doi:10.1126/science.1064890
27. Le Bon A, Thompson C, Kamphuis E, et al. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J Immunol. 2006;176(4):2074–2078. doi:10.4049/jimmunol.176.4.2074
28. Lambers WM, Westra J, Bootsma H, de Leeuw K. From incomplete to complete systemic lupus erythematosus; A review of the predictive serological immune markers. Semin Arthritis Rheum. 2021;51(1):43–48. doi:10.1016/j.semarthrit.2020.11.006
29. Mosca M, Neri R, Bombardieri S. Undifferentiated connective tissue diseases (UCTD): a review of the literature and a proposal for preliminary classification criteria. Clin Exp Rheumatol. 1999;17(5):615–620.
30. LeRoy EC, Maricq HR, Kahaleh MB. Undifferentiated connective tissue syndromes. Arthritis Rheum. 1980;23(3):341–343. doi:10.1002/art.1780230312
31. Pistiner M, Wallace DJ, Nessim S, Metzger AL, Klinenberg JR. Lupus erythematosus in the 1980s: a survey of 570 patients. Semin Arthritis Rheum. 1991;21(1):55–64. doi:10.1016/0049-0172(91)90057-7
32. Wallace DJ, Podell T, Weiner J, Klinenberg JR, Forouzesh S, Dubois EL. Systemic lupus erythematosus–survival patterns. Experience with 609 patients. JAMA. 1981;245(9):934–938. doi:10.1001/jama.1981.03310340024021
33. Doria A, Iaccarino L, Ghirardello A, et al. Long-term prognosis and causes of death in systemic lupus erythematosus. Am J Med. 2006;119(8):700–706. doi:10.1016/j.amjmed.2005.11.034
34. Doria A, Rinaldi S, Ermani M, et al. Health-related quality of life in Italian patients with systemic lupus erythematosus. II. Role of clinical, immunological and psychological determinants. Rheumatology. 2004;43(12):1580–1586. doi:10.1093/rheumatology/keh392
35. Wijeyesinghe U, Russell AS. Outcome of high titer antinuclear antibody positivity in individuals without connective tissue disease: a 10-year follow-up. Clin Rheumatol. 2008;27(11):1399–1402. doi:10.1007/s10067-008-0932-y
36. Arbuckle MR, McClain MT, Rubertone MV, et al. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–1533. doi:10.1056/NEJMoa021933
37. Eriksson C, Kokkonen H, Johansson M, Hallmans G, Wadell G, Rantapää-Dahlqvist S. Autoantibodies predate the onset of systemic lupus erythematosus in northern Sweden. Arthritis Res Ther. 2011;13(1):R30. doi:10.1186/ar3258
38. McClain MT, Heinlen LD, Dennis GJ, Roebuck J, Harley JB, James JA. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med. 2005;11(1):85–89. doi:10.1038/nm1167
39. Yoshimi R, Ueda A, Ozato K, Ishigatsubo Y. Clinical and pathological roles of Ro/SSA autoantibody system. Clin Dev Immunol. 2012;2012:606195. doi:10.1155/2012/606195
40. Calvo-Alen J, Alarcon GS, Burgard SL, Burst N, Bartolucci AA, Williams HJ. Systemic lupus erythematosus: predictors of its occurrence among a cohort of patients with early undifferentiated connective tissue disease: multivariate analyses and identification of risk factors. J Rheumatol. 1996;23(3):469–475.
41. Al Daabil M, Massarotti EM, Fine A, et al. Development of SLE among “potential SLE” patients seen in consultation: long-term follow-up. Int J Clin Pract. 2014;68(12):1508–1513. doi:10.1111/ijcp.12466
42. Vilá LM, Mayor AM, Valentín AH, García-Soberal M, Vilá S. Clinical outcome and predictors of disease evolution in patients with incomplete lupus erythematosus. Lupus. 2000;9(2):110–115. doi:10.1191/096120300678828073
43. Ståhl Hallengren C, Nived O, Sturfelt G. Outcome of incomplete systemic lupus erythematosus after 10 years. Lupus. 2004;13(2):85–88. doi:10.1191/0961203304lu477oa
44. Swaak AJ, van de Brink H, Smeenk RJ, et al. Incomplete lupus erythematosus: results of a multicentre study under the supervision of the EULAR Standing Committee on International Clinical Studies Including Therapeutic Trials (ESCISIT). Rheumatology. 2001;40(1):89–94. doi:10.1093/rheumatology/40.1.89
45. Greer JM, Panush RS. Incomplete lupus erythematosus. Arch Intern Med. 1989;149(11):2473–2476. doi:10.1001/archinte.1989.00390110061013
46. Olsen NJ, Li QZ, Quan J, Wang L, Mutwally A, Karp DR. Autoantibody profiling to follow evolution of lupus syndromes. Arthritis Res Ther. 2012;14(4):R174. doi:10.1186/ar3927
47. Mosca M, Tani C, Vagnani S, Carli L, Bombardieri S. The diagnosis and classification of undifferentiated connective tissue diseases. J Autoimmun. 2014;48:50–52. doi:10.1016/j.jaut.2014.01.019
48. Ganczarczyk L, Urowitz MB, Gladman DD. Latent lupus. J Rheumatol. 1989;16(4):475–478.
49. Aberle T, Bourn RL, Chen H, et al. Use of SLICC criteria in a large, diverse lupus registry enables SLE classification of a subset of ACR-designated subjects with incomplete lupus. Lupus sci med. 2017;4(1):e000176. doi:10.1136/lupus-2016-000176
50. Cancro MP, D’Cruz DP, Khamashta MA. The role of B lymphocyte stimulator (BLyS) in systemic lupus erythematosus. J Clin Invest. 2009;119(5):1066–1073. doi:10.1172/JCI38010
51. Lenert A, Niewold TB, Lenert P. Spotlight on blisibimod and its potential in the treatment of systemic lupus erythematosus: evidence to date. Drug Des Devel Ther. 2017;11:747–757. doi:10.2147/DDDT.S114552
52. Furie R, Petri M, Zamani O, et al. A Phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011;63(12):3918–3930. doi:10.1002/art.30613
53. Slight-Webb S, Smith M, Bylinska A, et al. Autoantibody-positive healthy individuals with lower lupus risk display a unique immune endotype. J Allergy Clin Immunol. 2020;146(6):1419–1433. doi:10.1016/j.jaci.2020.04.047
54. Munroe ME, Young KA, Kamen DL, et al. Discerning Risk of Disease Transition in Relatives of Systemic Lupus Erythematosus Patients Utilizing Soluble Mediators and Clinical Features. Arthritis rheumatol. 2017;69(3):630–642. doi:10.1002/art.40004
55. Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979;301(1):5–8. doi:10.1056/NEJM197907053010102
56. Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8(6):492–502. doi:10.1038/sj.gene.6364408
57. Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100(5):2610–2615. doi:10.1073/pnas.0337679100
58. Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003;197(6):711–723. doi:10.1084/jem.20021553
59. Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. 2003;36(8):481–490. doi:10.1080/08916930310001625952
60. Kirou KA, Lee C, George S, et al. Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum. 2004;50(12):3958–3967. doi:10.1002/art.20798
61. Li QZ, Zhou J, Lian Y, et al. Interferon signature gene expression is correlated with autoantibody profiles in patients with incomplete lupus syndromes. Clin Exp Immunol. 2010;159(3):281–291. doi:10.1111/j.1365-2249.2009.04057.x
62. Lu R, Munroe ME, Guthridge JM, et al. Dysregulation of innate and adaptive serum mediators precedes systemic lupus erythematosus classification and improves prognostic accuracy of autoantibodies. J Autoimmun. 2016;74:182–193. doi:10.1016/j.jaut.2016.06.001
63. Kim T, Kanayama Y, Negoro N, Okamura M, Takeda T, Inoue T. Serum levels of interferons in patients with systemic lupus erythematosus. Clin Exp Immunol. 1987;70(3):562–569.
64. Munroe ME, Lu R, Zhao YD, et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann Rheum Dis. 2016;75(11):2014–2021. doi:10.1136/annrheumdis-2015-208140
65. Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum. 2008;58(8):2481–2487. doi:10.1002/art.23613
66. Salloum R, Franek BS, Kariuki SN, et al. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-alpha activity in lupus patients. Arthritis Rheum. 2010;62(2):553–561. doi:10.1002/art.27182
67. Md Yusof MY, Psarras A, El-Sherbiny YM, et al. Prediction of autoimmune connective tissue disease in an at-risk cohort: prognostic value of a novel two-score system for interferon status. Ann Rheum Dis. 2018;77(10):1432–1439. doi:10.1136/annrheumdis-2018-213386
68. Lambers WM, De leeuw K. Doornbos-van der Meer B, Diercks GFH, Bootsma H, Westra J. Interferon score is increased in incomplete systemic lupus erythematosus and correlates with myxovirus-resistance protein A in blood and skin. Arthritis Res Ther. 2019;21(1):260. doi:10.1186/s13075-019-2034-4
69. Ghodke-Puranik Y, Imgruet M, Dorschner JM, et al. Novel genetic associations with interferon in systemic lupus erythematosus identified by replication and fine-mapping of trait-stratified genome-wide screen. Cytokine. 2020;132:154631. doi:10.1016/j.cyto.2018.12.014
70. Der E, Suryawanshi H, Morozov P, et al. Tubular cell and keratinocyte single-cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol. 2019;20(7):915–927. doi:10.1038/s41590-019-0386-1
71. Biesen R, Demir C, Barkhudarova F, et al. Sialic acid-binding Ig-like lectin 1 expression in inflammatory and resident monocytes is a potential biomarker for monitoring disease activity and success of therapy in systemic lupus erythematosus. Arthritis rheumatol. 2008;58(4):1136–1145. doi:10.1002/art.23404
72. Rose T, Grützkau A, Klotsche J, et al. Are interferon-related biomarkers advantageous for monitoring disease activity in systemic lupus erythematosus? A longitudinal benchmark study. Rheumatology. 2017;56(9):1618–1626. doi:10.1093/rheumatology/kex220
73. Oliveira JJ, Karrar S, Rainbow DB, et al. The plasma biomarker soluble SIGLEC-1 is associated with the type I interferon transcriptional signature, ethnic background and renal disease in systemic lupus erythematosus. Arthritis Res Ther. 2018;20(1):152. doi:10.1186/s13075-018-1649-1
74. Datta SK. Production of pathogenic antibodies: cognate interactions between autoimmune T and B cells. Lupus. 1998;7(9):591–596. doi:10.1191/096120398678920703
75. Kil LP, Hendriks RW. Aberrant B cell selection and activation in systemic lupus erythematosus. Int Rev Immunol. 2013;32(4):445–470.
76. Munroe ME, James JA. Genetics of Lupus Nephritis: clinical Implications. Semin Nephrol. 2015;35(5):396–409. doi:10.1016/j.semnephrol.2015.08.002
77. Munroe ME, Vista ES, Guthridge JM, Thompson LF, Merrill JT, James JA. Proinflammatory adaptive cytokine and shed tumor necrosis factor receptor levels are elevated preceding systemic lupus erythematosus disease flare. Arthritis rheumatol. 2014;66(7):1888–1899. doi:10.1002/art.38573
78. Henning S, Lambers WM. Doornbos-van der Meer B, et al. Proportions of B-cell subsets are altered in incomplete systemic lupus erythematosus and correlate with interferon score and IgG levels. Rheumatology. 2020;59(9):2616–2624. doi:10.1093/rheumatology/keaa114
79. Toro-Domínguez D, Martorell-Marugán J, Goldman D, Petri M, Carmona-Sáez P, Alarcón-Riquelme ME. Stratification of systemic lupus erythematosus patients into three groups of disease activity progression according to longitudinal gene expression. Arthritis rheumatol. 2018;70(12):2025–2035. doi:10.1002/art.40653
80. Ramsey-Goldman R, Alexander RV, Massarotti EM, et al. Complement Activation in Patients With Probable Systemic Lupus Erythematosus and Ability to Predict Progression to American College of Rheumatology-Classified Systemic Lupus Erythematosus. Arthritis rheumatol. 2020;72(1):78–88. doi:10.1002/art.41093
81. Aringer M, Johnson SR. Classifying and diagnosing systemic lupus erythematosus in the 21st century. Rheumatology. 2020;59(Suppl5):v4–v11. doi:10.1093/rheumatology/keaa379
82. Tan EM, Cohen AS, Fries JF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25(11):1271–1277. doi:10.1002/art.1780251101
83. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi:10.1002/art.1780400928
84. Petri M, Orbai AM, Alarcón GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677–2686. doi:10.1002/art.34473
85. Aringer M, Costenbader K, Daikh D, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. 2019;78(9):1151–1159. doi:10.1136/annrheumdis-2018-214819
86. Olsen NJ, James JA, Arriens C, et al. Study of Anti-Malarials in Incomplete Lupus Erythematosus (SMILE): study protocol for a randomized controlled trial. Trials. 2018;19(1):694. doi:10.1186/s13063-018-3076-7
87. James JA, Kim-Howard XR, Bruner BF, et al. Hydroxychloroquine sulfate treatment is associated with later onset of systemic lupus erythematosus. Lupus. 2007;16(6):401–409. doi:10.1177/0961203307078579
88. Lambers WM, Westra J, Bootsma H, de Leeuw K. Hydroxychloroquine Suppresses Interferon-inducible Genes and B Cell Activating Factor in Patients With Incomplete and New-onset Systemic Lupus Erythematosus. J Rheumatol. 2021;48(6):847–851. doi:10.3899/jrheum.200726
89. Olsen NJ, McAloose C, Carter J, et al. Clinical and Immunologic Profiles in Incomplete Lupus Erythematosus and Improvement with Hydroxychloroquine Treatment. Autoimmune Dis. 2016;2016:8791629. doi:10.1155/2016/8791629
90. Sacre K, Criswell LA, McCune JM. Hydroxychloroquine is associated with impaired interferon-alpha and tumor necrosis factor-alpha production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res Ther. 2012;14(3):R155. doi:10.1186/ar3895
91. Singh N, Kumar B, Aluri V, Lenert P. Interfering with baffled B cells at the lupus tollway: promises, successes, and failed expectations. J Allergy Clin Immunol. 2016;137(5):1325–1333. doi:10.1016/j.jaci.2015.12.1326
92. Kalunian KC, Merrill JT, Maciuca R, et al. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis. 2016;75(1):196–202. doi:10.1136/annrheumdis-2014-206090
93. McBride JM, Jiang J, Abbas AR, et al. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: results of a Phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum. 2012;64(11):3666–3676. doi:10.1002/art.34632
94. Merrill JT, Wallace DJ, Petri M, et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon α monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann Rheum Dis. 2011;70(11):1905–1913. doi:10.1136/ard.2010.144485
95. Petri M, Wallace DJ, Spindler A, et al. Sifalimumab, a human anti-interferon-α monoclonal antibody, in systemic lupus erythematosus: a Phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 2013;65(4):1011–1021. doi:10.1002/art.37824
96. Lauwerys BR, Hachulla E, Spertini F, et al. Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon α-kinoid. Arthritis Rheum. 2013;65(2):447–456. doi:10.1002/art.37785
97. Morand EF, Furie R, Tanaka Y, et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med. 2020;382(3):211–221. doi:10.1056/NEJMoa1912196
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.